1. Introduction
The production and use of fossil fuels have long-standing negative impacts on the global climate, public health, local communities and ecosystem goods and services [
1]. This motivates the development of renewable energy sources, which include the production of bio-ethanol, biogas and biodiesel [
2]. With an increase in biodiesel production comes an increase in the production of crude glycerol, as it is a by-product of the transesterification of vegetable and animal fats/oils. Crude glycerol has little economic or industrial value due to the presence of methanol, inorganic salts, free fatty acids and unreacted esters. The purification process required to produce pure glycerol is very costly and involves many steps; thus, it would be beneficial to develop processes that make use of the unpurified compound directly [
3,
4,
5,
6].
Due to the presence of three hydroxyl groups in glycerol, its oxidation under certain specific conditions affords high-value chemicals such as glyceric acid, tartronic acid, mesoxalic acid and dihydroxyacetone. Tartronic acid is a very costly reagent (US
$1564.00 per gram), as well as a high-value chemical in the pharmaceutical industry for obesity and osteoporosis treatment. It is also used as an anti-corrosive protective agent in boilers and high temperature applications and as an oxygen scavenger in the food industry [
2]. Glyceric acid is also very costly to produce; thus, 10 mg of it can cost as much as US
$56.00. It is thus conceivable that developing inexpensive oxidation catalysts that can be applied to both pure and crude glycerol will potentially make these chemicals more widely available.
There are several ways in which catalytic oxidation of glycerol occurs, with essential features of the mechanism being the simultaneous dehydrogenation of the hydroxyl group and oxidation of the formed intermediates. The dehydrogenation of the primary alcoholic functions on the metal surface is dependent on the presence of a base, and the reaction rate is directly proportional to the concentration of the base used [
7]. The use of a basic medium enhances the selectivity of glyceric and tartronic acids as compared to the application of an acidic medium that yields dihydroxyacetone, hydroxypyruvic acid and mesoxalic acid [
8]. The production of three-carbon compounds from glycerol has been extensively investigated [
9,
10,
11,
12,
13]. Research has been conducted where noble-metal nanoparticles such as palladium (Pd), platinum (Pt) and gold (Au) in a basic medium have been used in the oxidation of glycerol [
14]. With the use of Pd and Pt as the catalysts, glyceric acid is the major product formed [
15] and tartronic acid and oxalate as the major over-oxidation by-products [
16]. Furthermore, the addition of bismuth to Pt catalyst resulted in the oxidation of the secondary hydroxyl group, improved catalytic performance, selectivity and stability [
8].
The use of Pt/CeO
2 catalysts was reported to promote the oxidation of the primary hydroxyl groups to produce a 40% tartronic acid yield [
16]. The main drawback of the application of both Pt and Pd is their decrease in catalytic performance at prolonged reaction times. One percent Au supported on activated carbon and graphite afforded 100% selectivity of glyceric acid under mild conditions of 333 K and 0.3–0.6 MPa of oxygen [
17]. The presence of Pt on the Au metal surface affected the selectivity of oxidation products; the use of the mole fraction of Pt between 0.25 and 0.33 increased the selectivity of dihydroxyacetone and decreased that of glyceric acid [
18]. Not only is the selectivity of glycerol oxidation products affected by the nature of metal catalyst used, the use of crude glycerol is found to affect both the selectivity and reaction rates of glycerol oxidation [
3]. Although limited thermochemical and electrochemical oxidation of crude glycerol has been conducted, one of the reported works shows that crude glycerol significantly decreased the reaction rates of Ag/Al
2O
3, Au/Al
2O
3, Pd/Al
2O
3 and Pt/Al
2O
3 and also affected the selectivity of formic acid and glyceric acid [
3]. The reason for this is not unconnected with the impurities found in crude glycerol including methanol, ash (sodium sulfate), organic sulfur derivatives (OSD) and non-methanol organic matter derivatives (MONG-NM). The noble metal-based catalysts are prone to poison by sulfur; thus, interest in the development of non-noble metal-based catalysts [
7] that can tolerate sulfur becomes attractive. Hence, catalytic systems developed for glycerol oxidation must have enhanced sulfur and MONG-NM resistance [
3,
19].
A drawback of the aforementioned approaches is the price of the noble metal used in the catalysts. A potential cost-effective approach would involve the use of non-noble metal-based catalyst for the thermochemical oxidation of glycerol. Elendu et al. [
20] synthesized low-cost transition metal-based catalyst containing copper, nickel, molybdenum and phosphorus for the electrochemical oxidation of glycerol. The major electrochemical oxidation products of the electroless catalysts were not provided in [
20], but were discussed extensively in [
21]. Our interest in this work is to evaluate the as-deposited electroless CuNiMoP catalyst for its thermochemical oxidation capability for pure glycerol and compare its thermochemical oxidation products’ selectivity versus its electrochemical oxidation products’ selectivity for pure glycerol as reported in [
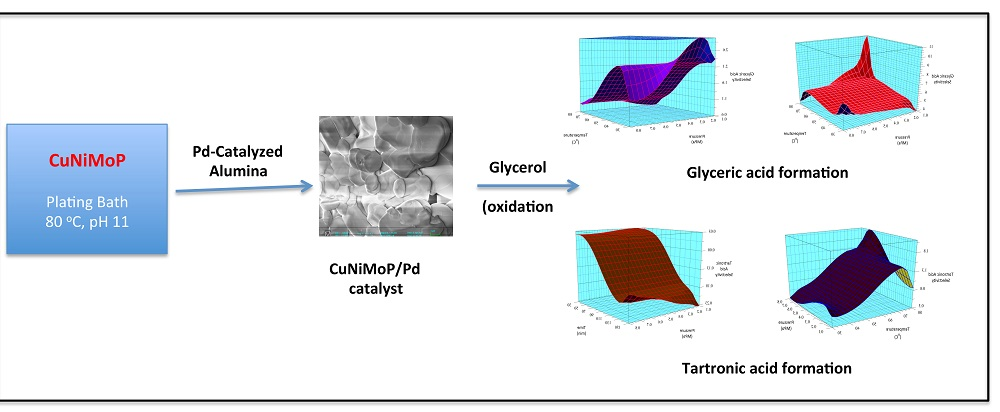
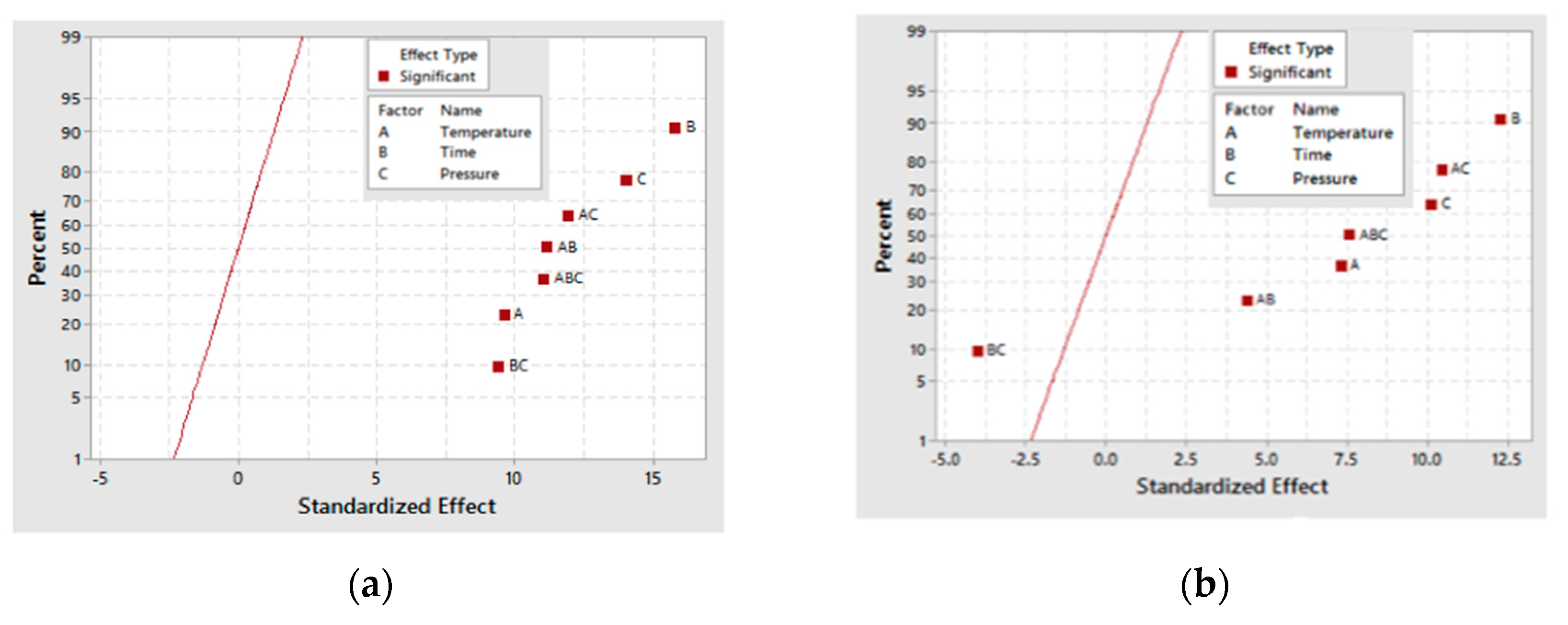
21]. A qualitative comparison of the electrochemical and thermochemical oxidation products of the catalyst could provide insight into the mechanism that is operating at different electrochemical oxidation potentials. The effect of deposition time of CuNiMoP onto the catalyst support was also evaluated. A 2
3 full factorial design of experiments was performed to assess the influence of temperature, time and pressure on the oxidation products from pure glycerol.
2. Materials and Methods
All chemicals were used as received without any further purification. CuSO4·5H2O (≥98%), NiSO4·6H2O (≥98%), H2NaO2P·H2O (≥99%), Na2MoO4·2H2O (≥99%) and HCOOH (ACS, 37 wt% in H2O, contains 10–15% methanol as a stabilizer solution) were purchased from Sigma-Aldrich. Sodium citrate dihydrate (99%), gluconic acid potassium salt (99%) and potassium sodium L-tartrate tetrahydrate (ACS, 99%–102.0%) were purchased from Alfa Aesar. Degassed deionized water was used throughout the experiment.
2.1. Catalyst Preparation via Electroless Plating
Based on studies conducted by Elendu et al. [
20], a CuNiMoP bath was prepared using the reagents shown in
Table 1. The plating bath was allowed to reach a temperature of 80 °C, and a few drops of concentrated NaOH were added to bring the pH to 11. The catalyst support, Pd catalyzed alumina, was then introduced, and the mixture was continuously stirred. Plating was conducted for 30 and 60 min. At the end of each plating time, the plating mixture was placed in an ice bath for approximately 2 min. The reaction mixture was then centrifuged for 15 min, and the solution was pipetted out, leaving the solid catalyst, which was subsequently washed with distilled water and 2-propanol and dried for 24 h at 60 °C. Subsequently, the catalyst was subjected to physical characterization.
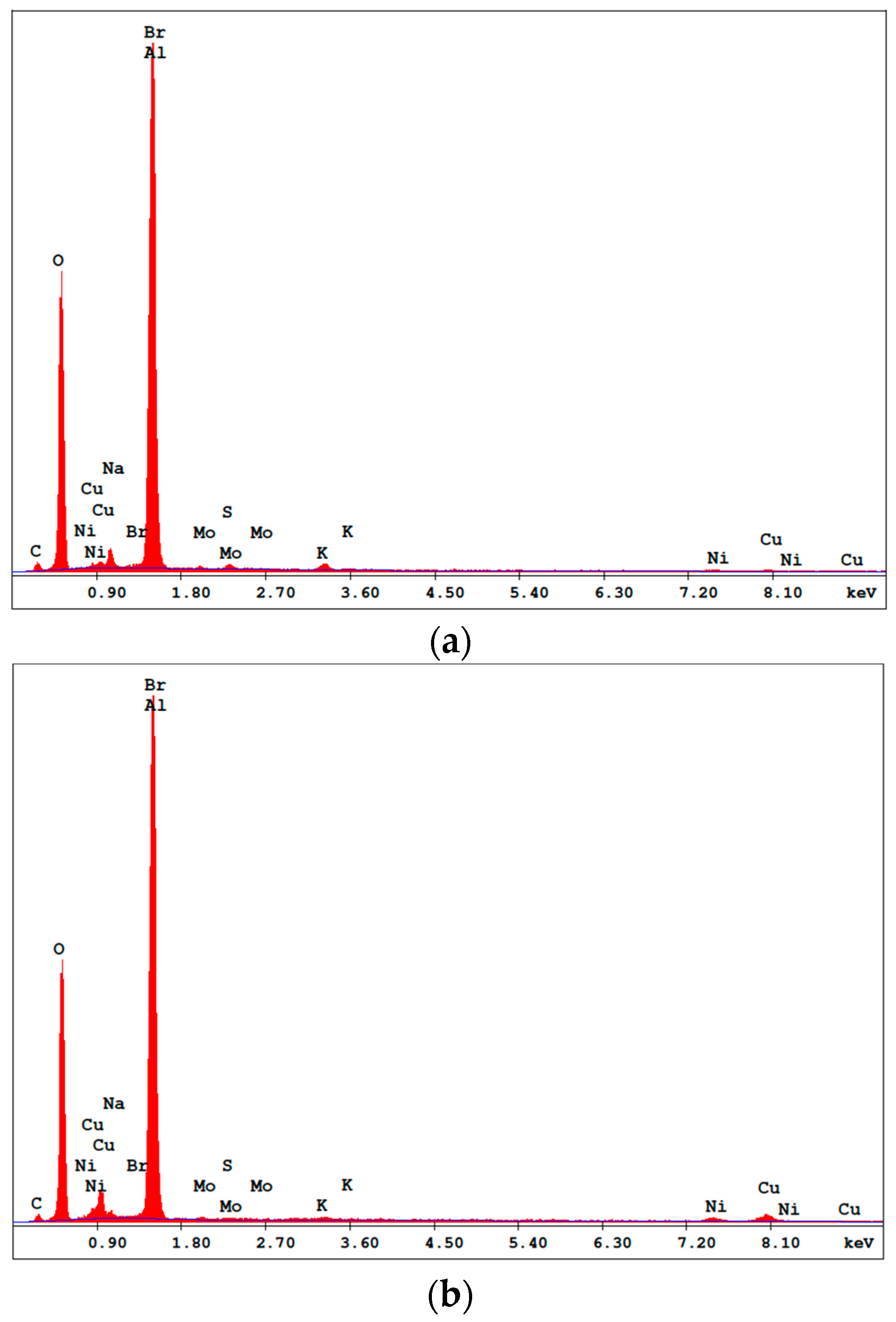
2.2. Characterization of CuNiMoP/Al2O3
The prepared catalyst was characterized by three methods: scanning electron microscopy (SEM), X-ray diffraction (XRD) and energy dispersive spectroscopy (EDS). Zeiss 1540 XB Cross Beam SEM was conducted to determine the change in morphology of the alumina surface after electroless deposition. XRD was performed to determine the crystallinity of the deposited catalyst on the alumina surface, while EDS was done to identify the elemental composition of the catalyst.
2.3. Oxidation Experiments
The glycerol oxidations were carried out in a 250-mL high-pressure chemical reactor equipped with pressure, temperature and speed controllers (Yudian 508/509). The 2
3 full factorial experiments were designed to investigate the influence of temperature, time and pressure on glycerol oxidation as shown in
Table 2. Each reaction was conducted twice at the prescribed conditions. The reactions were performed with 100 mL of an aqueous glycerol/NaOH (0.3/1.2 M) solution, 0.1 g of the respective catalyst and a stirring rate of 205 rpm. Samples were taken after each experiment and subjected to analysis via high performance liquid chromatography (HPLC).
2.4. Product Analysis
The quantitative analysis of each reaction mixture was conducted by HPLC. The chromatograph (Schimadzu Technologies) was equipped with a Hi-plex column (7.7 mm × 300 mm, Agilent), a refractive index and a UV (190 nm) detector. Dilute H2SO4 (5 mM) was used as the eluent. Three hundred microliters of each sample were diluted with distilled water with a dilution factor of 5/3. An injection volume of 10 μL, a measuring time of 20 min and a flow rate of 0.7 mL min−1 were adjusted.
4. Conclusions
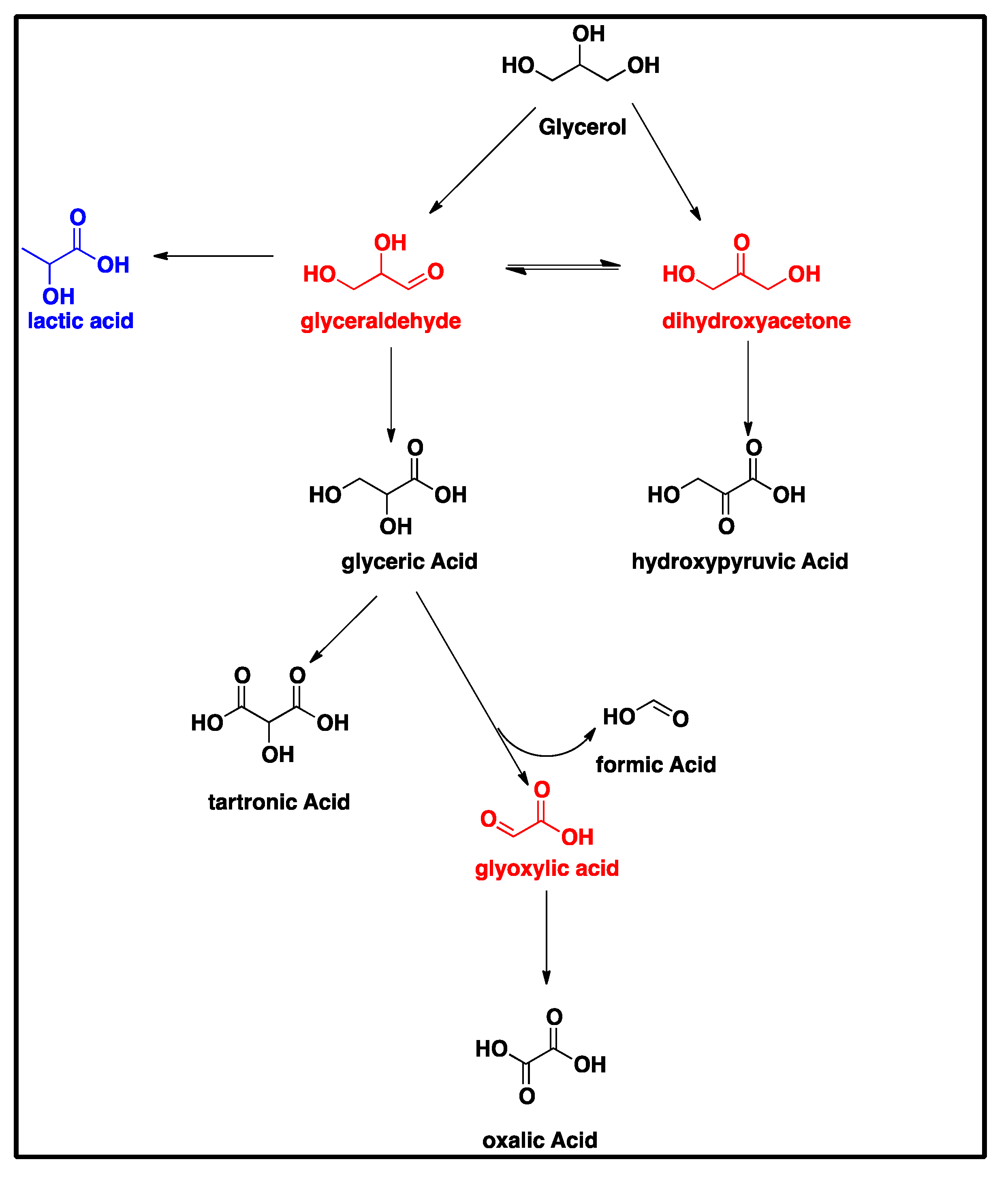
As-deposited electroless CuNiMoP/Al2O3 catalyst was successful synthesized, and its thermochemical oxidation ability for glycerol was tested and the comparison made between its thermochemical oxidation products and those of its electrochemical oxidation of glycerol. The SEM and EDS characterization results showed that the increase in plating time favored more active material deposition of CuNiMoP. HPLC analysis showed that except some minor differences in product distributions, two catalysts deposited for different lengths of time produced the following oxidation by-products: glyceric acid, tartronic acid, hydroxypyruvic acid, oxalic acid and formic acid. Lactic acid was observed only for the 60-min as-deposited electroless sample. Our results indicate that this low-cost non-noble metal-based CuNiMoP/Al2O3 catalyst is able to oxidize glycerol. However, more research has to be conducted to optimize its catalytic capability. A comparison of the thermochemical oxidation products with those formed from electrochemistry shows that in addition to organic acid by-products, the electrochemical process resulted in the production of some intermediate glycerol oxidation products, such as aldehydes and ketones, thereby suggesting that the electrochemical process is milder than the thermochemical oxidation process. No intermediate oxidation products such as aldehydes and ketones were recovered from the thermochemical oxidation. The electrochemical reaction mechanism for the oxidation of glycerol using the synthesized electroless CuNiMoP catalyst is suggested to have some important difference between it and the thermochemical oxidation mechanism for glycerol. The thermochemical oxidation mechanism for glycerol oxidation on the CuNiMoP/Al2O3 catalyst is suggested. The reason for the observed low conversion of glycerol with the catalyst is explained as being due to the presence of oxides, the amorphous nature of the deposit and the lack of catalyst activation by annealing under a hydrogen reductive environment. While the presence of oxides on the surface of CuNiMo catalyst does not seem to hinder the electron flow during electrocatalytic oxidation of glycerol on CuNiMo, it appears not favorable for electron exchange for the thermochemical oxidation of glycerol on the CuNiMo catalyst.
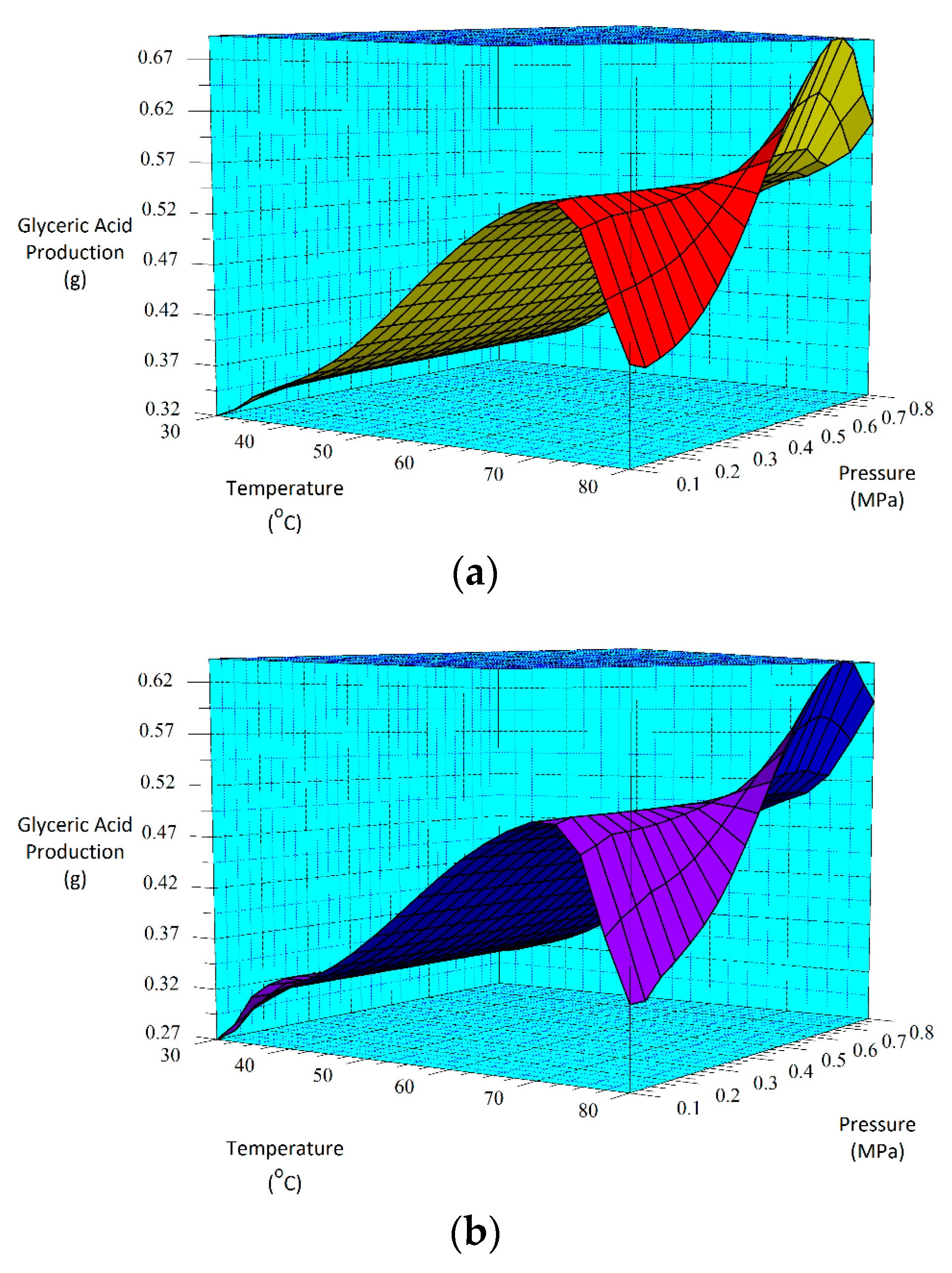
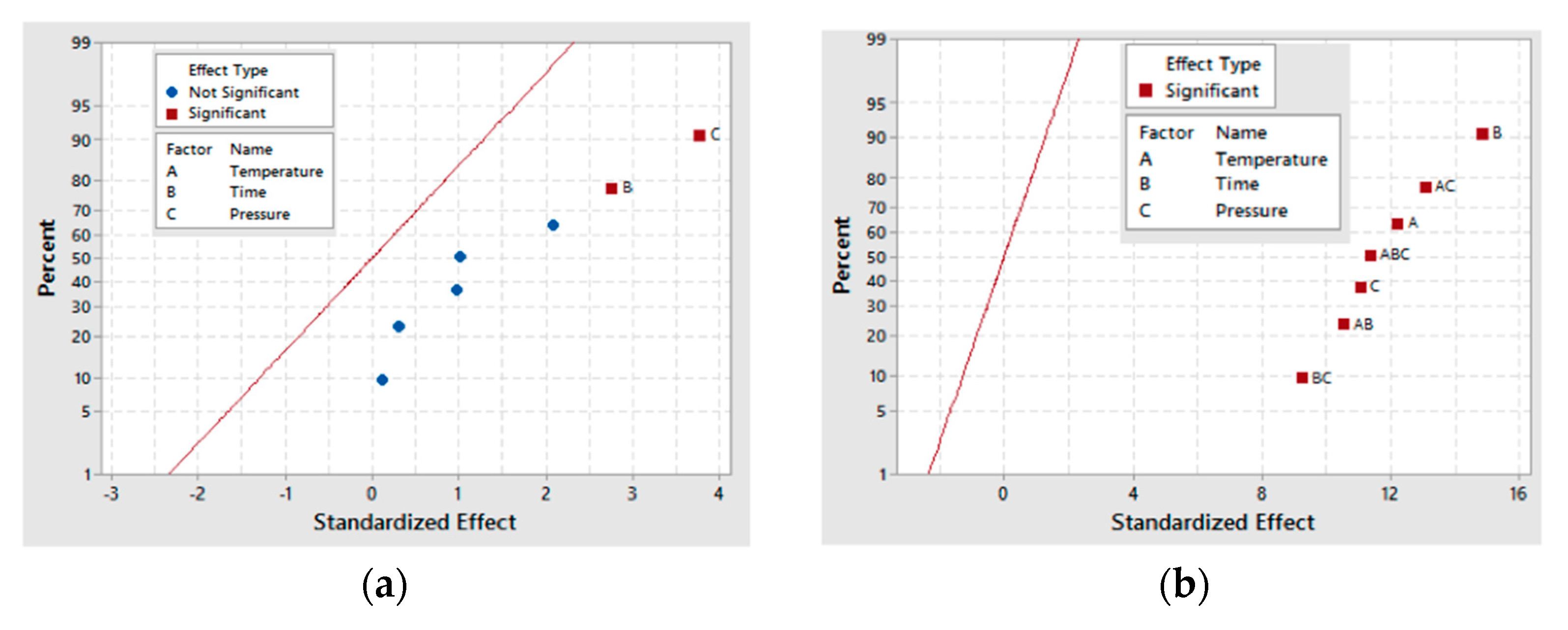
From the three-carbon compounds produced, the full factorial design demonstrated that the increase in each factor (A, B and C) enhances the formation of glyceric and tartronic acids. Additionally, the statistical analysis of the model emphasized the significance of temperature (A) and pressure (C) and the interactions (AC, BC and AB) on their production.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}