


Interconversion and Removal of Inorganic Nitrogen Compounds via UV Irradiation


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Irradiation Experiments
2.3. Analytical Techniques
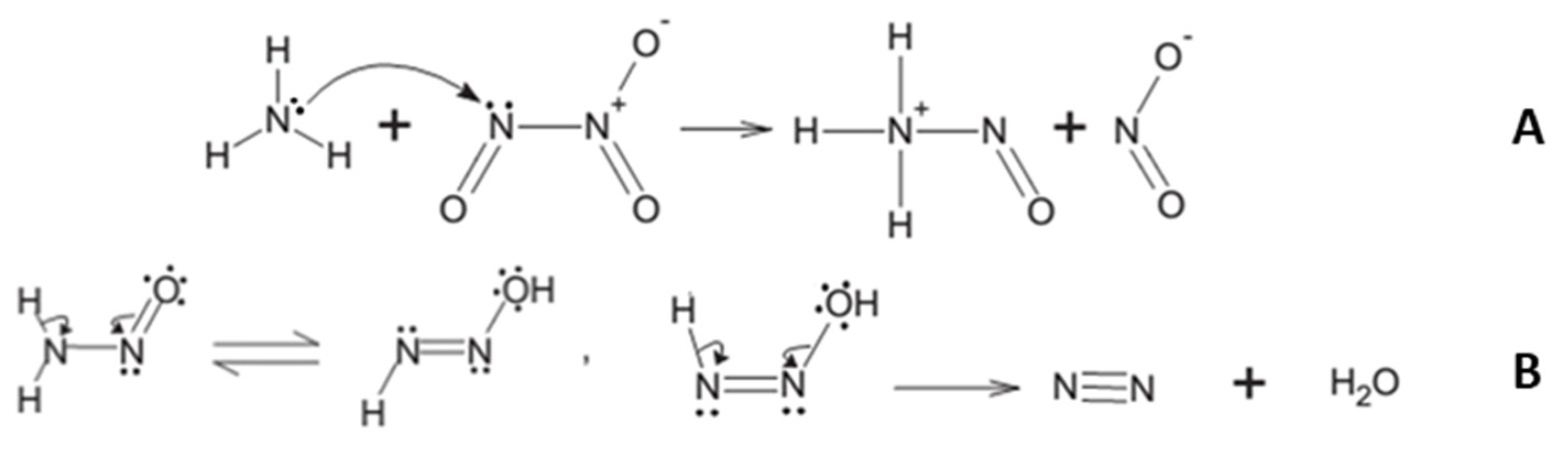
3. Results and Discussion
3.1. Irradiation of
3.2. Irradiation of
3.3. Irradiation of /
3.4. Simultaneous Irradiation of and /
3.5. Irradiation of in the Presence of Formic Acid
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galloway, J.N.; Aber, J.D.; Erisman, J.W.; Seitzinger, S.P.; Howarth, R.W.; Cowling, E.B.; Cosby, B.J. The Nitrogen Cascade. BioScience 2003, 53, 341–356. [Google Scholar] [CrossRef]
- Nolan, B.T.; Hitt, K.J.; Ruddy, B.C. Probability of Nitrate Contamination of Recently Recharged Groundwaters in the Conterminous United States. Environ. Sci. Technol. 2002, 36, 2138–2145. [Google Scholar] [CrossRef]
- Galloway, J.N.; Schlesinger, W.H.; Levy, H.; Michaels, A.; Schnoor, J.L. Nitrogen fixation: Anthropogenic enhancement-environmental response. Glob. Biogeochem. Cycles 1995, 9, 235–252. [Google Scholar] [CrossRef]
- Gruber, N.; Galloway, J.N. An Earth-system perspective of the global nitrogen cycle. Nature 2008, 451, 293–296. [Google Scholar] [CrossRef]
- Winkler, M.K.; Straka, L. New directions in biological nitrogen removal and recovery from wastewater. Curr. Opin. Biotechnol. 2019, 57, 50–55. [Google Scholar] [CrossRef]
- van der Star, W.R.L.; Abma, W.R.; Blommers, D.; Mulder, J.-W.; Tokutomi, T.; Strous, M.; Picioreanu, C.; van Loosdrecht, M.C.M. Startup of reactors for anoxic ammonium oxidation: Experiences from the first full-scale anammox reactor in Rotterdam. Water Res. 2007, 41, 4149–4163. [Google Scholar] [CrossRef]
- Tugaoen, H.O.; Garcia-Segura, S.; Hristovski, K.; Westerhoff, P. Challenges in photocatalytic reduction of nitrate as a water treatment technology. Sci. Total. Environ. 2017, 599–600, 1524–1551. [Google Scholar] [CrossRef]
- Mack, J.; Bolton, J.R. Photochemistry of nitrite and nitrate in aqueous solution: A review. J. Photochem. Photobiol. A Chem. 1999, 128, 1–13. [Google Scholar] [CrossRef]
- Vione, D.; Maurino, V.; Minero, C.; Pelizzetti, E. Reactions Induced in Natural Waters by Irradiationof Nitrate and Nitrite Ions. In The Handbook of Environmental Chemistry; Environmental Photochemistry Part II; Boule, P., Bahnemann, D.W., Robertson, P.K.J., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 221–253. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, D.D.; Huang, W.; Liu, B.; Chen, Q.; Huang, R.; Gen, M.; Mabato, B.R.G.; Chan, C.K.; Li, X.; et al. Enhanced Nitrite Production from the Aqueous Photolysis of Nitrate in the Presence of Vanillic Acid and Implications for the Roles of Light-Absorbing Organics. Environ. Sci. Technol. 2021, 55, 15694–15704. [Google Scholar] [CrossRef]
- Shibuya, S.; Aoki, S.; Sekine, Y.; Mikami, I. Influence of oxygen addition on photocatalytic oxidation of aqueous ammonia over platinum-loaded TiO2. Appl. Catal. B Environ. 2013, 138–139, 294–298. [Google Scholar] [CrossRef]
- Ren, H.-T.; Liang, Y.; Han, X.; Liu, Y.; Wu, S.-H.; Bai, H.; Jia, S.-Y. Photocatalytic oxidation of aqueous ammonia by Ag2O/TiO2 (P25): New insights into selectivity and contributions of different oxidative species. Appl. Surf. Sci. 2020, 504, 144433. [Google Scholar] [CrossRef]
- Mokhtar, B.; Ahmed, A.Y.; Kandiel, T.A. Revisiting the mechanisms of nitrite ions and ammonia removal from aqueous solutions: Photolysis versus photocatalysis. Photochem. Photobiol. Sci. 2022, 21, 1833–1843. [Google Scholar] [CrossRef]
- Zhou, S.; Li, L.; Wu, Y.; Zhu, S.; Zhu, N.; Bu, L.; Dionysiou, D.D. UV365 induced elimination of contaminants of emerging concern in the presence of residual nitrite: Roles of reactive nitrogen species. Water Res. 2020, 178, 115829. [Google Scholar] [CrossRef]
- Wang, J.; Song, M.; Chen, B.; Wang, L.; Zhu, R. Effects of pH and H2O2 on ammonia, nitrite, and nitrate transformations during UV254nm irradiation: Implications to nitrogen removal and analysis. Chemosphere 2017, 184, 1003–1011. [Google Scholar] [CrossRef]
- APHA; AWWA; WEF (Eds.) Method 4500-NO3- E. Cadmium reduction method. In Standard Methods for the Examination of Water and Wastewater, 21st ed.; American Public Health Association/American Water Works Association/Water Environment Federation: Washington, DC, USA, 2005. [Google Scholar]
- APHA; AWWA; WEF (Eds.) Method 4500-NO2- B. Colorimetric method. In Standard Methods for the Examination of Water and Wastewater, 21st ed.; American Public Health Association/American Water Works Association/Water Environment Federation: Washington, DC, USA, 2005. [Google Scholar]
- APHA; AWWA; WEF (Eds.) Method 4500-NH3 F. Phenate Method. In Standard Methods for the Examination of Water and Wastewater, 21st ed.; American Public Health Association/American Water Works Association/Water Environment Federation: Washington, DC, USA, 2005. [Google Scholar]
- Molina, C.; Kissner, R.; Koppenol, W.H. Decomposition kinetics of peroxynitrite: Influence of pH and buffer. Dalton Trans. 2013, 42, 9898–9905. [Google Scholar] [CrossRef]
- Chu, L.; Anastasio, C. Quantum Yields of Hydroxyl Radical and Nitrogen Dioxide from the Photolysis of Nitrate on Ice. J. Phys. Chem. A 2003, 107, 9594–9602. [Google Scholar] [CrossRef]
- Tomanová, K.; Precek, M.; Múčka, V.; Vyšín, L.; Juha, L.; Čuba, V. At the crossroad of photochemistry and radiation chemistry: Formation of hydroxyl radicals in diluted aqueous solutions exposed to ultraviolet radiation. Phys. Chem. Chem. Phys. 2017, 19, 29402–29408. [Google Scholar] [CrossRef]
- Huang, L.; Li, L.; Dong, W.; Liu, Y.; Hou, H. Removal of Ammonia by OH Radical in Aqueous Phase. Environ. Sci. Technol. 2008, 42, 8070–8075. [Google Scholar] [CrossRef]
- Wang, A.; Edwards, J.G.; Davies, J.A. Photooxidation of aqueous ammonia with titania-based heterogeneous catalysts. Sol. Energy 1994, 52, 459–466. [Google Scholar] [CrossRef]
- Nguyen, D.A.; Iwaniw, M.A.; Fogler, H. Kinetics and mechanism of the reaction between ammonium and nitrite ions: Experimental and theoretical studies. Chem. Eng. Sci. 2003, 58, 4351–4362. [Google Scholar] [CrossRef]
- Harrison, C.C.; Malati, M.A.; Smetham, N.B. The UV-enhanced decomposition of aqueous ammonium nitrite. J. Photochem. Photobiol. A Chem. 1995, 89, 215–219. [Google Scholar] [CrossRef]
- Arakaki, T.; Miyake, T.; Hirakawa, T.; Sakugawa, H. pH Dependent Photoformation of Hydroxyl Radical and Absorbance of Aqueous-Phase N(III) (HNO2 and NO2−). Environ. Sci. Technol. 1999, 33, 2561–2565. [Google Scholar] [CrossRef]
- Vione, D.; Maurino, V.; Minero, C.; Pelizzetti, E. Phenol photonitration upon UV irradiation of nitrite in aqueous solution I: Effects of oxygen and 2-propanol. Chemosphere 2001, 45, 893–902. [Google Scholar] [CrossRef]
- Chen, G.; Hanukovich, S.; Chebeir, M.; Christopher, P.; Liu, H. Nitrate Removal via a Formate Radical-Induced Photochemical Process. Environ. Sci. Technol. 2019, 53, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Miller, D.N.; Brooks, M.H.; Widdowson, M.A.; Killingstad, M.W. In Situ Stimulation of Groundwater Denitrification with Formate to Remediate Nitrate Contamination. Environ. Sci. Technol. 2001, 35, 196–203. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purge | pH | Kinetic Rate Constant (min−1) | Correlation Coefficient (R2) | Nitrogen Recovery (%) |
|---|---|---|---|---|
| Argon | 3 | 0.003 | 0.9980 | 83 |
| 6 | 0.024 | 0.9917 | 93 | |
| 7 | 0.048 | 0.9879 | 92 | |
| 11 | 0.121 | 0.9995 | 99 | |
| Oxygen | 10 | 0.084 | 0.9919 | 92 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Senn, A.M.; Quici, N. Interconversion and Removal of Inorganic Nitrogen Compounds via UV Irradiation. ChemEngineering 2023, 7, 79. https://doi.org/10.3390/chemengineering7050079
Senn AM, Quici N. Interconversion and Removal of Inorganic Nitrogen Compounds via UV Irradiation. ChemEngineering. 2023; 7(5):79. https://doi.org/10.3390/chemengineering7050079
Chicago/Turabian StyleSenn, Alejandro M., and Natalia Quici. 2023. "Interconversion and Removal of Inorganic Nitrogen Compounds via UV Irradiation" ChemEngineering 7, no. 5: 79. https://doi.org/10.3390/chemengineering7050079
APA StyleSenn, A. M., & Quici, N. (2023). Interconversion and Removal of Inorganic Nitrogen Compounds via UV Irradiation. ChemEngineering, 7(5), 79. https://doi.org/10.3390/chemengineering7050079