Mitigation of Cr(VI) Aqueous Pollution by the Reuse of Iron-Contaminated Water Treatment Residues



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Procedure
2.2.1. Fe(II) Treatability Experiments
2.2.2. Recovery and Activation of Exhausted Bentonite
2.2.3. Cr(VI) Treatability Experiments
2.2.4. Analytical Procedure
3. Results and Discussion
3.1. Mitigation of Fe(II) Pollution
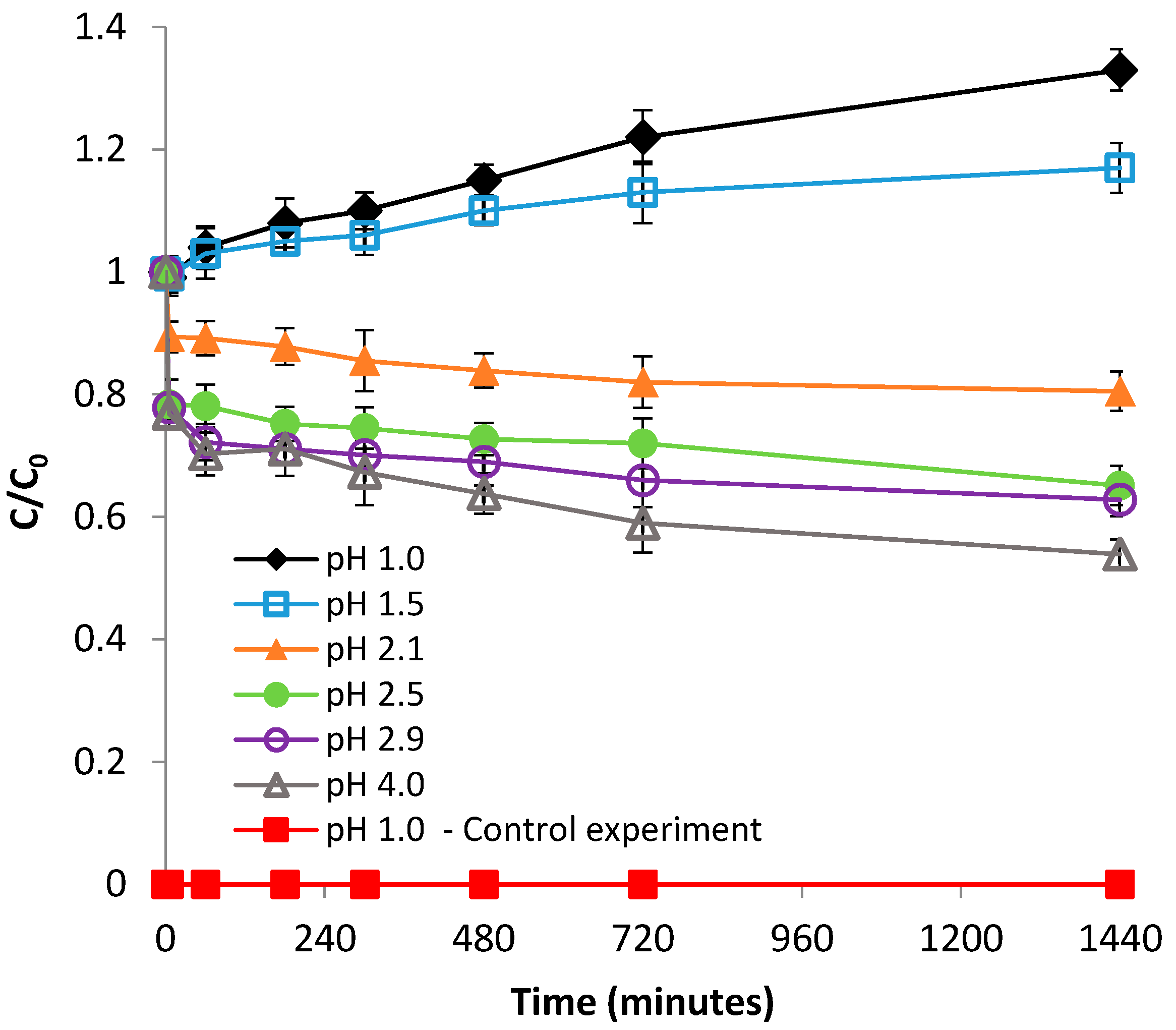
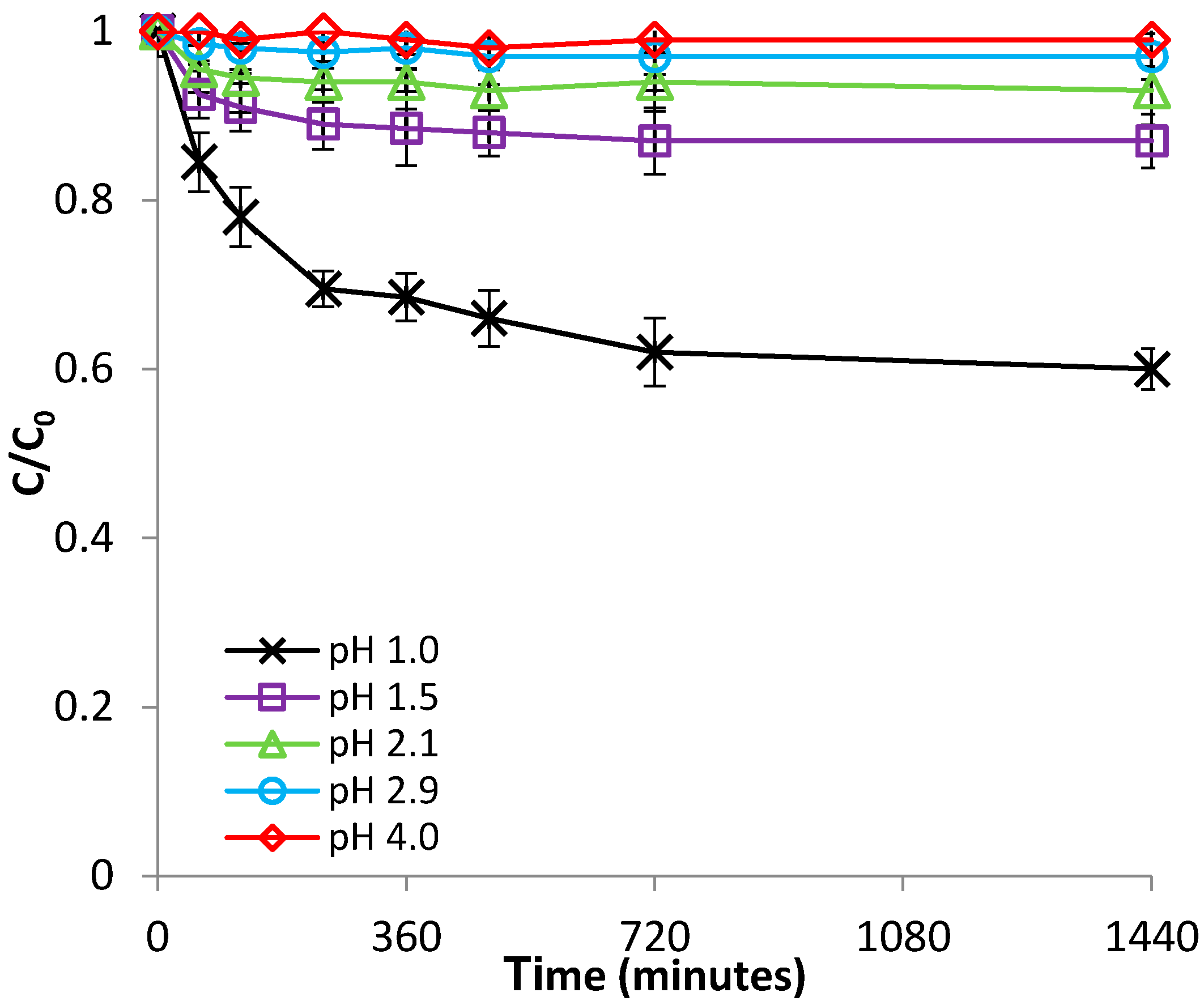
3.1.1. Effect of pH
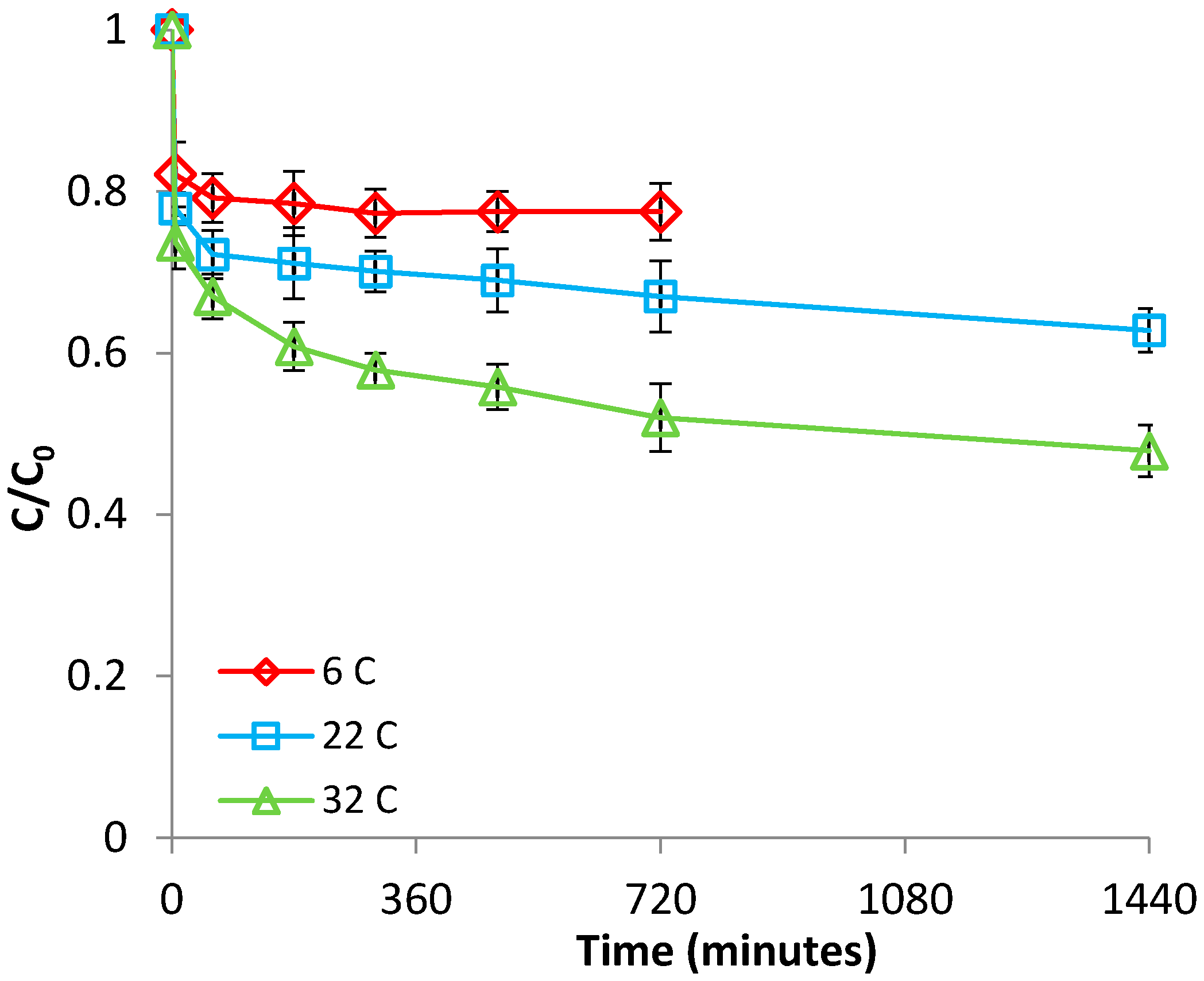
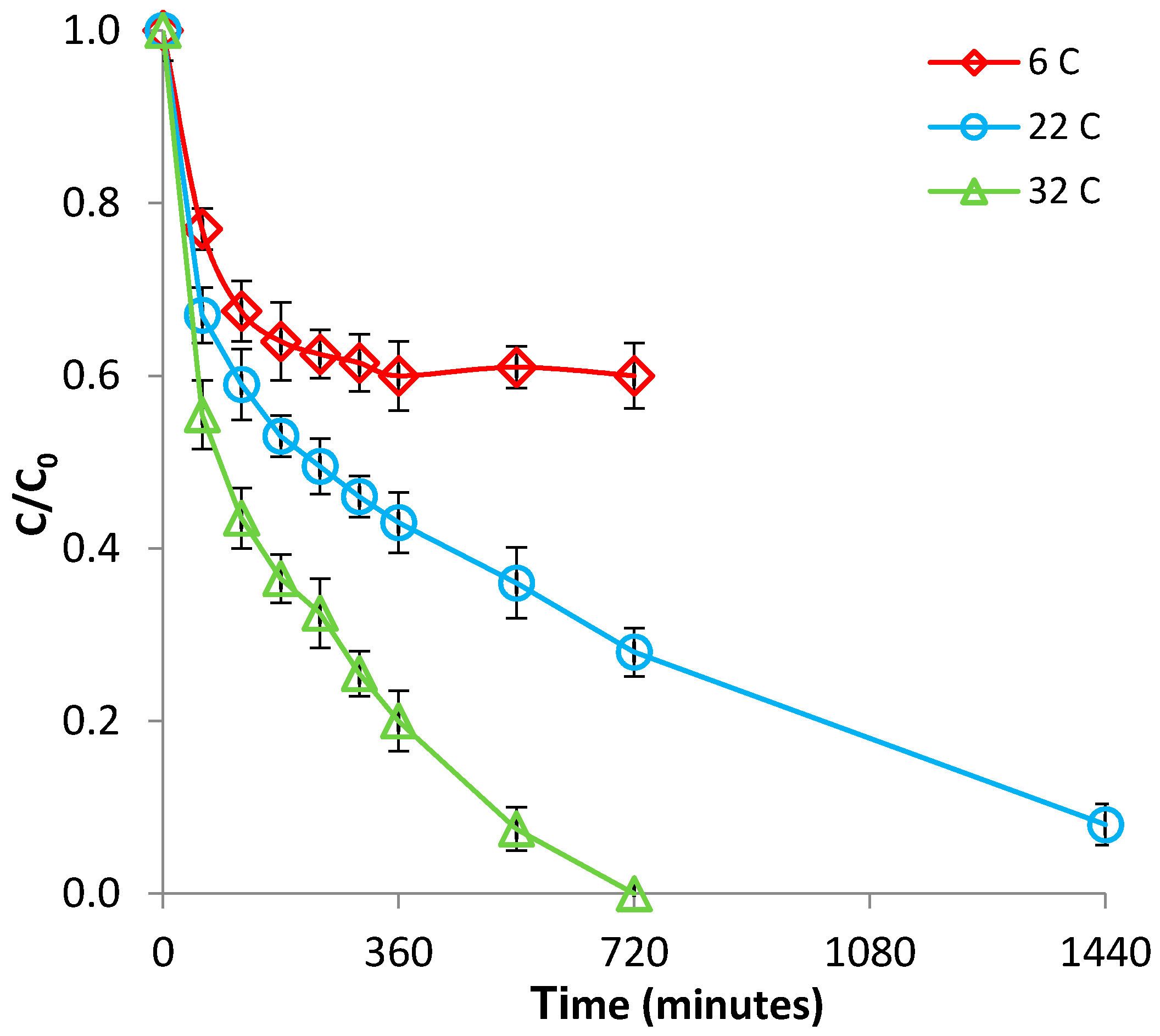
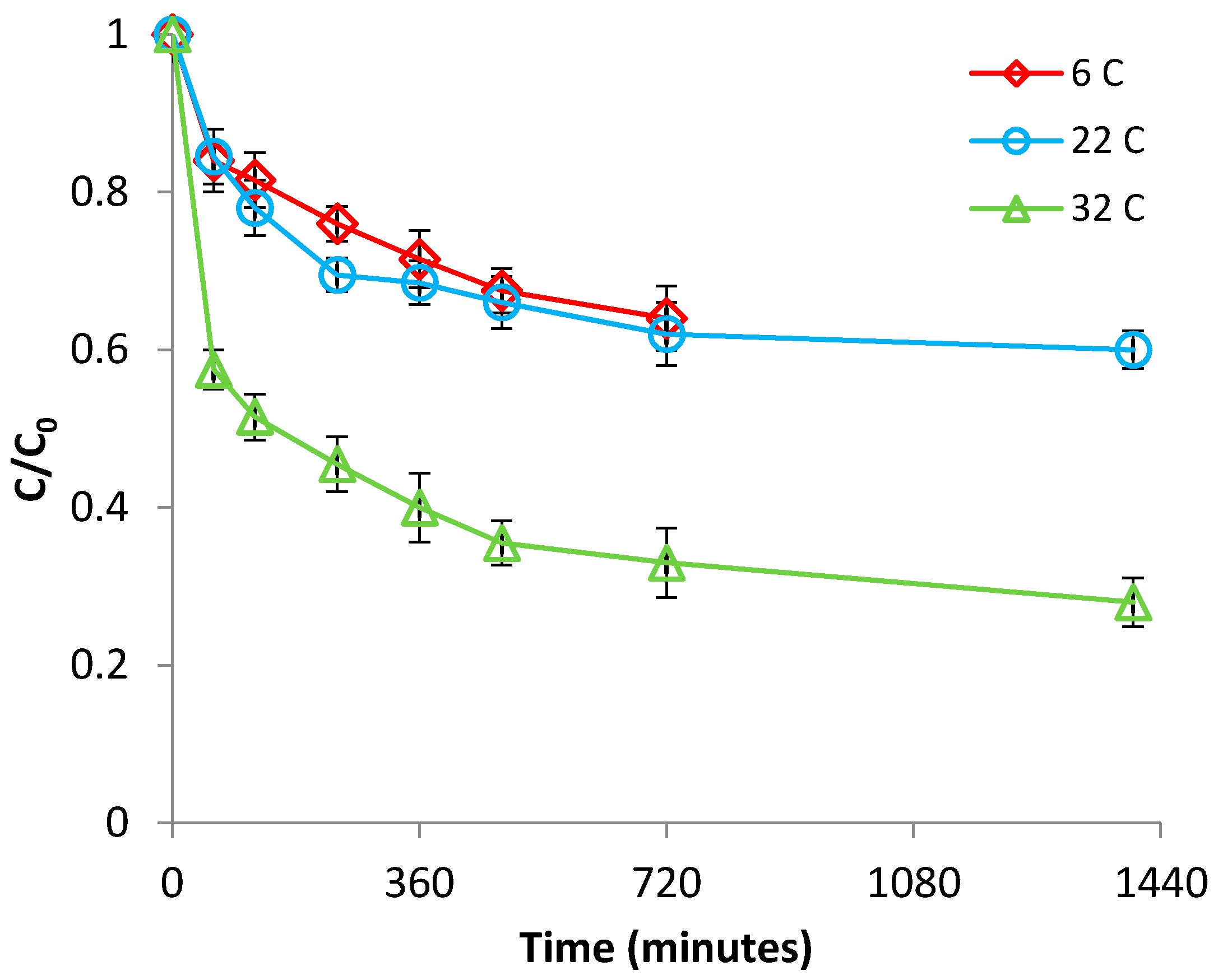
3.1.2. Effect of Temperature
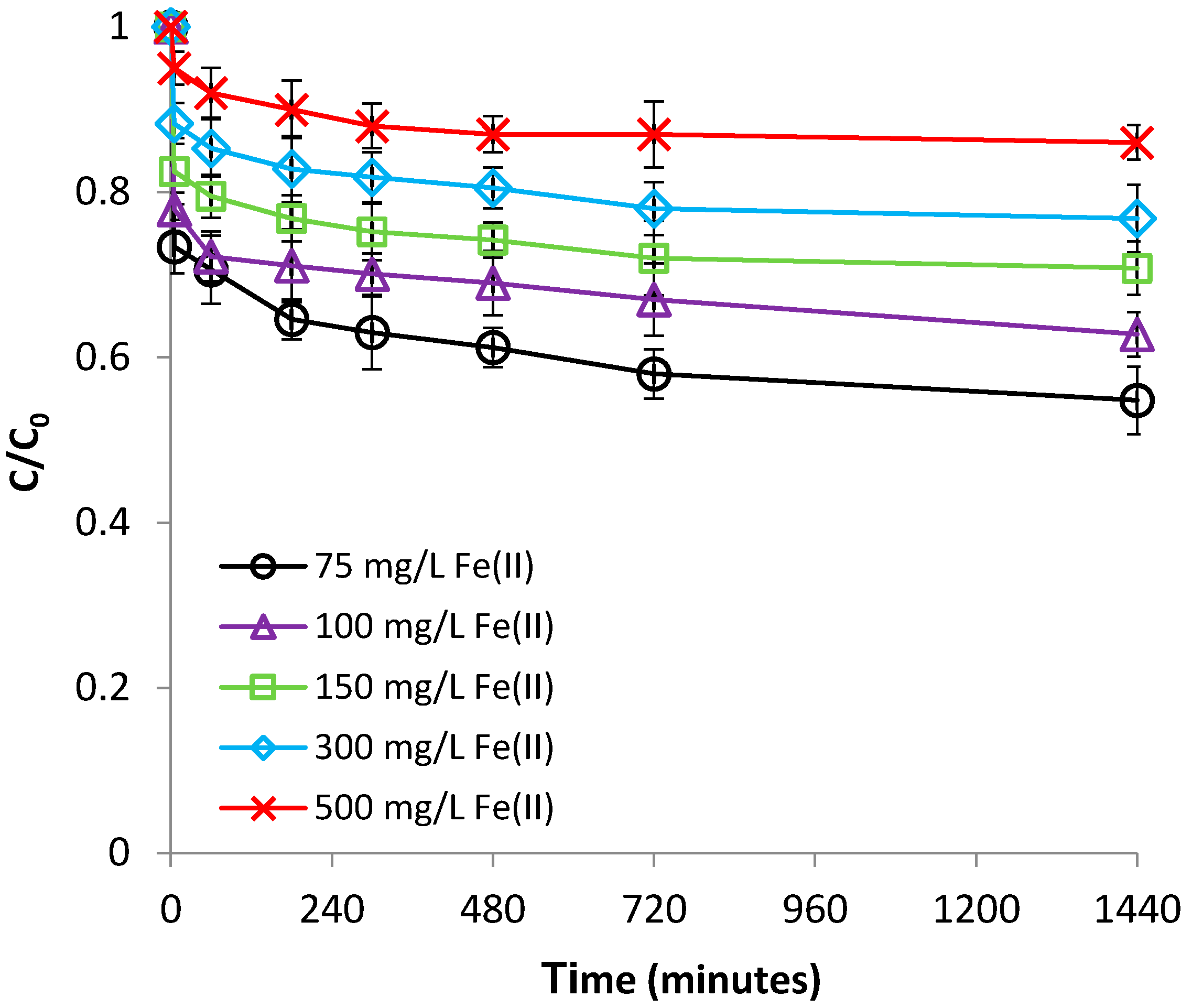
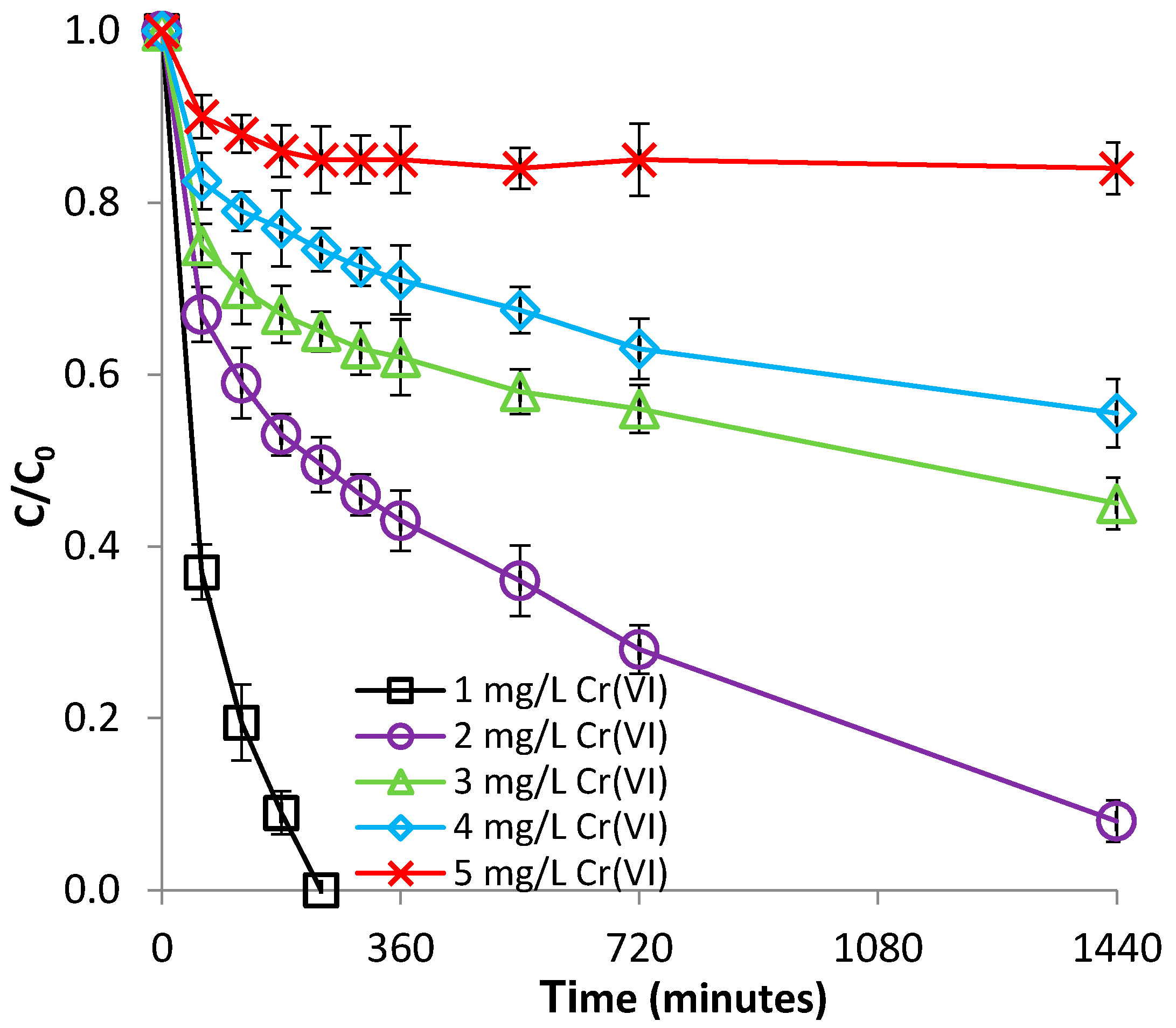
3.1.3. Effect of Fe(II) Concentration
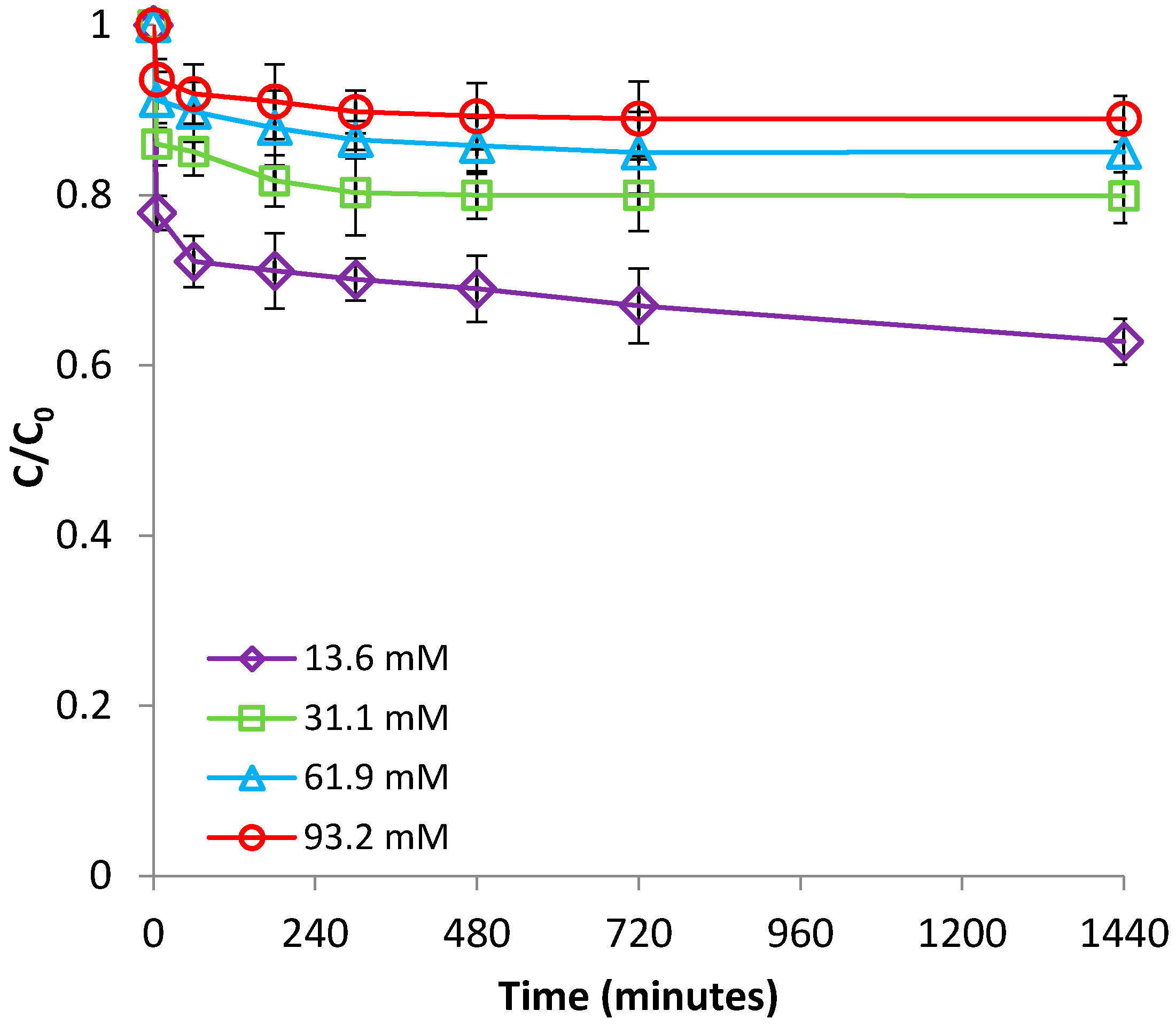
3.1.4. Effect of Ionic Strength
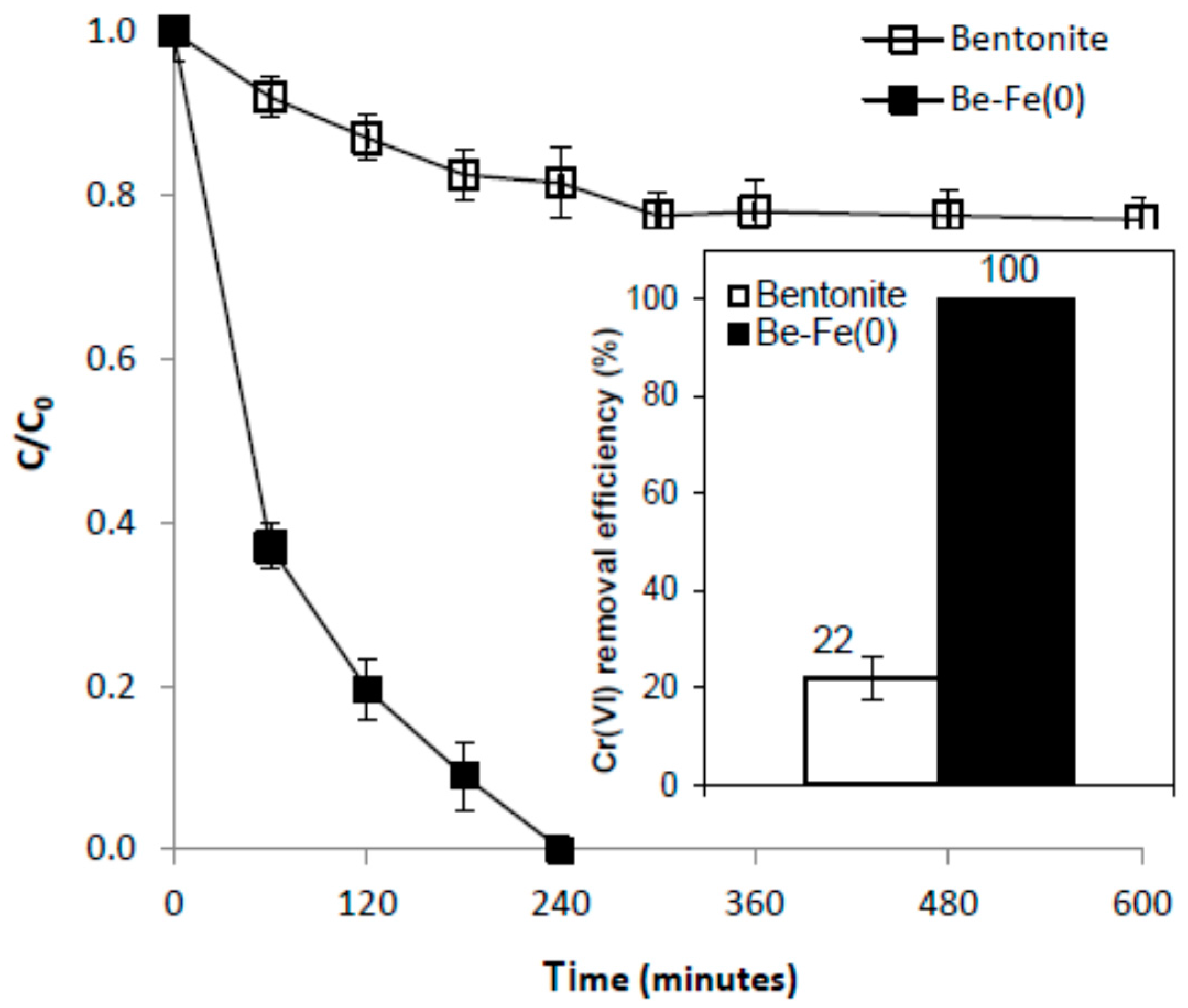
3.2. Mitigation of Cr(VI) Pollution
3.2.1. Effect of pH
3.2.2. Effect of Temperature
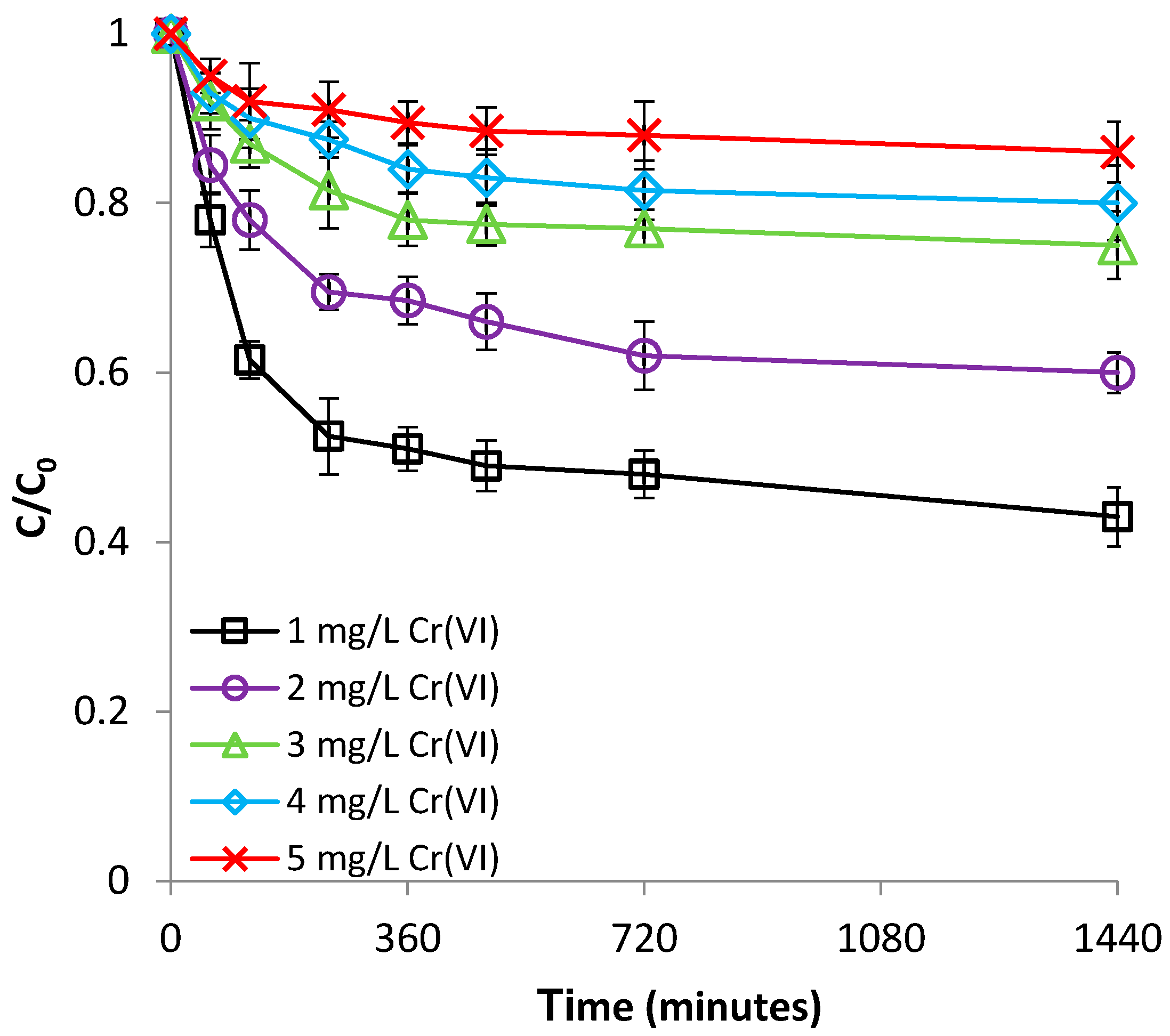
3.2.3. Effect of Cr(VI) Concentration
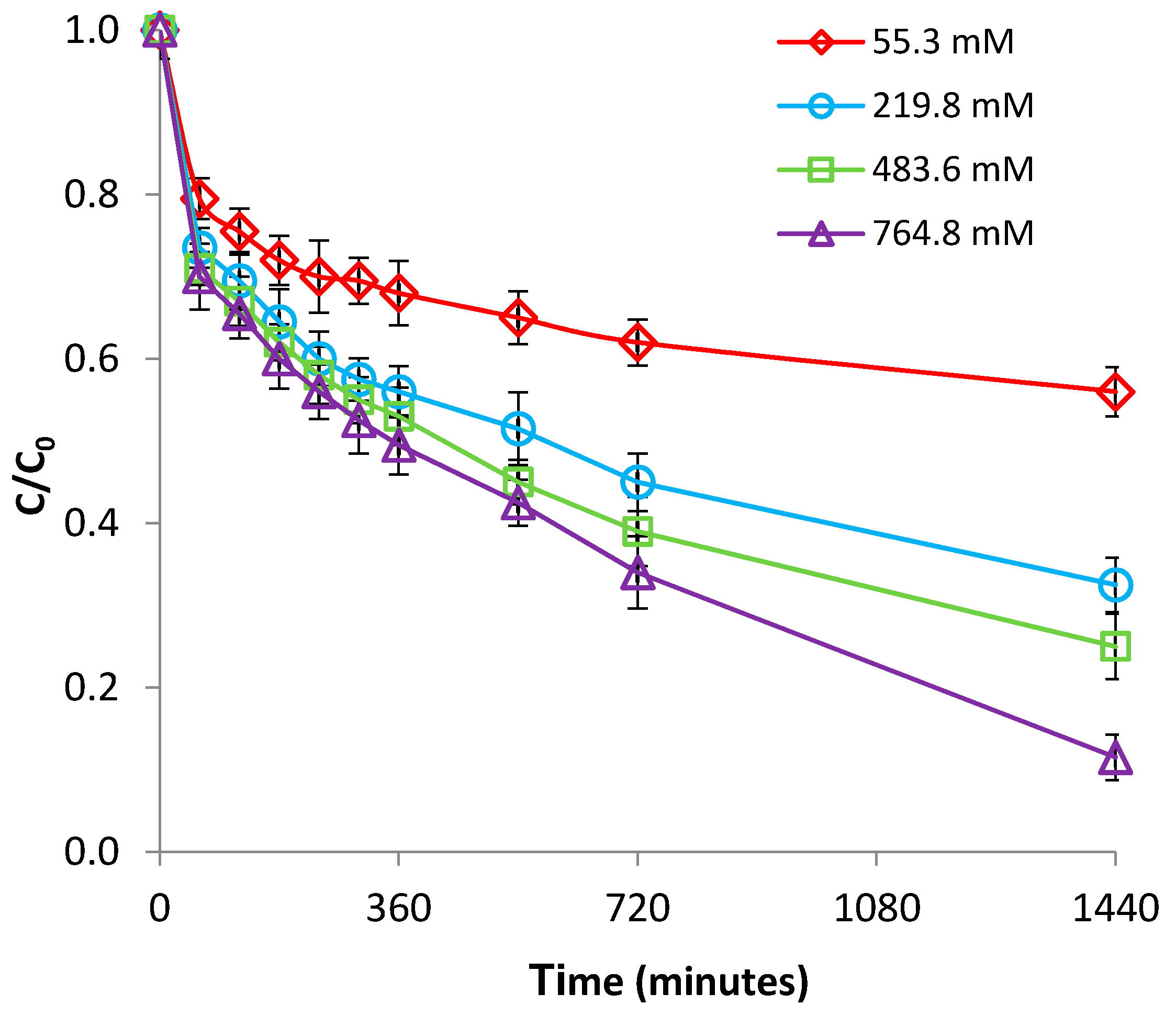
3.2.4. Effect of Ionic Strength
3.3. Solid Phase Characterization
3.4. Mechanism of Cr(VI) Removal with Be-Fe(0)
3SO42− + 6Fe(OH)3
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shrivastava, R.; Upreti, R.K.; Chaturvedi, U.C. Various cells of the immune system and intestine differ in their capacity to reduce hexavalent chromium. FEMS Immunol. Med. Microbiol. 2003, 1555, 1–6. [Google Scholar] [CrossRef]
- Keepax, R.E.; Moyes, L.; Livens, F.R. Speciation of heavy metals and radioisotopes. In Environmental and Ecological Chemistry; Sabljic, A., Ed.; Eolss Publishers Co.: Oxford, UK, 2009; Volume 2, pp. 165–199. [Google Scholar]
- Gheju, M. Hexavalent chromium reduction with zero-valent iron (ZVI) in aquatic systems. Water Air Soil Pollut. 2011, 222, 103–148. [Google Scholar]
- Vaiopoulou, E.; Gikas, P. Effects of chromium on activated sludge and on the performance of wastewater treatment plants: A review. Water Res. 2012, 46, 549–570. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Orvig, C. Biosorbents for hexavalent chromium elimination from industrial and municipal effluents. Coord. Chem. Rev. 2010, 254, 2959–2972. [Google Scholar] [CrossRef]
- Saha, R.; Nandi, R.; Saha, B. Sources and toxicity of hexavalent chromium. J. Coord. Chem. 2011, 64, 1782–1806. [Google Scholar] [CrossRef]
- Wise, S.S.; Holmes, A.L.; Liou, L.; Adam, R.M.; Wise, J.P. Hexavalent chromium induces chromosome instability in human urothelial cells. Toxicol. Appl. Pharmacol. 2016, 296, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Choppala, G.; Bolan, N.; Park, J.H. Chromium contamination and its risk management in complex environmental settings. Adv. Agron. 2013, 120, 129–172. [Google Scholar]
- Costa, M. Potential hazards of hexavalent chromate in our drinking water. Toxicol. Appl. Pharmacol. 2003, 188, 1–5. [Google Scholar] [CrossRef]
- Stefansson, A.; Seward, T. A spectrophotometric study of iron (III) hydrolysis in aqueous solutions to 200 °C. Chem. Geol. 2008, 249, 227–235. [Google Scholar] [CrossRef]
- Davison, W.; Seed, G. The kinetics of the oxidation of ferrous iron in synthetic and natural waters. Geochim. Cosmochim. Acta 1983, 47, 67–79. [Google Scholar] [CrossRef]
- Davison, W. Iron and manganese in lakes. Earth Sci. Rev. 1993, 34, 119–163. [Google Scholar] [CrossRef]
- Lieu, P.; Heiskala, M.; Peterson, P.; Yang, Y. The roles of iron in health and disease. Mol. Asp. Med. 2001, 22, 1–87. [Google Scholar] [CrossRef]
- Nair, M.; Iyengar, V. Iron content, bioavailability & factors affecting iron status of Indians. Indian J. Med. Res. 2009, 130, 634–645. [Google Scholar] [PubMed]
- Fontecave, M.; Pierre, J.L. Iron: Metabolism, toxicity and therapy. Biochimie 1993, 75, 767–773. [Google Scholar] [CrossRef]
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar] [PubMed]
- Andrews, N.C. Iron metabolism: Iron deficiency and iron overload. Annu. Rev. Genom. Hum. Genet. 2000, 1, 75–98. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, F.; Rocha, S.; Fernandes, R. Iron metabolism: From health to disease. J. Clin. Lab. Anal. 2014, 28, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Yingxin, Z.; Wenfang, Q.; Guanyi, C.; Min, J.; Zhenya, Z. Behavior of Cr(VI) removal from wastewater by adsorption onto HCl activated Akadama clay. J. Taiwan Inst. Chem. Eng. 2015, 50, 190–197. [Google Scholar]
- Te, B.; Wichitsathian, B.; Yossapol, C. Modification of natural common clays as low cost adsorbents for arsenate adsorption. Int. J. Environ. Sci. Dev. 2015, 6, 799–804. [Google Scholar] [CrossRef]
- Potgieter, J.H.; Potgieter-Vermaak, S.S.; Kalibantonga, P.D. Heavy metals removal from solution by palygorskite clay. Miner. Eng. 2006, 19, 463–470. [Google Scholar] [CrossRef]
- Noubactep, C. Metallic iron for environmental remediation: A review of reviews. Water Res. 2015, 85, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Mwakabona, H.; Ndé-Tchoupé, A.; Njau, K.; Noubactep, C.; Wydra, K. Metallic iron for safe drinking water provision: Considering a lost knowledge. Water Res. 2017, 117, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Suteerapataranon, S.; Bouby, M.; Geckeis, H.; Fanghanel, T.; Grudpan, K. Interaction of trace elements in acid mine drainage solution with humic acid. Water Res. 2006, 40, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Fiore, S.; Zanetti, M.C. Preliminary test concerning zero-valent iron efficiency in inorganic pollutants remediation. Am. J. Environ Sci. 2009, 5, 555–560. [Google Scholar] [CrossRef]
- Yunfei, X.; Megharaj, M.; Ravendra, N. Reduction and adsorption of Pb2+ in aqueous solution by nano-zero-valent iron—A SEM, TEM and XPS study. Mater. Res. Bull. 2010, 45, 1361–1367. [Google Scholar]
- Gheju, M.; Iovi, A.; Balcu, I. Hexavalent chromium reduction with scrap iron in continuous-flow system. Part 1: Effect of feed solution pH. J. Hazard. Mater. 2008, 153, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Zach-Maor, A.; Semiat, R.; Shemer, H. Synthesis, performance, and modeling of immobilized nano-sized magnetite layer for phosphate removal. J. Colloid Interface Sci. 2011, 357, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Reckhow, D.A. Analytical Chemistry for Environmental Engineers and Scientists. Lecture Notes to Accompany CEE 572 and CEE 772. Department of Civil Engineering University of Massachusetts: Amherst, MA 01003 Chapter XVIII: Electrochemical Methods. Available online: http://www.ecs.umass.edu/cee/reckhow/courses/572/572BKI.html (accessed on 8 September 2017).
- Espana, J.S.; Pamo, E.L.; Pastor, E.S. The oxidation of ferrous iron in acidic mine effluents from the Iberian Pyrite Belt (Odiel Basin, Huelva, Spain): Field and laboratory rates. J. Geochem. Explor. 2007, 92, 120–132. [Google Scholar] [CrossRef]
- Espana, J.S. Acid mine drainage in the Iberian pyrite belt: An overview with special emphasis on generation mechanisms, aqueous composition and associated mineral phases. Macla 2008, 10, 34–43. [Google Scholar]
- Karthikeyan, T.; Rajgopal, S.; Miranda, L.R. Chromium (VI) adsorption from aqueous solution by Hevea Brasilinesis sawdust activated carbon. J. Hazard. Mater. 2005, B124, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Granados-Correa, F.; Jimenez-Becerril, J. Chromium (VI) adsorption on boehmite. J. Hazard. Mater. 2009, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Baral, S.S.; Das, S.N.; Rath, P. Hexavalent chromium removal from aqueous solution by adsorption on treated sawdust. Biochem. Eng. J. 2006, 31, 216–222. [Google Scholar] [CrossRef]
- Brown, G., Jr.; Parks, G. Sorption of trace elements on mineral surfaces: Modern perspectives from spectroscopic studies, and comments on sorption in the marine environment. Int. Geol. Rev. 2001, 43, 963–1073. [Google Scholar] [CrossRef]
- Flury, B.; Eggenberger, U.; Mader, U. First results of operating and monitoring an innovative design of a permeable reactive barrier for the remediaton of chromate contaminated groundwater. J. Appl. Geochem. 2009, 24, 687–697. [Google Scholar] [CrossRef]
- Traubenberg, S.E.; Foley, R.T. The influence of chloride and sulfate ions on the corrosion of iron in sulfuric acid. J. Electrochem. Soc. 1971, 18, 1066–1070. [Google Scholar] [CrossRef]
- Tepong-Tsindé, R.; Phukan, M.; Nassi, A.; Noubactep, C.; Ruppert, H. Validating the efficiency of the MB discoloration method for the characterization of Fe0/H2O systems using accelerated corrosion by chloride ions. Chem. Eng. J. 2015, 279, 353–362. [Google Scholar] [CrossRef]
- White, A.F.; Paterson, M.L. Reduction of aqueous transition metal species on the surface of Fe(II)-containing oxides. Geochim. Cosmochim. Acta 1996, 60, 3799–3814. [Google Scholar] [CrossRef]













© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gheju, M.; Balcu, I. Mitigation of Cr(VI) Aqueous Pollution by the Reuse of Iron-Contaminated Water Treatment Residues. ChemEngineering 2017, 1, 9. https://doi.org/10.3390/chemengineering1020009
Gheju M, Balcu I. Mitigation of Cr(VI) Aqueous Pollution by the Reuse of Iron-Contaminated Water Treatment Residues. ChemEngineering. 2017; 1(2):9. https://doi.org/10.3390/chemengineering1020009
Chicago/Turabian StyleGheju, Marius, and Ionel Balcu. 2017. "Mitigation of Cr(VI) Aqueous Pollution by the Reuse of Iron-Contaminated Water Treatment Residues" ChemEngineering 1, no. 2: 9. https://doi.org/10.3390/chemengineering1020009
APA StyleGheju, M., & Balcu, I. (2017). Mitigation of Cr(VI) Aqueous Pollution by the Reuse of Iron-Contaminated Water Treatment Residues. ChemEngineering, 1(2), 9. https://doi.org/10.3390/chemengineering1020009