Early Stage Glycosylation Biomarkers in Alzheimer’s Disease

Abstract
:1. Introduction
1.1. Alzheimer’s Disease (AD)—A Cause for Concern
1.2. AD Pathogenesis
1.3. Treatments for AD and the Urgency of An Early Diagnosis
1.4. AD Diagnosis—Invasive and Inconclusive
2. Glycosylation Overview
2.1. What is Glycosylation?
2.2. Approaches to Glycoanalysis
2.3. Glycosylation and Disease
3. Glycosylation and AD
3.1. Glycosylation of Proteins Implicated in AD in the Brain
3.2. Glycosylation Biomarkers of AD in the CSF and Blood
3.3. The Direct and Indirect Impact of BACE1 Glycosylation in AD Pathology
3.4. APP Glycans as Protective Mechanisms in AD
3.5. Tau Phosphorylation is Directed by its Glycosylation
3.6. Presenilin and Transmembrane Protein 59 (TMEM59) are Regulators of Protein Glycosylation
3.7. Apolipoprotein Glycosylation Changes in AD
3.8. AD-Associated Glycosylation Changes to Transferrin are Different in CSF to Serum
3.9. Other Glycoproteins Associated with AD Pathology
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Morris, M.C. The role of nutrition in Alzheimer’s disease: Epidemiological evidence. Eur. J. Neurol. 2009, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dementia. Available online: http://www.who.int/en/news-room/fact-sheets/detail/dementia (accessed on 1 March 2018).
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. What Is Alzheimer’s? Available online: https://www.alz.org/alzheimers_disease_what_is_alzheimers.asp (accessed on 15 January 2018).
- Montgomery, W.; Goren, A.; Kahle-Wrobleski, K.; Nakamura, T.; Ueda, K. Detection, diagnosis, and treatment of Alzheimer’s disease dementia stratified by severity as reported by caregivers in Japan. Neuropsychiatr. Dis. Treat. 2018, 14, 1843–1854. [Google Scholar] [CrossRef]
- Musicco, M.; Palmer, K.; Salamone, G.; Lupo, F.; Perri, R.; Mosti, S.; Spalletta, G.; di Iulio, F.; Pettenati, C.; Cravello, L.; et al. Predictors of progression of cognitive decline in Alzheimer’s disease: The role of vascular and sociodemographic factors. J. Neurol. 2009, 256, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Gotz, J. Amyloid-beta and tau complexity—Towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Waser, M.; Garn, H.; Schmidt, R.; Benke, T.; Dal-Bianco, P.; Ransmayr, G.; Schmidt, H.; Seiler, S.; Sanin, G.; Mayer, F.; et al. Quantifying synchrony patterns in the EEG of Alzheimer’s patients with linear and non-linear connectivity markers. J. Neural Transm. 2016, 123, 297–316. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Sabatini, B.; Südhof, T.C. Synapses and Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2012, 4, a005777. [Google Scholar] [CrossRef] [PubMed]
- Mora-Bermúdez, F.; Badsha, F.; Kanton, S.; Camp, J.G.; Vernot, B.; Köhler, K.; Voigt, B.; Okita, K.; Maricic, T.; He, Z.; et al. Differences and similarities between human and chimpanzee neural progenitors during cerebral cortex development. ELife 2016, 5, e18683. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S. Hippocampus-dependent Task Improves the Cognitive Function after Ovariectomy in Rats. Osong Public Health Res. Perspect. 2017, 8, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Ridha, B.H.; Barnes, J.; Bartlett, J.W.; Godbolt, A.; Pepple, T.; Rossor, M.N.; Fox, N.C. Tracking atrophy progression in familial Alzheimer’s disease: A serial MRI study. Lancet Neurol. 2006, 5, 828–834. [Google Scholar] [CrossRef]
- Frisoni, G.B.; Fox, N.C.; Jack, C.R.; Scheltens, P.; Thompson, P.M. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef]
- Malik, G.A.; Robertson, N.P. Treatments in Alzheimer’s disease. J. Neurol. 2017, 264, 416–418. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef]
- Coimbra, J.R.M.; Marques, D.F.F.; Baptista, S.J.; Pereira, C.M.F.; Moreira, P.I.; Dinis, T.C.P.; Santos, A.E.; Salvador, J.A.R. Highlights in BACE1 Inhibitors for Alzheimer’s Disease Treatment. Front. Chem. 2018, 6, 1–10. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Gilman, S.; Fox, N.C.; Blennow, K.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Doody, R.; van Dyck, C.H.; et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 2009, 73, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Black, R.S.; Sperling, R.A.; Safirstein, B.; Motter, R.N.; Pallay, A.; Nichols, A.; Grundman, M. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Staff, R.T.; Wischik, D.J.; Bentham, P.; Murray, A.D.; Storey, J.M.; Kook, K.A.; Harrington, C.R. Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.S.; Wang, Y.Y.; Song, J.T.; Yu, J.H. Acylphloroglucinols as kinase inhibitors from Sargassum nigrifoloides. J. Asian Nat. Prod. Res. 2018, 21, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.; Anand, R.; Messina, J., Jr.; Hartman, R.; Veach, J. A 52-Week Study of the Efficacy of Rivastigmine in Patients with Mild to Moderately Severe Alzheimer’s Disease. Eur. Neurol. 2000, 44, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Almkvist, O.; Darreh-Shori, T.; Stefanova, E.; Spiegel, R.; Nordberg, A. Preserved cognitive function after 12 months of treatment with rivastigmine in mild Alzheimer’s disease in comparison with untreated AD and MCI patients. Eur. J. Neurol. 2004, 11, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Meraz-Ríos, M.A.; Franco-Bocanegra, D.; Toral Rios, D.; Campos-Peña, V. Early onset Alzheimer’s disease and oxidative stress. Oxidative Med. Cell. Longev. 2014, 2014, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sassi, C.; Guerreiro, R.; Gibbs, R.; Ding, J.; Lupton, M.K.; Troakes, C.; Lunnon, K.; Al-Sarraj, S.; Brown, K.S.; Medway, C.; et al. Exome sequencing identifies 2 novel presenilin 1 mutations (p. L166V and p. S230R) in British early-onset Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2422-e13. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Correia, S. Alzheimer disease: Time to improve its diagnosis and treatment. Clevel. Clin. J. Med. 2009, 76, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neugroschl, J.; Wang, S. Alzheimer’s Disease: Diagnosis and Treatment Across the Spectrum of Disease Severity. Mt. Sinai J. Med. N. Y. 2011, 78, 596–612. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.C.; Schott, J.M. Imaging cerebral atrophy: Normal ageing to Alzheimer’s disease. Lancet 2004, 363, 392–394. [Google Scholar] [CrossRef]
- Szot, P. Common factors among Alzheimer’s disease, Parkinson’s disease, and epilepsy: Possible role of the noradrenergic nervous system. Epilepsia 2012, 53, 61–66. [Google Scholar] [CrossRef]
- Cuttler, J.M.; Moore, E.R.; Hosfeld, V.D.; Nadolski, D.L. Treatment of Alzheimer Disease with CT Scans: A Case Report. Dose Response Int. J. 2016, 14, 1559325816640073. [Google Scholar] [CrossRef]
- van de Pol, L.A.; Hensel, A.; van der Flier, W.M.; Visser, P.J.; Pijnenburg, Y.A.L.; Barkhof, F.; Gertz, H.J.; Scheltens, P. Hippocampal atrophy on MRI in frontotemporal lobar degeneration and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2006, 77, 439–442. [Google Scholar] [CrossRef]
- Montoya, A.; Price, B.H.; Menear, M.; Lepage, M. Brain imaging and cognitive dysfunctions in Huntington’s disease. J. Psychiatry Neurosci. 2006, 31, 21–29. [Google Scholar]
- Cruz de Souza, L.; Chupin, M.; Bertoux, M.; Lehéricy, S.; Dubois, B.; Lamari, F.; Le Ber, I.; Bottlaender, M.; Colliot, O.; Sarazin, M. Is Hippocampal Volume a Good Marker to Differentiate Alzheimer’s Disease from Frontotemporal Dementia? J. Alzheimer’s Dis. 2013, 36, 57–66. [Google Scholar] [CrossRef]
- Fodero, L.R.; Sáez-Valero, J.; Barquero, M.S.; Marcos, A.; McLean, C.A.; Small, D.H. Wheat germ agglutinin-binding glycoproteins are decreased in Alzheimer’s disease cerebrospinal fluid. J. Neurochem. 2001, 79, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Schilling, L.P.; Zimmer, E.R.; Shin, M.; Leuzy, A.; Pascoal, T.A.; Benedet, A.L.; Borelli, W.V.; Palmini, A.; Gauthier, S.; Rosa-Neto, P. Imaging Alzheimer’s disease pathophysiology with PET. Dement. Neuropsychol. 2016, 10, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s disease. Clin. Nucl. Med. 2014, 39, e413–e426. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Berti, V.; Glodzik, L.; Pupi, A.; De Santi, S.; de Leon, M.J. Pre-Clinical Detection of Alzheimer’s Disease Using FDG-PET, with or without Amyloid Imaging. J. Alzheimer’s Dis. 2010, 20, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mucke, L. Alzheimer Mechanisms and Therapeutic Strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [Green Version]
- Rabinovici, G.D.; Jagust, W.J. Amyloid Imaging in Aging and Dementia: Testing the Amyloid Hypothesis In Vivo. Behav. Neurol. 2009, 21, 117–128. [Google Scholar] [CrossRef]
- Okamura, N.; Harada, R.; Ishiki, A.; Kikuchi, A.; Nakamura, T.; Kudo, Y. The development and validation of tau PET tracers: Current status and future directions. Clin. Transl. Imaging 2018, 6, 305–316. [Google Scholar] [CrossRef]
- Khan, T.; Alkon, D. Alzheimer’s Disease Cerebrospinal Fluid and Neuroimaging Biomarkers: Diagnostic Accuracy and Relationship to Drug Efficacy. J. Alzheimer’s Dis. 2015, 46, 817–836. [Google Scholar] [CrossRef]
- Liu, L.; Xu, Y.-X.; Hirschberg, C.B. The role of nucleotide sugar transporters in development of eukaryotes. Semin. Cell Dev. Biol. 2010, 21, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Aebi, M. N-linked protein glycosylation in the ER. Biochim. Biophys. Acta 2013, 1833, 2430–2437. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Vijayan, M. Influence of glycosidic linkage on the nature of carbohydrate binding in β-prism I fold lectins: An X-ray and molecular dynamics investigation on banana lectin–carbohydrate complexes. Glycobiology 2011, 21, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Lairson, L.; Henrissat, B.; Davies, G.J.; Withers, S. Glycosyltransferases: Structures, Functions, and Mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, D.H. Chapter Two—Structure-Guided Directed Evolution of Glycosidases: A Case Study in Engineering a Blood Group Antigen-Cleaving Enzyme. In Methods in Enzymology; Imperiali, B., Ed.; Academic Press: New York, NY, USA, 2017; Volume 597, pp. 25–53. [Google Scholar]
- An, H.J.; Froehlich, J.W.; Lebrilla, C.B. Determination of Glycosylation Sites and Site-specific Heterogeneity in Glycoproteins. Curr. Opin. Chem. Biol. 2009, 13, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Li, S.; Shao, F. Sweet Talk: Protein Glycosylation in Bacterial Interaction with the Host. Trends Microbiol. 2015, 23, 630–641. [Google Scholar] [CrossRef]
- Jensen, P.H.; Kolarich, D.; Packer, N.H. Mucin-type O-glycosylation—Putting the pieces together. FEBS J. 2010, 277, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Kizuka, Y.; Kitazume, S.; Taniguchi, N. N-glycan and Alzheimer’s disease. Biochim. Biophys. Acta 2017, 1861, 2447–2454. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281, 46–62. [Google Scholar] [CrossRef]
- Frenkel-Pinter, M.; Stempler, S.; Tal-Mazaki, S.; Losev, Y.; Singh-Anand, A.; Escobar-Alvarez, D.; Lezmy, J.; Gazit, E.; Ruppin, E.; Segal, D. Altered protein glycosylation predicts Alzheimer’s disease and modulates its pathology in disease model Drosophila. Neurobiol. Aging 2017, 56, 159–171. [Google Scholar] [CrossRef]
- Taniguchi, N.; Takahashi, M.; Kizuka, Y.; Kitazume, S.; Shuvaev, V.V.; Ookawara, T.; Furuta, A. Glycation vs. glycosylation: A tale of two different chemistries and biology in Alzheimer’s disease. Glycoconj. J. 2016, 33, 487–497. [Google Scholar] [CrossRef]
- Lassen, P.S.; Thygesen, C.; Larsen, M.R.; Kempf, S.J. Understanding Alzheimer’s disease by global quantification of protein phosphorylation and sialylated N-linked glycosylation profiles: A chance for new biomarkers in neuroproteomics? J. Proteom. 2017, 161, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Mohorko, E.; Glockshuber, R.; Aebi, M. Oligosaccharyltransferase: The central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis. 2011, 34, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Higel, F.; Seidl, A.; Sörgel, F.; Friess, W. N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur. J. Pharm. Biopharm. 2016, 100, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, P.; Ungar, D. Bridging the Gap between Glycosylation and Vesicle Traffic. Front. Cell Dev. Biol. 2016, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Roth, Z.; Yehezkel, G.; Khalaila, I. Identification and Quantification of Protein Glycosylation. Int. J. Carbohydr. Chem. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.Y.; Majewska, N.I.; Wang, Q.; Paul, J.T.; Betenbaugh, M.J. SnapShot: N-Glycosylation Processing Pathways across Kingdoms. Cell 2017, 171, 258. [Google Scholar] [CrossRef]
- Zhang, L.; Luo, S.; Zhang, B. Glycan analysis of therapeutic glycoproteins. MAbs 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Geyer, H.; Geyer, R. Strategies for analysis of glycoprotein glycosylation. Biochim. Biophys. Acta 2006, 1764, 1853–1869. [Google Scholar] [CrossRef]
- Tarentino, A.L.; Gomez, C.M.; Plummer, T.H. Deglycosylation of asparagine-linked glycans by peptide: N-glycosidase F. Biochemistry 1985, 24, 4665–4671. [Google Scholar] [CrossRef]
- Mucha, E.; Stuckmann, A.; Marianski, M.; Struwe, W.B.; Meijer, G.; Pagel, K. In-depth structural analysis of glycans in the gas phase. Chem. Sci. 2019, 10, 1272–1284. [Google Scholar] [CrossRef] [Green Version]
- Mechref, Y. Analysis of glycans derived from glycoconjugates by capillary electrophoresis-mass spectrometry. Electrophoresis 2011, 32, 3467–3481. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nie, Y.; Boyes, B.; Orlando, R. Resolving Isomeric Glycopeptide Glycoforms with Hydrophilic Interaction Chromatography (HILIC). J. Biomol. Tech. 2016, 27, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bones, J.; McLoughlin, N.; Hilliard, M.; Wynne, K.; Karger, B.L.; Rudd, P.M. 2D-LC Analysis of BRP 3 Erythropoietin N-Glycosylation using Anion Exchange Fractionation and Hydrophilic Interaction UPLC Reveals Long Poly-N-Acetyl Lactosamine Extensions. Anal. Chem. 2011, 83, 4154–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Walsh, I.; Abrahams, J.L.; Royle, L.; Nguyen-Khuong, T.; Spencer, D.; Fernandes, D.L.; Packer, N.H.; Rudd, P.M.; Campbell, M.P. GlycoStore: A database of retention properties for glycan analysis. Bioinformatics 2018, 34, 3231–3232. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Bones, J.; Yu, Y.Q.; Rudd, P.M.; Gilar, M. Separation of 2-aminobenzamide labeled glycans using hydrophilic interaction chromatography columns packed with 1.7 μm sorbent. J. Chromatogr. B 2010, 878, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Grumbach, E.; Diehl, D.; D Neue, U. The application of novel 1.7 μm ethylene bridged hybrid particles for hydrophilic interaction chromatography. J. Sep. Sci. 2008, 31, 1511–1518. [Google Scholar] [CrossRef]
- Wilson, I.D.; Nicholson, J.K.; Castro-Perez, J.; Granger, J.H.; Johnson, K.A.; Smith, B.W.; Plumb, R.S. High Resolution “Ultra Performance” Liquid Chromatography Coupled to oa-TOF Mass Spectrometry as a Tool for Differential Metabolic Pathway Profiling in Functional Genomic Studies. J. Proteome Res. 2005, 4, 591–598. [Google Scholar] [CrossRef]
- Pu, Y.; Ridgeway, M.E.; Glaskin, R.S.; Park, M.A.; Costello, C.E.; Lin, C. Separation and Identification of Isomeric Glycans by Selected Accumulation-Trapped Ion Mobility Spectrometry-Electron Activated Dissociation Tandem Mass Spectrometry. Anal. Chem. 2016, 88, 3440–3443. [Google Scholar] [CrossRef] [Green Version]
- Mauko, L.; Lacher, N.A.; Pelzing, M.; Nordborg, A.; Haddad, P.R.; Hilder, E.F. Comparison of ZIC-HILIC and graphitized carbon-based analytical approaches combined with exoglycosidase digestions for analysis of glycans from monoclonal antibodies. J. Chromatogr. B 2012, 911, 93–104. [Google Scholar] [CrossRef]
- Gotz, L.; Abrahams, J.L.; Mariethoz, J.; Rudd, P.M.; Karlsson, N.G.; Packer, N.H.; Campbell, M.P.; Lisacek, F. GlycoDigest: A tool for the targeted use of exoglycosidase digestions in glycan structure determination. Bioinformatics 2014, 30, 3131–3133. [Google Scholar] [CrossRef]
- Kobata, A. Exo- and endoglycosidases revisited. Proc. Jpn. Acad. Ser. B 2013, 89, 97–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, K.; Bones, J.; Kattla, J.J.; Rudd, P.M. A systematic approach to protein glycosylation analysis: A path through the maze. Nat. Chem. Biol. 2010, 6, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Silveyra, M.X.; Evin, G.; Montenegro, M.F.; Vidal, C.J.; Martínez, S.; Culvenor, J.G.; Sáez-Valero, J. Presenilin 1 Interacts with Acetylcholinesterase and Alters Its Enzymatic Activity and Glycosylation. Mol. Cell. Biol. 2008, 28, 2908–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara, J.; Dilhuydy, H.; Espinosa, B.; Delacourte, A.; Quirion, R.; Mena, R.; Joanette, Y.; Zenteno, E.; Robitaille, Y. Coexistence of reactive plasticity and neurodegeneration in Alzheimer diseased brains. Histol. Histopathol. 2004, 19, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Belický, Š.; Katrlík, J.; Tkáč, J. Glycan and lectin biosensors. Essays Biochem. 2016, 60, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Yoshida, M.; Nagai-Okatani, C.; Iwaki, J.; Matsuda, A.; Tan, B.; Hagiwara, K.; Sato, T.; Itakura, Y.; Noro, E.; et al. A standardized method for lectin microarray-based tissue glycome mapping. Sci. Rep. 2017, 7, 43560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Vega, E.; Tengattini, S.; Peintner, C.; van Angeren, J.; Temporini, C.; Haselberg, R.; Massolini, G.; Somsen, G.W. High-resolution glycoform profiling of intact therapeutic proteins by hydrophilic interaction chromatography-mass spectrometry. Talanta 2018, 184, 375–381. [Google Scholar] [CrossRef]
- Nwosu, C.; Yau, H.K.; Becht, S. Assignment of Core versus Antenna Fucosylation Types in Protein N-Glycosylation via Procainamide Labeling and Tandem Mass Spectrometry. Anal. Chem. 2015, 87, 5905–5913. [Google Scholar] [CrossRef]
- Tsai, T.H.; Wang, M.; Di Poto, C.; Hu, Y.; Zhou, S.; Zhao, Y.; Varghese, R.S.; Luo, Y.; Tadesse, M.G.; Ziada, D.H.; et al. LC-MS profiling of N-Glycans derived from human serum samples for biomarker discovery in hepatocellular carcinoma. J. Proteome Res. 2014, 13, 4859–4868. [Google Scholar] [CrossRef]
- Zhou, S.; Wooding, K.; Mechref, Y. Analysis of Permethylated Glycan by Liquid Chromatography (LC) and Mass Spectrometry (MS). Methods Mol. Biol. 2017, 1503, 83–96. [Google Scholar] [CrossRef]
- Leymarie, N.; Zaia, J. Effective Use of Mass Spectrometry for Glycan and Glycopeptide Structural Analysis. Anal. Chem. 2012, 84, 3040–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumer-Bayraktar, Z.; Nguyen-Khuong, T.; Jayo, R.; Chen, D.D.; Ali, S.; Packer, N.H.; Thaysen-Andersen, M. Micro- and macroheterogeneity of N-glycosylation yields size and charge isoforms of human sex hormone binding globulin circulating in serum. Proteomics 2012, 12, 3315–3327. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Nguyen-Khuong, T.; Wongtrakul-Kish, K.; Tay, S.J.; Chew, D.; Tasha, J.; Taron, C.; Rudd, P. GlycanAnalyzer: Software for Automated Interpretation of N-Glycan Profiles after Exoglycosidase Digestions. Bioinformatics 2018, 35, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Ioffe, E.; Stanley, P. Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc. Natl. Acad. Sci. USA 1994, 91, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Marek, K.W.; Vijay, I.K.; Marth, J.D. A recessive deletion in the GlcNAc-1-phosphotransferase gene results in peri-implantation embryonic lethality. Glycobiology 1999, 9, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.E.; Hsieh-Wilson, L.C. Glycan Engineering for Cell and Developmental Biology. Cell Chem. Biol. 2016, 23, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.Y.; Takahashi, M.; Gu, J.G.; Miyoshi, E.; Matsumoto, A.; Kitazume, S.; Taniguchi, N. Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 2008, 99, 1304–1310. [Google Scholar] [CrossRef]
- Ding, N.; Nie, H.; Sun, X.; Sun, W.; Qu, Y.; Liu, X.; Yao, Y.; Liang, X.; Chen, C.C.; Li, Y. Human serum N-glycan profiles are age and sex dependent. Age Ageing 2011, 40, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Selman, M.H.; Derks, R.J.; Bondt, A.; Palmblad, M.; Schoenmaker, B.; Koeleman, C.A.; van de Geijn, F.E.; Dolhain, R.J.; Deelder, A.M.; Wuhrer, M. Fc specific IgG glycosylation profiling by robust nano-reverse phase HPLC-MS using a sheath-flow ESI sprayer interface. J. Proteom. 2012, 75, 1318–1329. [Google Scholar] [CrossRef]
- Adamczyk, B.; Tharmalingam, T.; Rudd, P.M. Glycans as cancer biomarkers. Biochim. Biophys. Acta 2012, 1820, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Saldova, R.; Asadi Shehni, A.; Haakensen, V.D.; Steinfeld, I.; Hilliard, M.; Kifer, I.; Helland, A.; Yakhini, Z.; Borresen-Dale, A.L.; Rudd, P.M. Association of N-glycosylation with breast carcinoma and systemic features using high-resolution quantitative UPLC. J. Proteome Res. 2014, 13, 2314–2327. [Google Scholar] [CrossRef] [PubMed]
- Bones, J.; Byrne, J.C.; O’Donoghue, N.; McManus, C.; Scaife, C.; Boissin, H.; Nastase, A.; Rudd, P.M. Glycomic and glycoproteomic analysis of serum from patients with stomach cancer reveals potential markers arising from host defense response mechanisms. J. Proteome Res. 2011, 10, 1246–1265. [Google Scholar] [CrossRef] [PubMed]
- Block, T.M.; Comunale, M.A.; Lowman, M.; Steel, L.F.; Romano, P.R.; Fimmel, C.; Tennant, B.C.; London, W.T.; Evans, A.A.; Blumberg, B.S.; et al. Use of targeted glycoproteomics to identify serum glycoproteins that correlate with liver cancer in woodchucks and humans. Proc. Natl. Acad. Sci. USA 2005, 102, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, S.; Unwin, L.; Muniyappa, M.; Rudd, P. Glycosylation as a marker for inflammatory arthritis. Cancer Biomark. 2014, 14, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Peanne, R. What is new in CDG? J. Inherit. Metab. Dis. 2017, 40, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Quelhas, D.; Critchley, A.J.; Carchon, H.; Hebestreit, H.F.; Hibbert, R.G.; Vilarinho, L.; Teles, E.; Matthijs, G.; Schollen, E.; et al. Detailed glycan analysis of serum glycoproteins of patients with congenital disorders of glycosylation indicates the specific defective glycan processing step and provides an insight into pathogenesis. Glycobiology 2003, 13, 601–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A. Human total serum N-glycome. Adv. Clin. Chem. 2008, 46, 51–85. [Google Scholar]
- Kizuka, Y.; Kitazume, S.; Fujinawa, R.; Saito, T.; Iwata, N.; Saido, T.C.; Nakano, M.; Yamaguchi, Y.; Hashimoto, Y.; Staufenbiel, M.; et al. An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease. EMBO Mol. Med. 2015, 7, 175. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease β-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef]
- Kizuka, Y.; Nakano, M.; Kitazume, S.; Saito, T.; Saido, T.C.; Taniguchi, N. Bisecting GlcNAc modification stabilizes BACE1 protein under oxidative stress conditions. Biochem. J. 2016, 473, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Chun, Y.S.; Kwon, O.H.; Chung, S. O-GlcNAcylation of amyloid-beta precursor protein at threonine 576 residue regulates trafficking and processing. Biochem. Biophys. Res. Commun. 2017, 490, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Akasaka-Manya, K.; Endo, T.; Manya, H.; Kawamura, M.; Hisanaga, S.I.; Tsumoto, H.; Miura, Y.; Hatsuta, H.; Murayama, S.; Saito, Y.; et al. Excess APP O-glycosylation by GalNAc-T6 decreases Aβ production. J. Biochem. 2016, 161, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Kitazume, S.; Tachida, Y.; Kato, M.; Yamaguchi, Y.; Honda, T.; Hashimoto, Y.; Wada, Y.; Saito, T.; Iwata, N.; Saido, T.; et al. Brain endothelial cells produce amyloid {beta} from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J. Biol. Chem. 2010, 285, 40097–40103. [Google Scholar] [CrossRef] [PubMed]
- Akasaka-Manya, K.; Manya, H.; Sakurai, Y.; Wojczyk, B.S.; Kozutsumi, Y.; Saito, Y.; Taniguchi, N.; Murayama, S.; Spitalnik, S.L.; Endo, T. Protective effect of N -glycan bisecting GlcNAc residues on β-amyloid production in Alzheimer’s disease. Glycobiology 2010, 20, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zaidi, T.; Iqbal, K.; Grundke-Iqbal, I.; Merkle, R.K.; Gong, C.X. Role of glycosylation in hyperphosphorylation of tau in Alzheimer’s disease. FEBS Lett. 2002, 512, 101–106. [Google Scholar] [CrossRef]
- Sato, Y.; Naito, Y.; Grundke-Iqbal, I.; Iqbal, K.; Endo, T. Analysis of N-glycans of pathological tau: Possible occurrence of aberrant processing of tau in Alzheimer’s disease. FEBS Lett. 2001, 496, 152–160. [Google Scholar] [CrossRef]
- Crespo, C.; García-Mompó, C.; Sanchez-Mataredona, D.; Varea, E.; Castillo-Gómez, E.; Blasco-Ibáñez, J.M.; Nacher, J.; Gómez-Climent, M.Á.; Guirado, R.; Hernández, S.; et al. The Polysialylated Form of the Neural Cell Adhesion Molecule (PSA-NCAM) Is Expressed in a Subpopulation of Mature Cortical Interneurons Characterized by Reduced Structural Features and Connectivity. Cereb. Cortex 2010, 21, 1028–1041. [Google Scholar] [CrossRef] [Green Version]
- Murray, H.C.; Low, V.F.; Swanson, M.E.; Dieriks, B.V.; Turner, C.; Faull, R.L.; Curtis, M.A. Distribution of PSA-NCAM in normal, Alzheimer’s and Parkinson’s disease human brain. Neuroscience 2016, 330, 359–375. [Google Scholar] [CrossRef]
- Palmigiano, A.; Barone, R.; Sturiale, L.; Sanfilippo, C.; Bua, R.O.; Romeo, D.A.; Messina, A.; Capuana, M.L.; Maci, T.; Le Pira, F.; et al. CSF N-glycoproteomics for early diagnosis in Alzheimer’s disease. J. Proteom. 2016, 131, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; O’Bryant, S.E.; Hampel, H.; Trojanowski, J.Q.; Montine, T.J.; Jeromin, A.; Blennow, K.; Lönneborg, A.; Wyss-Coray, T.; Soares, H.; et al. The future of blood-based biomarkers for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2014, 10, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheng, X.; Zeng, J.; Yuan, J.; Wang, Z.; Zhou, W.; Zhang, Y. LW-AFC Effects on N-glycan Profile in Senescence-Accelerated Mouse Prone 8 Strain, a Mouse Model of Alzheimer’s Disease. Aging Dis. 2017, 8, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Gizaw, S.T.; Ohashi, T.; Tanaka, M.; Hinou, H.; Nishimura, S. Glycoblotting method allows for rapid and efficient glycome profiling of human Alzheimer’s disease brain, serum and cerebrospinal fluid towards potential biomarker discovery. Biochim. Biophys. Acta 2016, 1860, 1716–1727. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Sturiale, L.; Palmigiano, A.; Zappia, M.; Garozzo, D. Glycomics of pediatric and adulthood diseases of the central nervous system. J. Proteom. 2012, 75, 5123–5139. [Google Scholar] [CrossRef] [PubMed]
- Maguire, T.M.; Gillian, A.M.; O’Mahony, D.; Coughlan, C.M.; Dennihan, A.; Breen, K.C. A decrease in serum sialyltransferase levels in Alzheimer’s disease. Neurobiol. Aging 1994, 15, 99–102. [Google Scholar] [CrossRef]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.M.; Hooli, B.; Divito, J.; Ionita, I.; et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef]
- Vanhooren, V.; Desmyter, L.; Liu, X.E.; Cardelli, M.; Franceschi, C.; Federico, A.; Libert, C.; Laroy, W.; Dewaele, S.; Contreras, R.; et al. N-glycomic changes in serum proteins during human aging. Rejuvenation Res. 2007, 10, 521–531a. [Google Scholar] [CrossRef]
- Chen, C.C.; Engelborghs, S.; Dewaele, S.; Le Bastard, N.; Martin, J.J.; Vanhooren, V.; Libert, C.; De Deyn, P.P. Altered serum glycomics in Alzheimer disease: A potential blood biomarker? Rejuvenation Res. 2010, 13, 439–444. [Google Scholar] [CrossRef]
- Martínez-Cairo, S.; Zenteno, E.; Guzmán, A.; Espinosa, B.; Guevara, J.; Slomianny, M.C.; Hernández, P. Characterization of an O-Glycosylated Plaque-Associated Protein from Alzheimer Disease Brain. J. Neuropathol. Exp. Neurol. 2003, 62, 34–41. [Google Scholar] [CrossRef]
- Dos Santos, J.P.A.; Vizuete, A.; Hansen, F.; Biasibetti, R.; Goncalves, C.A. Early and Persistent O-GlcNAc Protein Modification in the Streptozotocin Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 61, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef] [PubMed]
- Forster, S.; Welleford, A.S.; Triplett, J.C.; Sultana, R.; Schmitz, B.; Butterfield, D.A. Increased O-GlcNAc levels correlate with decreased O-GlcNAcase levels in Alzheimer disease brain. Biochim. Biophys. Acta 2014, 1842, 1333–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaro, J.F.; Gong, C.X.; Monroe, M.E.; Aldrich, J.T.; Clauss, T.R.W.; Purvine, S.O.; Wang, Z.; Camp, D.G., 2nd; Shabanowitz, J.; Stanley, P.; et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. USA 2012, 109, 7280–7285. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, F.; Petyuk, V.A.; Shukla, A.K.; Monroe, M.E.; Gritsenko, M.A.; Rodland, K.D.; Smith, R.D.; Qian, W.J.; Gong, C.X.; et al. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J. Pathol. 2017, 243, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Akan, I.; Olivier-Van Stichelen, S.; Bond, M.R.; Hanover, J.A. Nutrient-driven O-GlcNAc in proteostasis and neurodegeneration. J. Neurochem. 2018, 144, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.Y.; Chatham, J.C.; Darley-Usmar, V.; McMahon, L.L.; Zhang, J. O-GlcNAcylation and neurodegeneration. Brain Res. Bull. 2017, 133, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martinez, I.; Martinez-Loustalot, P.; Lozano, L.; Issad, T.; Limon, D.; Diaz, A.; Perez-Torres, A.; Guevara, J.; Zenteno, E. Neuroinflammation induced by amyloid beta25-35 modifies mucin-type O-glycosylation in the rat’s hippocampus. Neuropeptides 2018, 67, 56–62. [Google Scholar] [CrossRef]
- Halim, A.; Brinkmalm, G.; Ruetschi, U.; Westman-Brinkmalm, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Larson, G.; Nilsson, J. Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid beta-peptides in human cerebrospinal fluid. Proc. Natl. Acad. Sci. USA 2011, 108, 11848–11853. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, B.; Culwell, A.R.; Esch, F.S.; Lieberburg, I.; Rydel, R.E. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron 1993, 10, 243–254. [Google Scholar] [CrossRef]
- Goodman, Y.; Mattson, M.P. Secreted Forms of β-Amyloid Precursor Protein Protect Hippocampal Neurons against Amyloid β-Peptide-Induced Oxidative Injury. Exp. Neurol. 1994, 128, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [PubMed]
- Charlwood, J.; Dingwall, C.; Matico, R.; Hussain, I.; Johanson, K.; Moore, S.; Powell, D.J.; Skehel, J.M.; Ratcliffe, S.; Clarke, B.; et al. Characterization of the glycosylation profiles of Alzheimer’s beta -secretase protein Asp-2 expressed in a variety of cell lines. J. Biol. Chem. 2001, 276, 16739–16748. [Google Scholar] [CrossRef] [PubMed]
- Kitazume, S.; Tachida, Y.; Oka, R.; Nakagawa, K.; Takashima, S.; Lee, Y.C.; Hashimoto, Y. Screening a series of sialyltransferases for possible BACE1 substrates. Glycoconj. J. 2006, 23, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Kitazume, S.; Nakagawa, K.; Oka, R.; Tachida, Y.; Ogawa, K.; Luo, Y.; Citron, M.; Shitara, H.; Taya, C.; Yonekawa, H.; et al. In Vivo Cleavage of α2,6-Sialyltransferase by Alzheimer β-Secretase. J. Biol. Chem. 2005, 280, 8589–8595. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Kitazume, S.; Oka, R.; Maruyama, K.; Saido, T.C.; Sato, Y.; Endo, T.; Hashimoto, Y. Sialylation enhances the secretion of neurotoxic amyloid-beta peptides. J. Neurochem. 2006, 96, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, I.; Futakawa, S.; Oka, R.; Ogawa, K.; Marth, J.D.; Miyoshi, E.; Taniguchi, N.; Hashimoto, Y.; Kitazume, S. Beta-galactoside alpha2,6-sialyltransferase I cleavage by BACE1 enhances the sialylation of soluble glycoproteins. A novel regulatory mechanism for alpha2,6-sialylation. J. Biol. Chem. 2007, 282, 34896–34903. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, I.; Breen, K.C.; Di Giamberardino, L.; Moya, K.L. Inhibition of N-glycan processing alters axonal transport of synaptic glycoproteins in vivo. Neuroreport 2000, 11, 1543–1547. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, I.; Georgopoulou, N.; Coughlan, C.M.; Gillian, A.M.; Breen, K.C. The role of the protein glycosylation state in the control of cellular transport of the amyloid beta precursor protein. Neuroscience 1999, 90, 15–25. [Google Scholar] [CrossRef]
- Tienari, P.J.; De Strooper, B.; Ikonen, E.; Simons, M.; Weidemann, A.; Czech, C.; Hartmann, T.; Ida, N.; Multhaup, G.; Masters, C.L.; et al. The beta-amyloid domain is essential for axonal sorting of amyloid precursor protein. EMBO J. 1996, 15, 5218–5229. [Google Scholar] [CrossRef] [PubMed]
- Akasaka-Manya, K.; Manya, H.; Sakurai, Y.; Wojczyk, B.S.; Spitalnik, S.L.; Endo, T. Increased bisecting and core-fucosylated N-glycans on mutant human amyloid precursor proteins. Glycoconj. J. 2008, 25, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Roßner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef] [PubMed]
- Malinverno, M.; Carta, M.; Epis, R.; Marcello, E.; Verpelli, C.; Cattabeni, F.; Sala, C.; Mulle, C.; Di Luca, M.; Gardoni, F. Synaptic Localization and Activity of ADAM10 Regulate Excitatory Synapses through N-Cadherin Cleavage. J. Neurosci. 2010, 30, 16343. [Google Scholar] [CrossRef] [PubMed]
- Colciaghi, F.; Borroni, B.; Pastorino, L.; Marcello, E.; Zimmermann, M.; Cattabeni, F.; Padovani, A.; Di Luca, M. α-Secretase ADAM10 as well as αAPPs is reduced in platelets and CSF of Alzheimer disease patients. Mol. Med. 2002, 8, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Escrevente, C.; Morais, V.A.; Keller, S.; Soares, C.M.; Altevogt, P.; Costa, J. Functional role of N-glycosylation from ADAM10 in processing, localization and activity of the enzyme. Biochim. Biophys. Acta 2008, 1780, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.S.; Johnson, G.V.W.; Cole, R.N.; Dong, D.L.Y.; Lee, M.; Hart, G.W. The Microtubule-associated Protein Tau Is Extensively Modified with O-linked N-acetylglucosamine. J. Biol. Chem. 1996, 271, 28741–28744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, T.; Ferreira, S.; Dupont-Wallois, L.; Bussière, T.; Dupire, M.J.; Delacourte, A.; Michalski, J.C.; Caillet-Boudin, M.L. Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins—A role in nuclear localization. Biochim. Biophys. Acta 2003, 1619, 167–176. [Google Scholar] [CrossRef]
- Hsu, Y.P.; Meng, X.; VanNieuwenhze, M.S. Chapter 1—Methods for visualization of peptidoglycan biosynthesis. In Methods in Microbiology; Harwood, C., Jensen, G.J., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 43, pp. 3–48. [Google Scholar]
- Saraswathy, N.; Ramalingam, P. 15—Phosphoproteomics. In Concepts and Techniques in Genomics and Proteomics; Woodhead Publishing: Cambridge, UK, 2012; pp. 203–211. [Google Scholar]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Grundke-Iqbal, I.; Iqbal, K. Glycosylation of microtubule–associated protein tau: An abnormal posttranslational modification in Alzheimer’s disease. Nat. Med. 1996, 2, 871. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P.; Taniguchi, N.; Aebi, N. Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017. [Google Scholar]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Tandon, A.; Chen, F.; Yu, G.; Yu, H.; Arawaka, S.; Hasegawa, H.; Duthie, M.; Schmidt, S.D.; Ramabhadran, T.V.; et al. Mature glycosylation and trafficking of nicastrin modulate its binding to presenilins. J. Biol. Chem. 2002, 277, 28135–28142. [Google Scholar] [CrossRef]
- Yu, G.; Nishimura, M.; Arawaka, S.; Levitan, D.; Zhang, L.; Tandon, A.; Song, Y.Q.; Rogaeva, E.; Chen, F.; Kawarai, T.; et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 2000, 407, 48–54. [Google Scholar] [CrossRef]
- Herreman, A.; Van Gassen, G.; Bentahir, M.; Nyabi, O.; Craessaerts, K.; Mueller, U.; Annaert, W.; De Strooper, B. Gamma-Secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J. Cell Sci. 2003, 116, 1127–1136. [Google Scholar] [CrossRef]
- Farquhar, M.J.; Gray, C.W.; Breen, K.C. The over-expression of the wild type or mutant forms of the presenilin-1 protein alters glycoprotein processing in a human neuroblastoma cell line. Neurosci. Lett. 2003, 346, 53–56. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Dolinoy, D.C.; Sartor, M.A.; Paulson, H.L.; Konen, J.R.; Lieberman, A.P.; Albin, R.L.; Hu, H.; Rozek, L.S. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J. Alzheimer’s Dis. 2012, 29, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Münch, A.; Neumann, S.; Kremmer, E.; Tatzelt, J.; Lichtenthaler, S.F. The novel membrane protein TMEM59 modulates complex glycosylation, cell surface expression, and secretion of the amyloid precursor protein. J. Biol. Chem. 2010, 285, 20664–20674. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.T.; Tan, L. The role of clusterin in Alzheimer’s disease: Pathways, pathogenesis, and therapy. Mol. Neurobiol. 2012, 45, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.B.; Hone, E.; Pedrini, S.; Doecke, J.; O’Bryant, S.; James, I.; Bush, A.I.; Rowe, C.C.; Villemagne, V.L.; Ames, D.; et al. Altered levels of blood proteins in Alzheimer’s disease longitudinal study: Results from Australian Imaging Biomarkers Lifestyle Study of Ageing cohort. Alzheimer’s Dement. 2017, 8, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.C.; Russell, C.; Mitra, V.; Chung, R.; Hye, A.; Bazenet, C.; Lovestone, S.; Pike, I.; Ward, M. Glycosylation of Human Plasma Clusterin Yields a Novel Candidate Biomarker of Alzheimer’s Disease. J. Proteome Res. 2015, 14, 5063–5076. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruberu, K.; Munoz, S.S.; Jenner, A.M.; Spiro, A.; Zhao, H.; Rassart, E.; Sanchez, D.; Ganfornina, M.D.; Karl, T.; et al. Apolipoprotein D modulates amyloid pathology in APP/PS1 Alzheimer’s disease mice. Neurobiol. Aging 2015, 36, 1820–1833. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruberu, K.; Karl, T.; Garner, B. Cerebral Apolipoprotein-D Is Hypoglycosylated Compared to Peripheral Tissues and Is Variably Expressed in Mouse and Human Brain Regions. PLoS ONE 2016, 11, e0148238. [Google Scholar] [CrossRef]
- Berg, D.; Youdim, M.B. Role of iron in neurodegenerative disorders. Top. Magn. Reson. Imaging 2006, 17, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Menzies, S.L.; St Martin, S.M.; Mufson, E.J. A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. J. Neurosci. Res. 1992, 31, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Okayama, Y.; Hashimoto, Y.; Kitaura, M.; Jimbo, D.; Wakutani, Y.; Wada-Isoe, K.; Nakashima, K.; Akatsu, H.; Furukawa, K.; et al. Sugar chains of cerebrospinal fluid transferrin as a new biological marker of Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2008, 26, 117–122. [Google Scholar] [CrossRef]
- van Rensburg, S.J.; Berman, P.; Potocnik, F.; MacGregor, P.; Hon, D.; de Villiers, N. 5- and 6-glycosylation of transferrin in patients with Alzheimer’s disease. Metab. Brain Dis. 2004, 19, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Atri, A.; Sherman, S.; Norman, K.A.; Kirchhoff, B.A.; Nicolas, M.M.; Greicius, M.D.; Cramer, S.C.; Breiter, H.C.; Hasselmo, M.E.; Stern, C.E. Blockade of central cholinergic receptors impairs new learning and increases proactive interference in a word paired-associate memory task. Behav. Neurosci. 2004, 118, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Kihara, T.; Shimohama, S. Alzheimer’s disease and acetylcholine receptors. Acta Neurobiologiae Experimentalis 2004, 64, 99–105. [Google Scholar] [PubMed]
- Sáez-Valero, J.; de Ceballos, M.A.L.; Small, D.H.; de Felipe, C. Changes in molecular isoform distribution of acetylcholinesterase in rat cortex and cerebrospinal fluid after intracerebroventricular administration of amyloid β-peptide. Neurosci. Lett. 2002, 325, 199–202. [Google Scholar] [CrossRef]
- Fodero, L.R.; Saez-Valero, J.; McLean, C.A.; Martins, R.N.; Beyreuther, K.; Masters, C.L.; Robertson, T.A.; Small, D.H. Altered glycosylation of acetylcholinesterase in APP (SW) Tg2576 transgenic mice occurs prior to amyloid plaque deposition. J. Neurochem. 2002, 81, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Saez-Valero, J.; Fodero, L.R.; Sjögren, M.; Andreasen, N.; Amici, S.; Gallai, V.; Vanderstichele, H.; Vanmechelen, E.; Parnetti, L.; Blennow, K.; et al. Glycosylation of acetylcholinesterase and butyrylcholinesterase changes as a function of the duration of Alzheimer’s disease. J. Neurosci. Res. 2003, 72, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Durakoglugil, M.S.; Chen, Y.; White, C.L.; Kavalali, E.T.; Herz, J. Reelin signaling antagonizes β-amyloid at the synapse. Proc. Natl. Acad. Sci. USA 2009, 106, 15938–15943. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Ballif, B.A.; Cooper, J.A. Regulation of Protein Tyrosine Kinase Signaling by Substrate Degradation during Brain Development. Mol. Cell. Biol. 2003, 23, 9293–9302. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Zhao, L.F.; Korwek, K.M.; Weeber, E.J. Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J. Neurosci. 2006, 26, 12943–12955. [Google Scholar] [CrossRef] [PubMed]
- Botella-López, A.; Burgaya, F.; Gavín, R.; García-Ayllón, M.S.; Gómez-Tortosa, E.; Peña-Casanova, J.; Ureña, J.M.; Del Río, J.A.; Blesa, R.; Soriano, E.; et al. Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5573–5578. [Google Scholar] [CrossRef] [PubMed]
- Bayes-Genis, A.; Barallat, J.; Richards, A.M. A Test in Context: Neprilysin: Function, Inhibition, and Biomarker. J. Am. Coll. Cardiol. 2016, 68, 639–653. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, D.; Wang, Y.; Huang, H.; Zhao, Y.; Zhou, H. Meta-analysis of expression and function of neprilysin in Alzheimer’s disease. Neurosci. Lett. 2017, 657, 69–76. [Google Scholar] [CrossRef]
- Lafrance, M.H.; Vézina, C.; Wang, Q.; Boileau, G.; Crine, P.; Lemay, G. Role of glycosylation in transport and enzymic activity of neutral endopeptidase-24.11. Biochem. J. 1994, 302, 451. [Google Scholar] [CrossRef] [PubMed]
- Sato, B.; Katagiri, Y.U.; Iijima, K.; Yamada, H.; Ito, S.; Kawasaki, N.; Okita, H.; Fujimoto, J.; Kiyokawa, N. The human CD10 lacking an N-glycan at Asn (628) is deficient in surface expression and neutral endopeptidase activity. Biochim. Biophys. Acta 2012, 1820, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, L. TREM2 in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Ji, I.J.; Kim, D.H.; An, H.J.; Yoon, S.Y. The Alzheimer’s Disease-Associated R47H Variant of TREM2 Has an Altered Glycosylation Pattern and Protein Stability. Front. Neurosci. 2016, 10, 618. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, S.L.; Yang, H.; Lyutvinskiy, Y.; Rutishauser, D.; Herukka, S.K.; Soininen, H.; Zubarev, R.A. Blood plasma IgG Fc glycans are significantly altered in Alzheimer’s disease and progressive mild cognitive impairment. J. Alzheimer’s Dis. 2014, 38, 567–579. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006, 313, 670–673. [Google Scholar] [CrossRef]
- Leeman, M.; Choi, J.; Hansson, S.; Storm, M.U.; Nilsson, L. Proteins and antibodies in serum, plasma, and whole blood-size characterization using asymmetrical flow field-flow fractionation (AF4). Anal. Bioanal. Chem. 2018, 410, 4867–4873. [Google Scholar] [CrossRef] [PubMed]



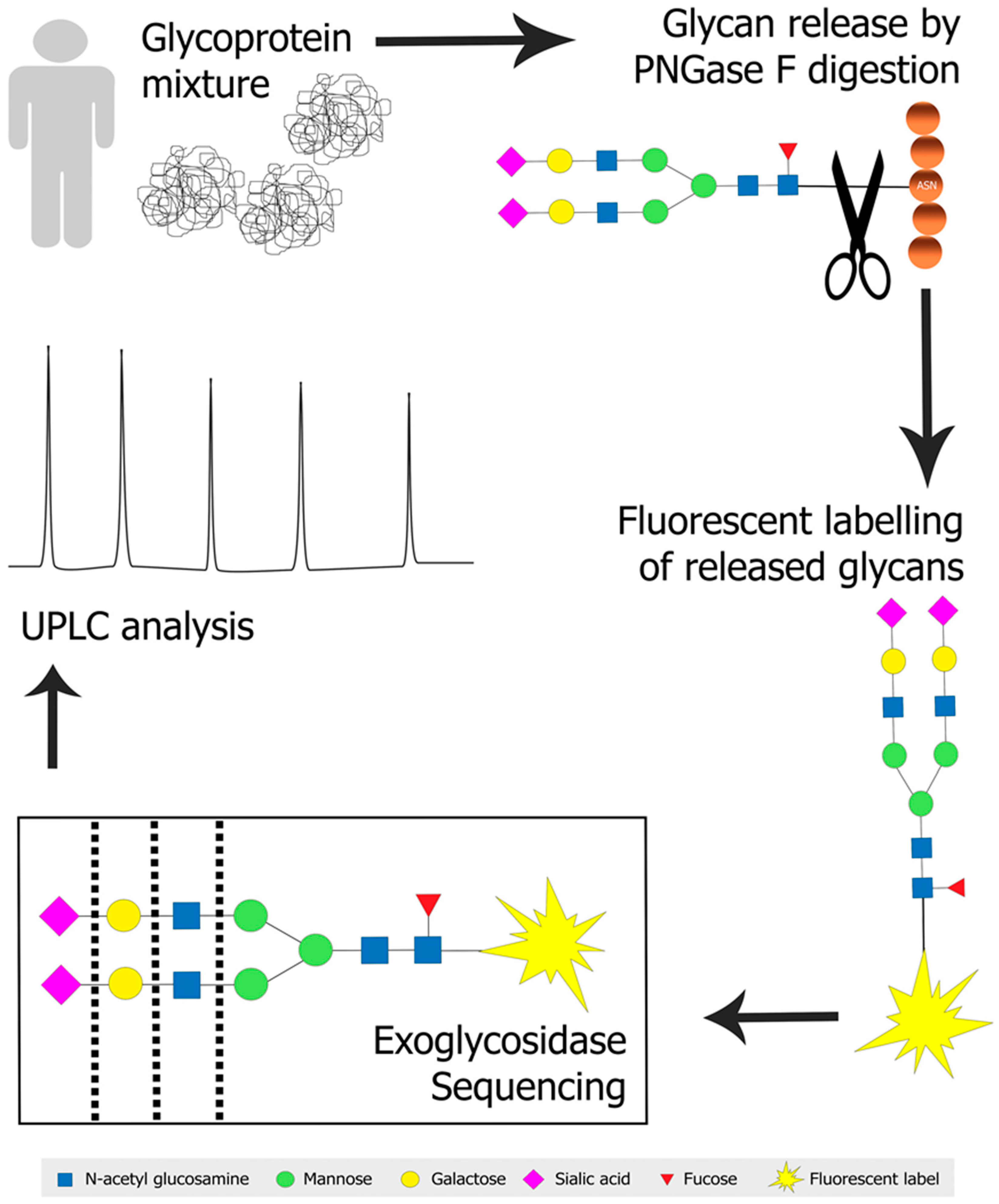{kind=link}
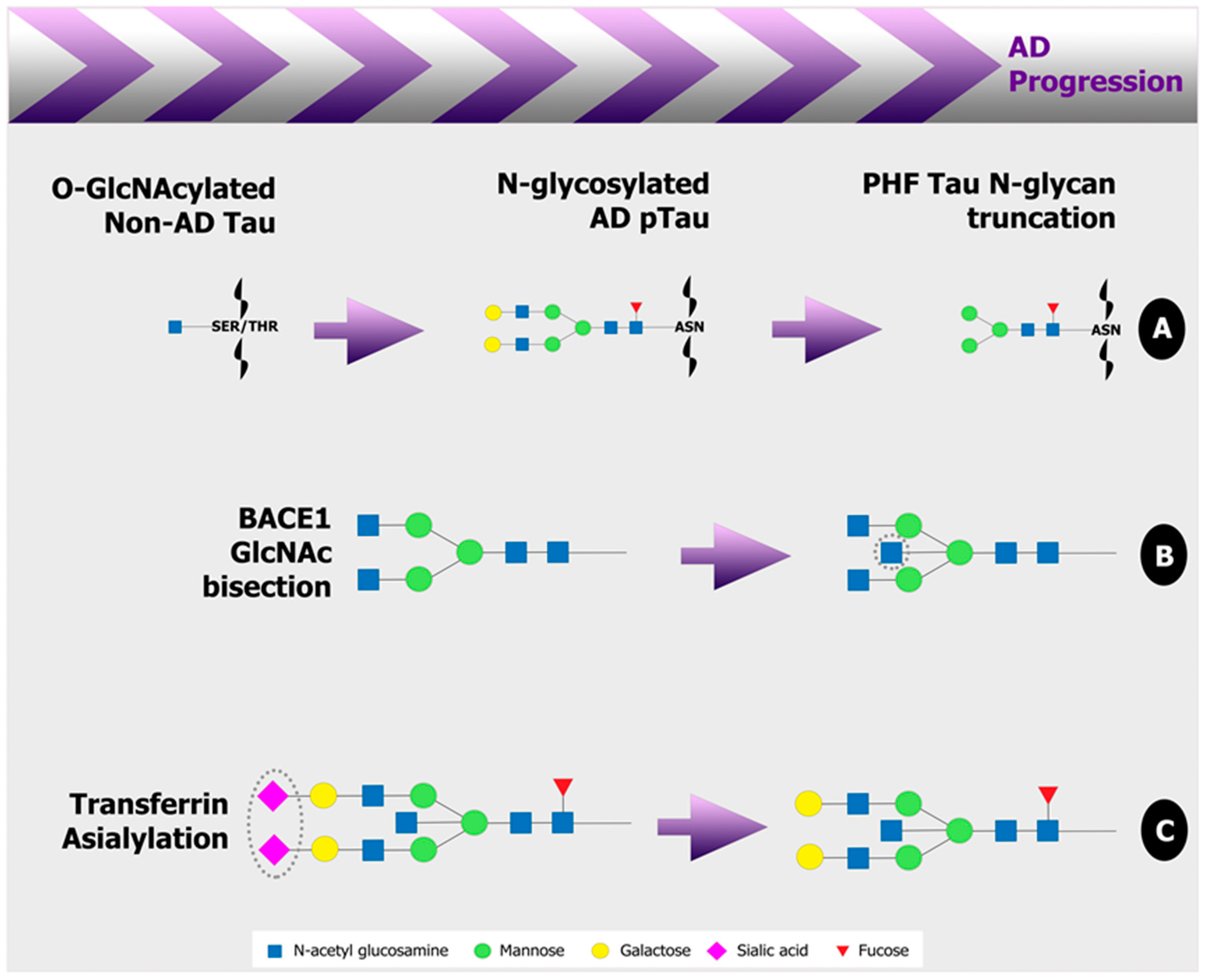{kind=link}
| Location | Analysis Method | Biomarker | Cohorts | Additional Comments |
|---|---|---|---|---|
| Serum and CSF | Glyco-blotting and MS | Increased bisect type, core fucosylated, highly branched species [127]. | AD patients (n = 2–3) versus sex-matched non-AD controls (n = 2–3). | |
| Serum | Radio-enzymatic assay | Decreased sialyltransferase activity [129]. | AD patients (n = 12) versus age and sex matched non-AD controls (n = 12). | Although both moderate and severe AD cases were assessed, there was no correlation between serum sialyltransferase activity and degree of AD. Considerable variation in the control group was observed. |
| Serum | DSA-FACE | Decreased bi-galactosylated core fucosylated bi-antennary glycan [132]. | Population of primarily moderate/severe AD patients (n = 48) versus age and sex matched healthy (n = 149) and non-AD (n = 31) controls. | Desialylated serum assessed. Difference not observed between non-AD patients and age and sex matched controls. Discriminated AD patients (n = 48) from non-AD patients and healthy controls (n = 180) with a diagnostic accuracy of 85.7% ± 2.8%, 92% specificity and 70% sensitivity. |
| CSF | Matrix-assisted laser de-sorption/ionization-MS | Increased bisect type species and decreased sialylated species [124]. | Pre-dementia (n = 11) and sporadic AD (n = 24) cases versus age matched healthy controls (n = 21). | 40–50% of the diseased patients had this altered glycoprofile versus controls. All pre-dementia cases that converted to AD displayed an altered glycoprofile. |
| CSF | LC-MS/MS | Increased ratio of tyrosine linked O-glycosylated Aβ peptides to corresponding unglycosylated peptides [142]. | AD patients (n = 6) versus non-AD patients (n = 7). | Patients not cognitively assessed in detail. Diagnosis based on sensitive and specific CSF biomarker detection of pathological tau and Aβ levels. |
| Plasma | LC-MS/MS | Decreased N-glycosylation of clusterin [181]. | Mild/moderate AD patients with high hippocampal atrophy (n = 14) versus those with low hippocampal atrophy (n = 13). | N-glycans modified with mannose, galactose, sialic acid and GlcNAc. Determined that decreased glycans all present at a common N-glycosylation site on clusterin. |
| CSF | Lectin blotting, isoelectric focusing and MS | Decreased sialylation of transferrin [186]. | Diagnosed probable AD patients (n = 43) versus non-AD (n = 13) and non-demented (n = 32) controls. | Combined with phosphorylated tau detection, specificity and sensitivity was 88.4% and 92.3%, respectively. CSF transferrin levels did not differ between groups. |
| Serum | Isoelectric focusing and immuno-blotting | Increased penta- and hexa-sialylation of transferrin [187]. | AD patients (n = 11) versus non-demented, age-matched controls (n = 14). | |
| CSF | Lectin blotting | Increased mannosylated glycans on reelin [196]. | AD patients (n = 11) versus non-demented, age- and sex-matched controls (n = 9). | Combining two lectin stains increased discrimination of AD from controls. 10 of 11 AD cases were below an arbitrary cutoff point, and 7 of 9 controls were above this cutoff. |
| Plasma | LC-MS/MS | Decreased complex, galactosylated and sialylated glycans on IgG [204]. | AD patients (n = 31) versus non-demented controls (n = 26). | One such bi-antennary, complex, bi-galactosylated glycan decreased in females (n = 93) steadily prior to disease onset from earlier to later stage cases, but an inverse trend was true for males (n = 65). |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Regan, P.; McClean, P.L.; Smyth, T.; Doherty, M. Early Stage Glycosylation Biomarkers in Alzheimer’s Disease. Medicines 2019, 6, 92. https://doi.org/10.3390/medicines6030092
Regan P, McClean PL, Smyth T, Doherty M. Early Stage Glycosylation Biomarkers in Alzheimer’s Disease. Medicines. 2019; 6(3):92. https://doi.org/10.3390/medicines6030092
Chicago/Turabian StyleRegan, Patricia, Paula L. McClean, Thomas Smyth, and Margaret Doherty. 2019. "Early Stage Glycosylation Biomarkers in Alzheimer’s Disease" Medicines 6, no. 3: 92. https://doi.org/10.3390/medicines6030092
APA StyleRegan, P., McClean, P. L., Smyth, T., & Doherty, M. (2019). Early Stage Glycosylation Biomarkers in Alzheimer’s Disease. Medicines, 6(3), 92. https://doi.org/10.3390/medicines6030092