Fabrication of Bioactive Surfaces by Functionalization of Electroactive and Surface-Active Block Copolymers

Abstract
:
1. Introduction
2. Experimental Section
2.1. Chemicals and Reagents
2.2. Preparation of Difunctional Initiator
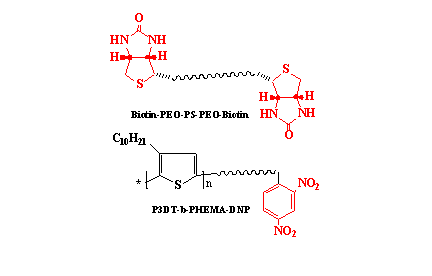
2.3. Synthesis of α, ω-bi-Biotin(Poly(Ethylene Oxide)-b-Poly(Styrene)-b-Poly(Ethylene Oxide)), (Biotin-PEO-PS-PEO-Biotin)

2.4. Synthesis of 2,5-Dibromo-3-Decylthiophene
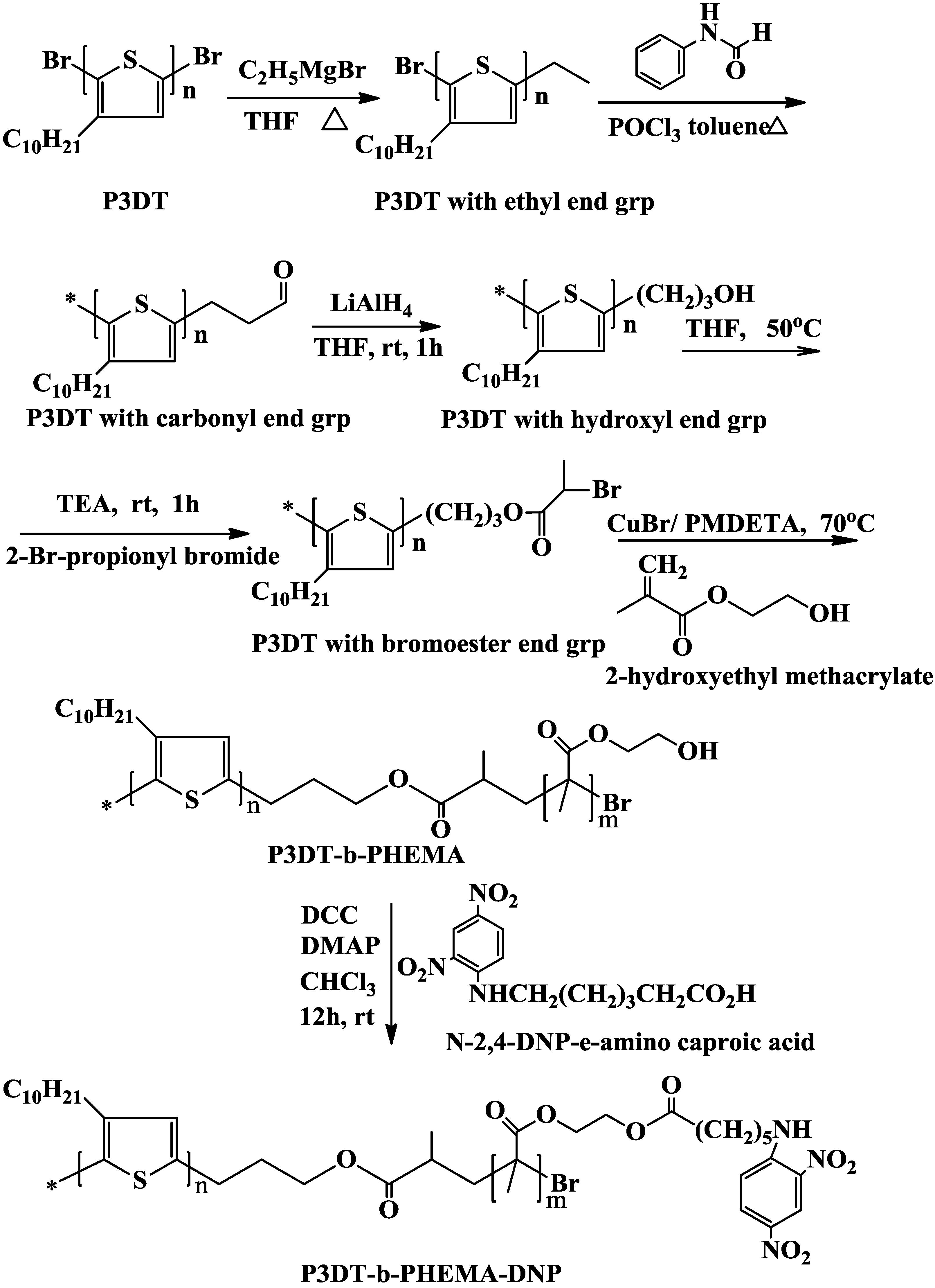
2.5. Synthesis of Regioregular Poly(3-Decylthiophene)
2.6. Preparation of P3DT with Proton End Group
2.7. Preparation of P3DT with Carbonyl End Group
2.8. Preparation of P3DT with Hydroxyl End Group
2.9. Preparation of Bromoester End Capped P3DT
2.10. ATRP of 2-Hydroxyethyl Methacrylate with P3DT Macroinitiator

2.11. Functionalization of P3DT-b-PHEMA with DNP Groups
2.12. Polymer Processing
2.13. Biofunctional Property Studies
2.13.1. Block Copolymers from 3-Decylthiophene and 2-Hydroxyethyl Methacrylate Functionalized with DNP
2.13.2. α, ω-bi-Biotin (Poly (Ethylene Oxide)-b-Poly(Styrene)-b-Poly (Ethylene Oxide)) Block Copolymers
2.14. Characterization/Instrumentation Techniques
3. Results and Discussion
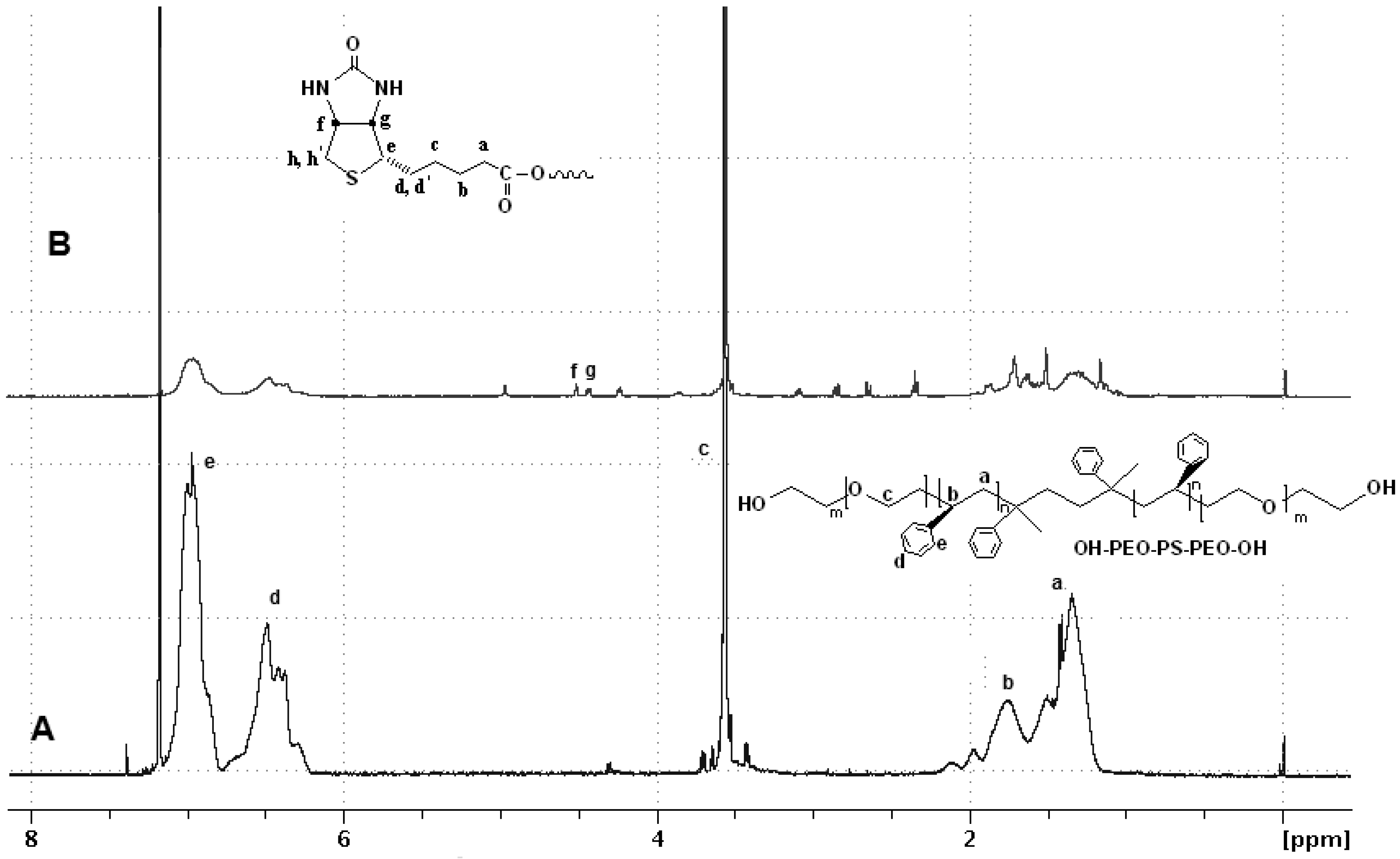
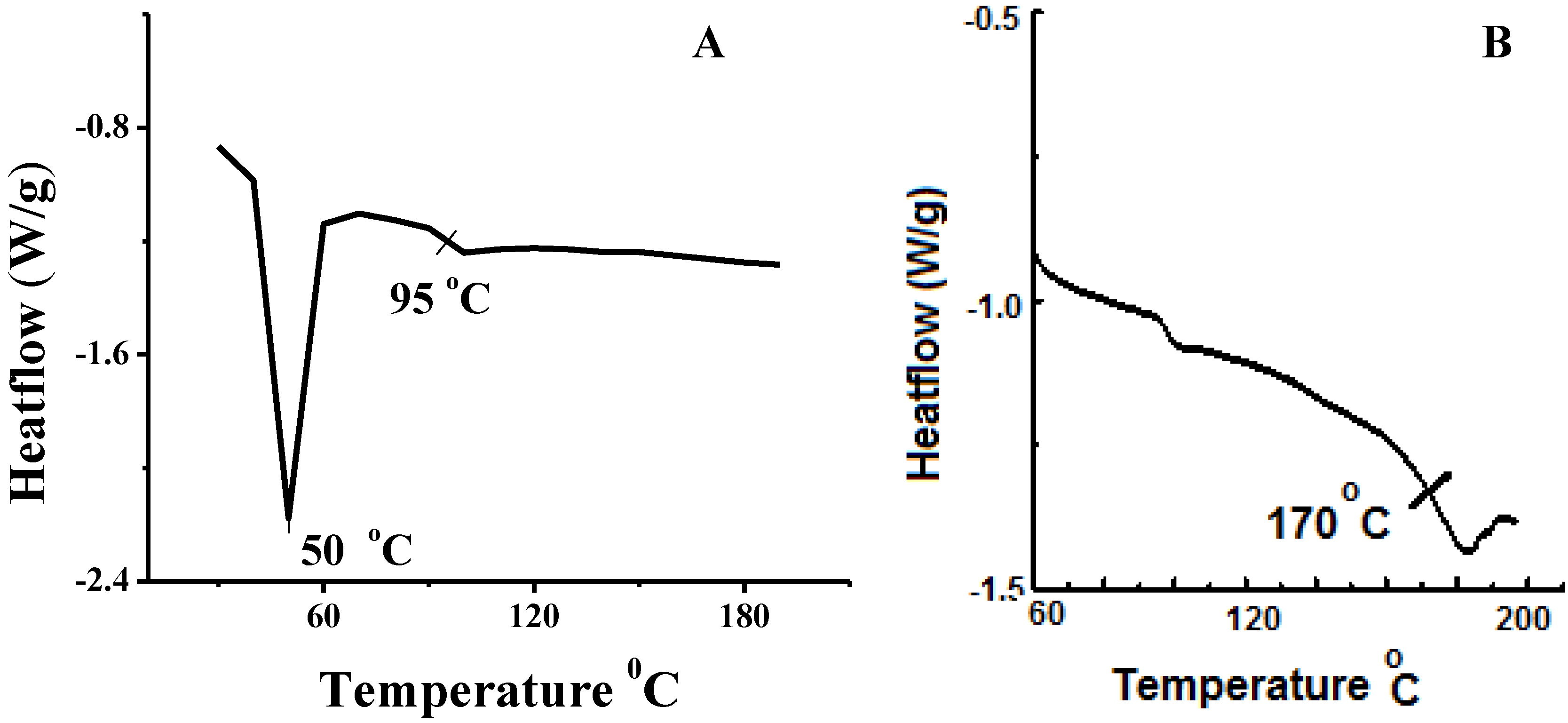
3.1. Characterization of α, ω-bi-Biotin(Poly(Ethylene Oxide)-b-Poly(Styrene)-b-Poly(Ethylene Oxide))

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | M1/M2 (1H NMR) | Mn (GPC) | Mw (GPC) | PDI | Mn (1HNMR) |
|---|---|---|---|---|---|
| Sample 1 | 21/79 | 15k | 20k | 1.34 | 15k |
| Sample 2 | 21/79 | 18k | 22k | 1.24 | 13k |
| Sample 3 | 31/69 | 13k | 25k | 1.91 | 13k |
| Sample 4 | 32/68 | - | - | - | 33k |


| Samples | Atom | Peak (eV) | FWHM (eV) | Atom % |
|---|---|---|---|---|
| 13k AuNPs fibers | O1s | 533.16 | 2.83 | 27.25 |
| Si2p | 100.22 | 2.61 | 43.72 | |
| C1s | 294.08 | 3.28 | 25.01 |

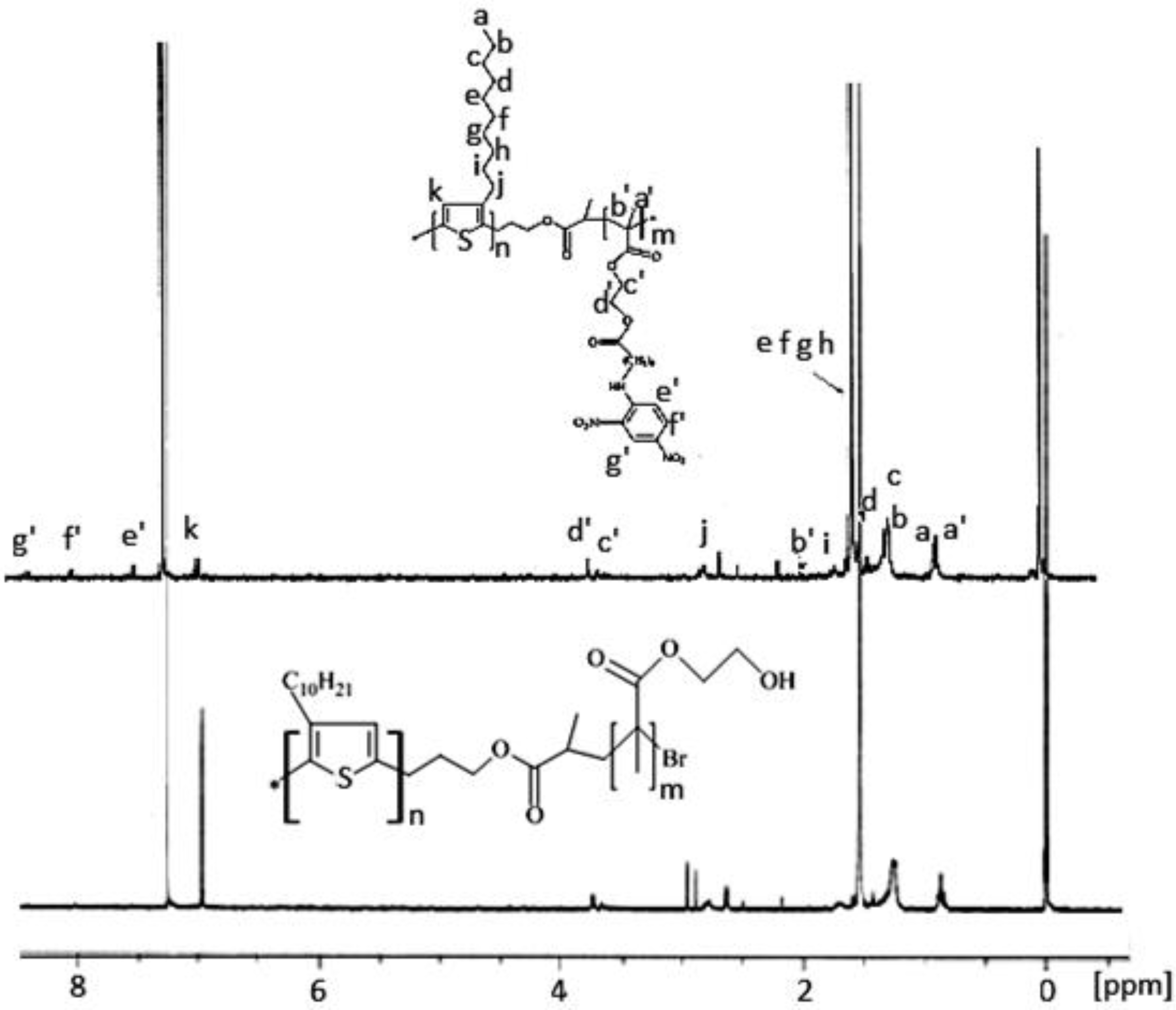
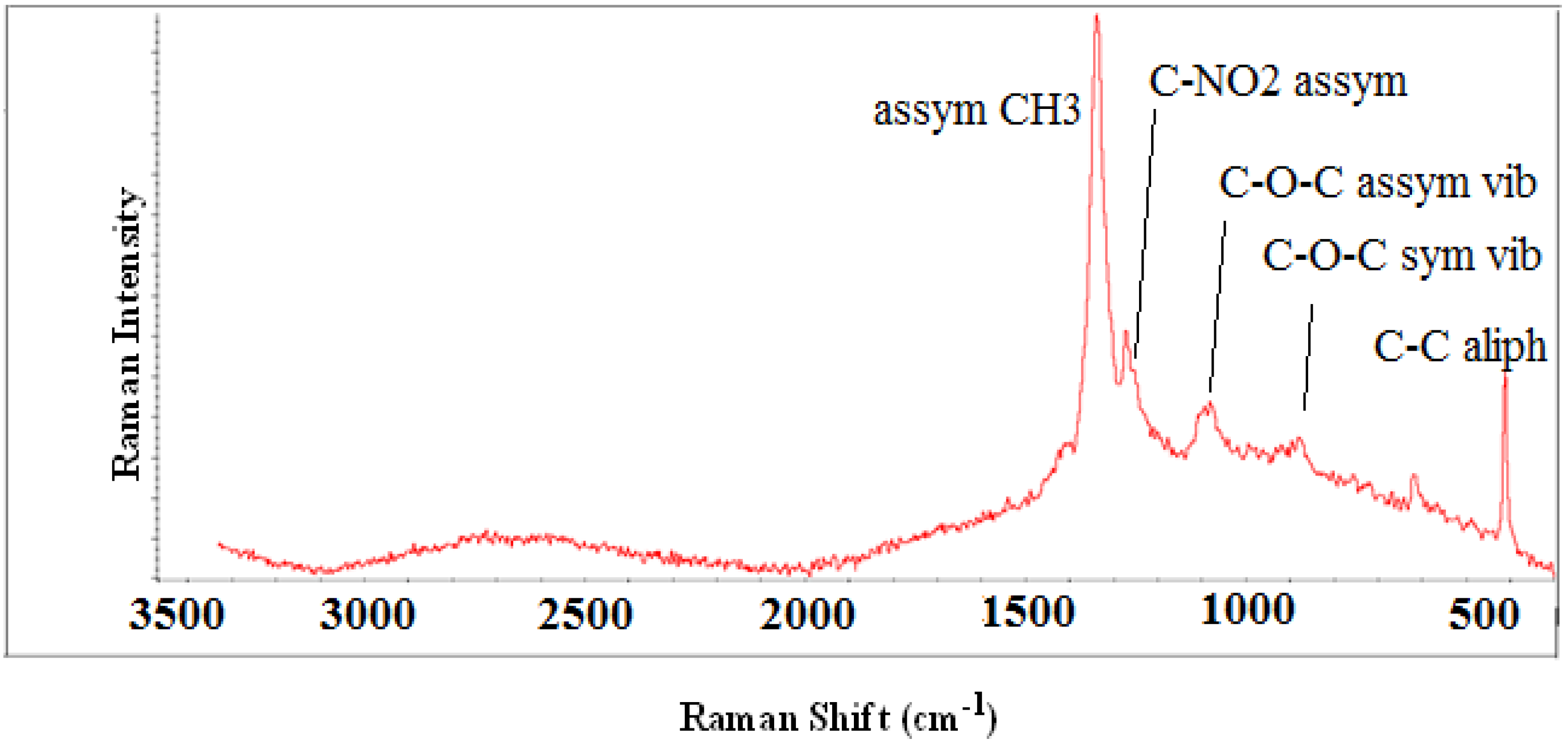
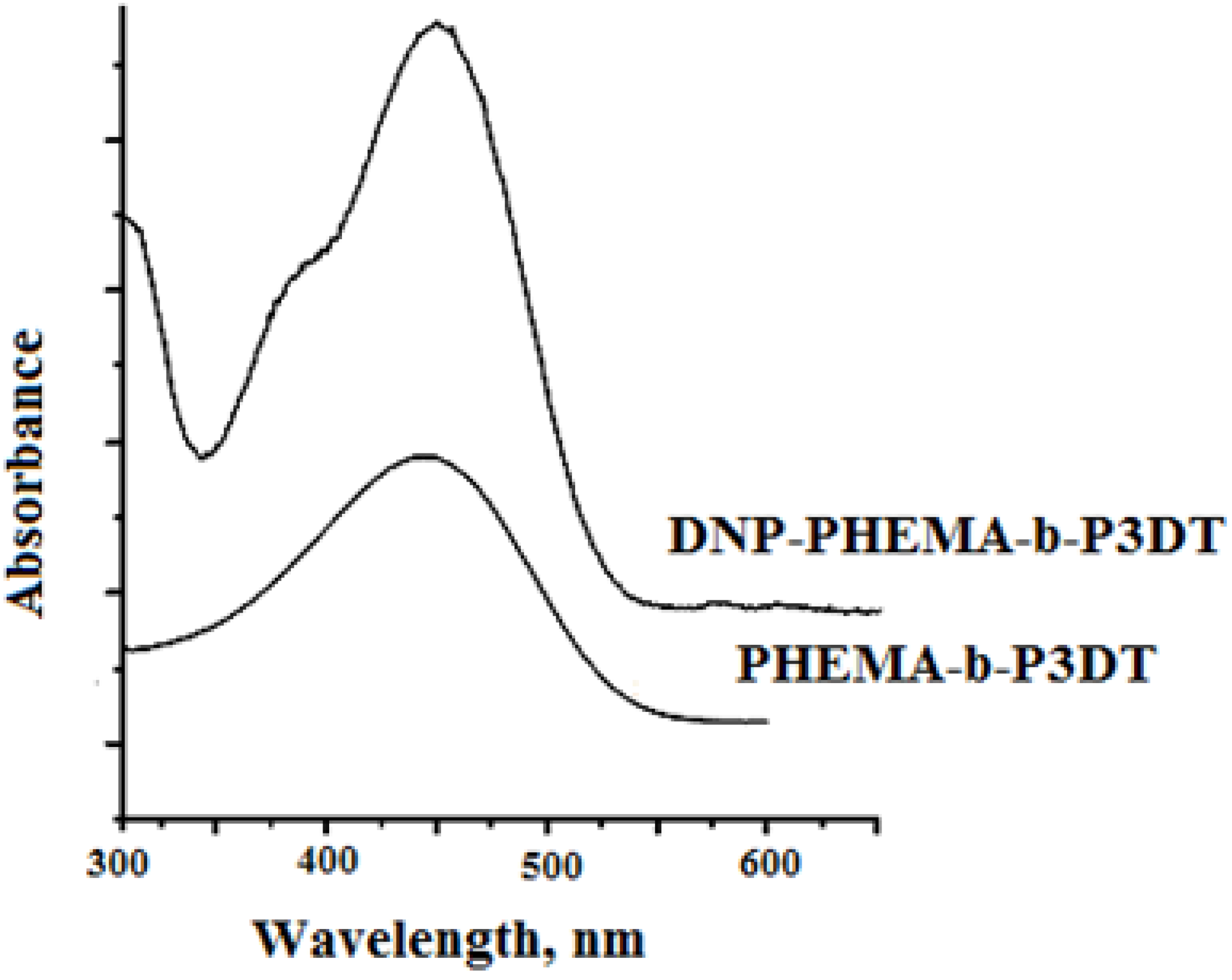
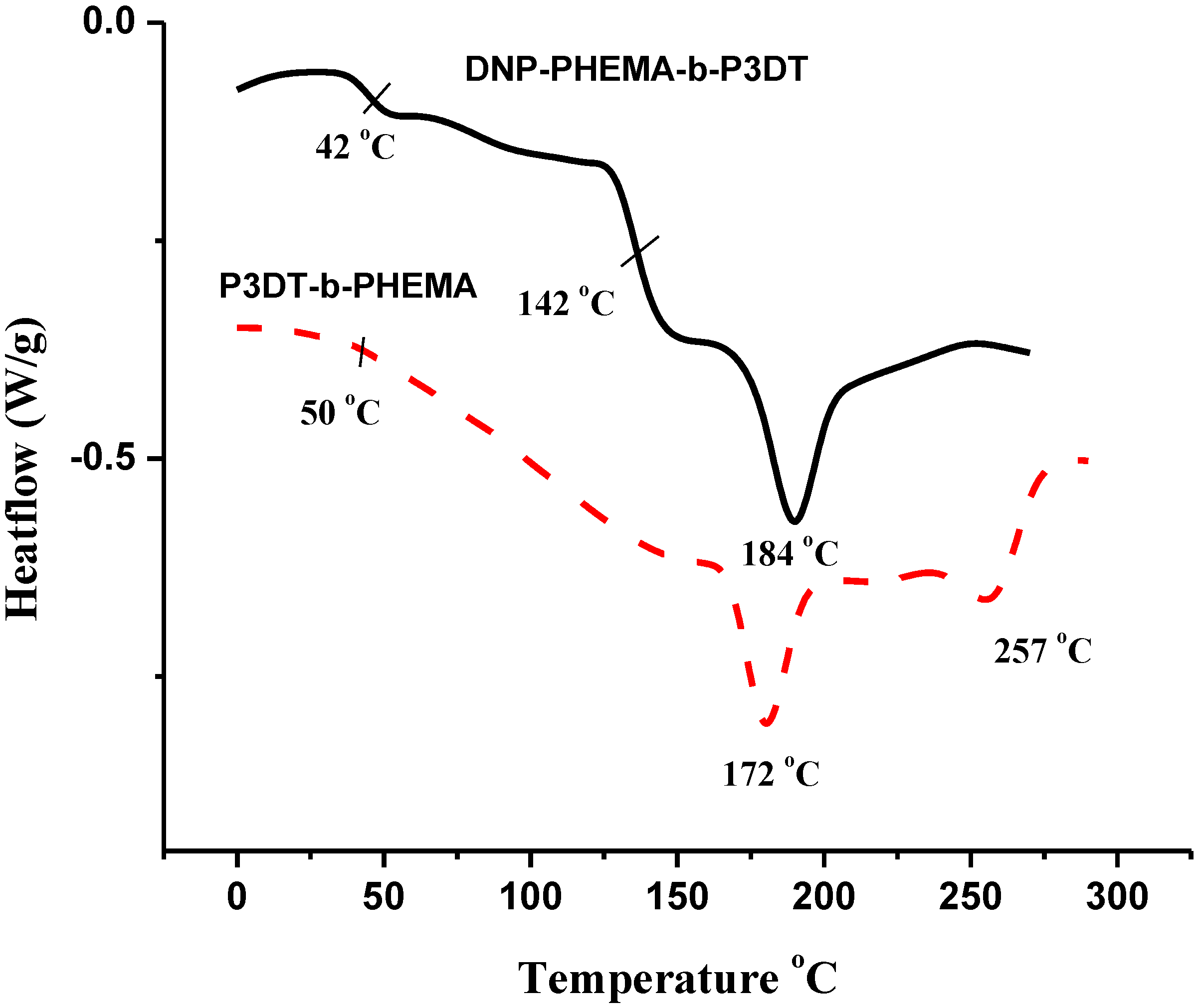
3.2. Characterization of Block Copolymers from 3-Decylthiophene and 2-Hydroxyethyl Methacrylate Functionalized with DNP






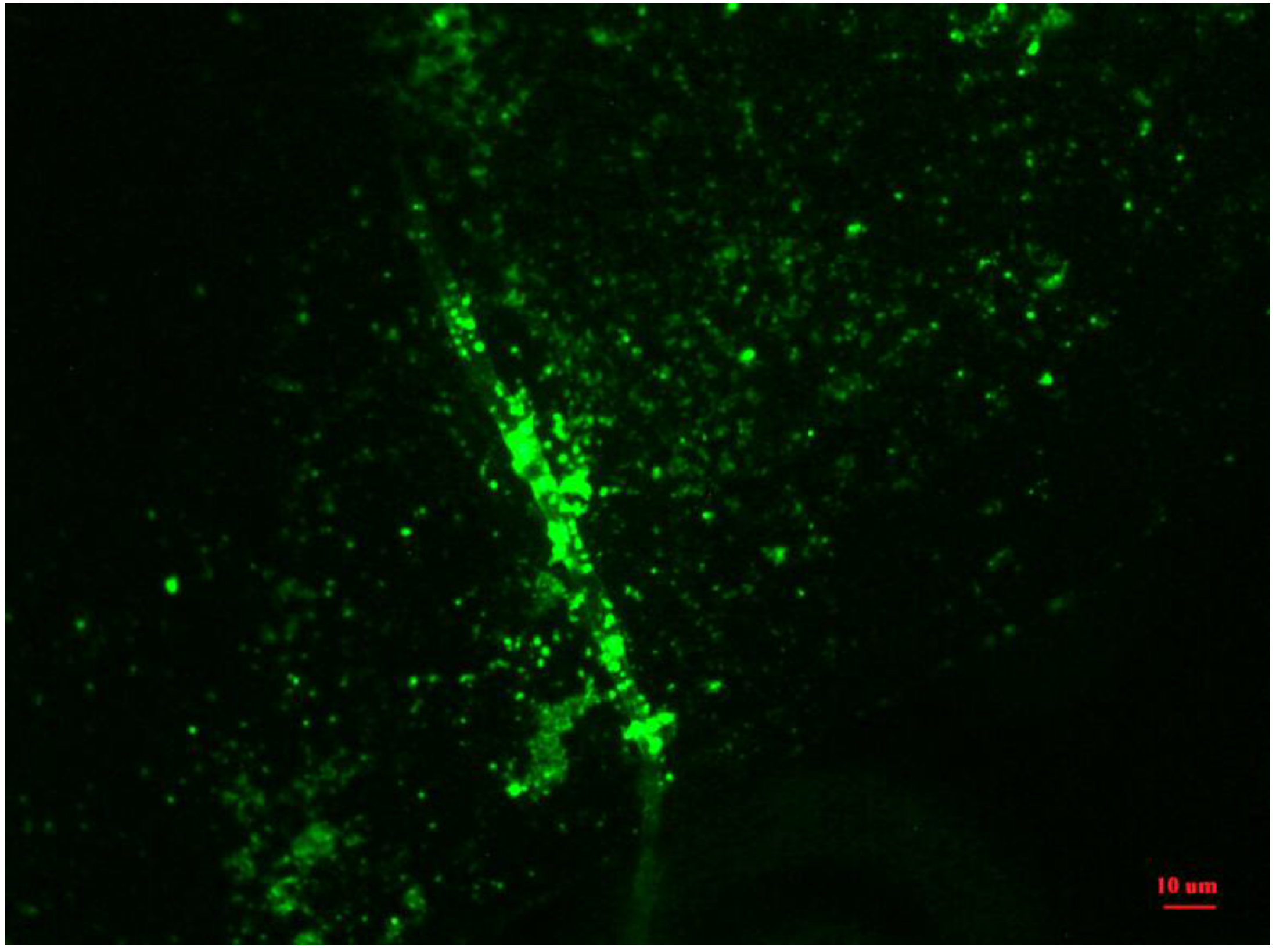
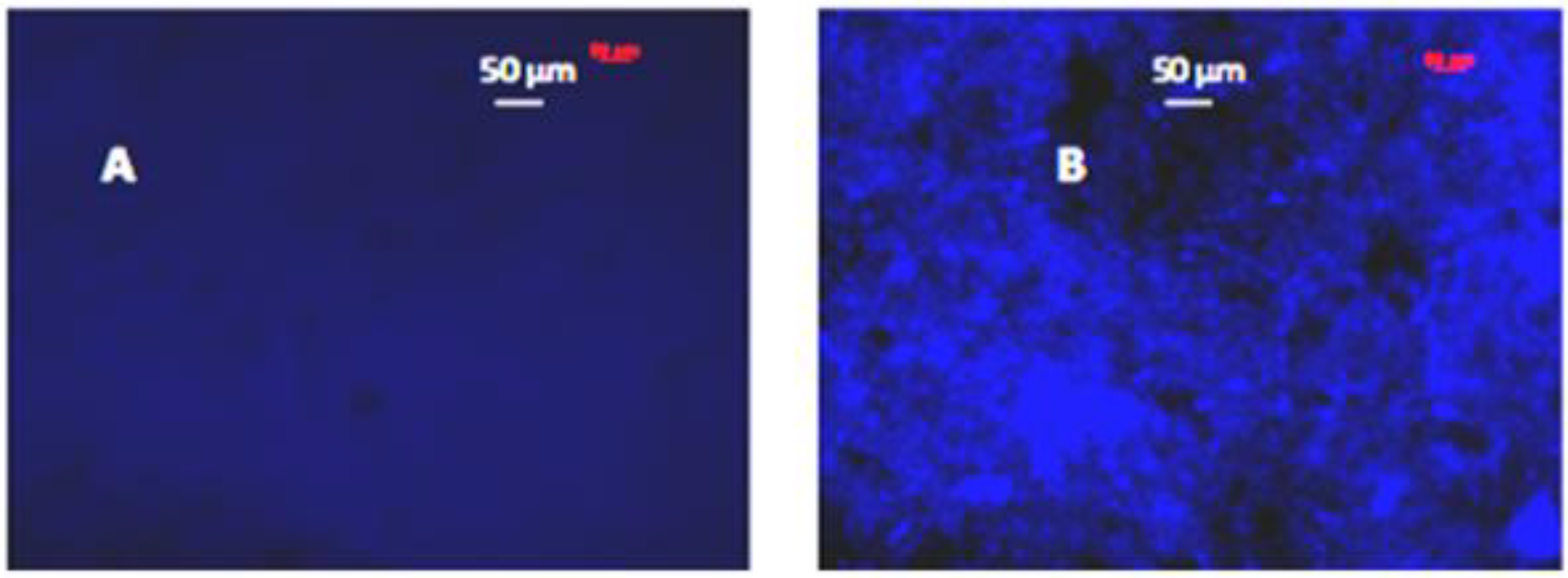
3.3. Functional Polymer-Protein Interactions
3.3.1. Block Copolymers from 3-Decylthiophene and 2-Hydroxyethyl Methacrylate Functionalized with DNP

3.3.2. Binding of Avidin with Surfaces Prepared from Block Copolymers with Biotin and without Biotin

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Goldberg, M.; Langer, R.; Jia, X. Nanostructured materials for applications in drug delivery and tissue engineering. J. Biomat. Sci. Polym. Ed. 2007, 18, 241–268. [Google Scholar] [CrossRef]
- Geeta, S.; Rao, C.R.K.; Vijayan, M.; Trivedi, D.C. Biosensing and drug delivery by polypyrrole. Anal. Chim. Acta 2006, 568, 119–125. [Google Scholar] [CrossRef]
- Sunoqrot, S.; Liu, Y.; Kim, D.-H.; Hog, S. In vitro evaluation of dendrimer-polymer hybrid nanoparticles on their controlled cellular targeting kinetics. Mol. Pharm. 2013, 10, 2157–2166. [Google Scholar] [CrossRef]
- Schiffman, J.D.; Schauer, C.L. A review: Electrospinning of biopolymer nanofibers and their applications. Polym. Rev. 2008, 48, 317–352. [Google Scholar] [CrossRef]
- Patil, Y.; Toti, U.; Khadir, A.; Panyam, M.L. Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials 2009, 30, 459–866. [Google Scholar]
- Zhang, Y.; Luo, Y.; Tian, J.; Asiri, A.M.; Ai-Youbi, A.O.; Sun, X. Rectangular coordination polymer nanoplates: Large scale, rapid synthesis and their application as a fluorescent sensing platform for DNA detection. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Yan, Y.; Martens, A.A.; Besseling, N.A.M.; de Wolf, F.A.; de Keizer, A.; Drechsler, M.; Cohen, S.M.A. Nanoribbons self-assembled from triblock peptide polymers and coordination polymers. Angew. Chem. Int. Edit. 2008, 47, 4192–4195. [Google Scholar] [CrossRef]
- Kirshenbaum, K.; Zuckermann, R.N.; Dill, K.A. Designing polymers that mimic biomolecules. Curr. Opin. Struc. Biol. 1999, 9, 530–535. [Google Scholar] [CrossRef]
- Gordon, K.; Sannigrahi, B.; McGeady, P.; Wang, X.Q.; Mendenhall, J.; Khan, I.M. Synthesis of optically active helical poly(2-methoxystyrene). Enhancement of Hela and Osteoblast cell growth on optically active helical poly(2-methoxystyrene) surfaces. J. Biomat. Sci. Polym. Ed. 2009, 20, 2055–2072. [Google Scholar] [CrossRef]
- Stigers, K.D.; Soth, M.J.; Nowick, J.J. Designed molecules that fold to mimic protein secondary structures. Curr. Opin. Chem. Biol. 1999, 3, 714–723. [Google Scholar]
- Wantanabe, J.; Eriguchi, T.; Ishihara, K. Stereocomplex formation by enantiomeric poly(lactic acid) graft-type phospholipid polymers for tissue engineering. Biomacromolecules 2002, 3, 1109–1114. [Google Scholar] [CrossRef]
- Johnson, G.; Jenkins, M.; McLean, K.M.; Griesser, H.J.; Kwak, J.; Goodman, M.; Steele, J.G. Peptide-containing collagen mimetics with cell binding activity. J. Biomed. Mater. Res. 2000, 51, 612–624. [Google Scholar]
- Khan, I.M. Synthetic macromolecules with higher structural order. ACS Symp. Ser. 2002, 812, 1–8. [Google Scholar] [CrossRef]
- Baird, E.J.; Holowka, D.; Coates, G.W.; Baird, B. Highly effective poly(ethylene glycol) architectures for specific inhibition of immune receptor activation. Biochemistry 2003, 42, 12739–12748. [Google Scholar]
- Acharya, S.; Dilnawaz, F.; Sahoo, S.K. Targeted epidermal growth factor receptor nanoparticle bioconjugates for breast cancer therapy. Biomaterials 2009, 30, 5737–5750. [Google Scholar] [CrossRef]
- Sannigrahi, B.; Khan, I.; Sil, D.; Baird, B. Synthesis and characterization of α,ω-bi(2,4-dinitrophenyl (DNP)) poly(2-methoxystyrene) functional polymers. Initial evaluation of the interaction of the functional polymers with RBL mast cells. J. Macromol. Sci. APure Appl. Chem. 2008, 45, 664–671. [Google Scholar]
- Mammen, M.; Choi, S.-K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implication for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Edit. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug. Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef]
- Poon, Z.; Chen, S.; Engler, A.C.; Lee, H.I.; Atas, E.; von Maltzahn, G.; Bhat, S.N.; Hammond, P.T. Ligand-clustered patchy nanoparticles for modulated cellular uptake and in vivo tumor targeting. Angew. Chem. Int. Edit. 2010, 49, 7266–7270. [Google Scholar]
- Baeumner, A.J. Biosensors for environmental pollutants and food contaminants. Anal. Bioanal. Chem. 2003, 377, 434–445. [Google Scholar] [CrossRef]
- Olubi, O.; Gadi, D.; Sannigrahi, B.; Williams, M.D.; Baird, B.; Khan, I. Fabrication of electroactive composite nanofibers of functionalized polymer and CNT capable of specifically binding with IgE antibody. Surf. Interface Anal. 2014, 46, 237–242. [Google Scholar]
- Reuven, D.G.; Sil, D.; Sannigrahi, B.; Wang, X.-Q.; Baird, B.; Khan, I.M. Archetypical conductive polymer structure for specific interaction with proteins. J. Macromol. Sci. APure Appl. Chem. 2012, 49, 330–338. [Google Scholar]
- Guiseppi-Elie, A. Electroactive hydragels: Synthesis, characterization and biomedical applications. Biomaterials 2010, 31, 2701–2716. [Google Scholar] [CrossRef]
- Kotanen, C.N.; Wison, N.A.; Dong, C.; Dinu, C.Z.; Justin, G.A.; Guiseppi-Elie, A. The effect of the physicochemical properties of bioactive electroconductive hydrogels on the growth and proliferation of attachment dependent cells. Biomaterials 2013, 34, 2701–2716. [Google Scholar] [CrossRef]
- Guiseppi-Elie, A.; Dong, C.; Dinu, C.Z. Crosslink density of a biomimetic poly(HEMA)-based hydrogel influences growth and proliferation of attachment dependent RMS 13 cells. J. Mater. Chem. 2012, 22, 19529–19539. [Google Scholar]
- Iraqi, A.; Barker, G.W.; Pickup, D.F. Synthesis and characterization of functionalized thiophene copolymer with electron donor and acceptor substituent. React. Funct. Polym. 2006, 66, 195–200. [Google Scholar]
- Loewe, R.S.; Khersonsky, S.M.; McCullough, R.D. A simple method to prepare head-to-tail coupled, regioregular poly(3-alkylthiophenes) using grignard metathesis. Adv. Mater. 1999, 11, 250–253. [Google Scholar] [CrossRef]
- Vargun, D. Living Radical Polymerization of Hydroxyethyl Methacrylate and Its Block Copolymerization with Poly(Dimethyl Siloxane) Macroazoinitiator. Ph.D. Thesis, Middle East Technica University, Ankara, Turkey, 2009. [Google Scholar]
- Mévellec, V.; Roussel, S.; Tessier, L.; Deniau, G.; Viel, P.; Palacin, S. Grafting polymers on surfaces: A new powerful and versatile diazonium salt-based one-step process in aqueous media. Chem. Mater. 2007, 19, 6323–6330. [Google Scholar] [CrossRef]
- Raman Spectroscopy for Analysis and Monitoring. Available online: http://www.horiba.com/fileadmin/uploads/Scientific/Documents/Raman/bands.pdf (accessed on 29 May 2014).
- Li, G.; Bhosale, S.; Tao, S.; Fuhthop, J.-H. Conducting polythiophenes with a broad spectrum of reactive groups. J. Polym. Sci. A Polym. Chem. 2005, 43, 4547–4558. [Google Scholar]
- Xia, L.; Li, S.-L.; Ai, X.-P.; Yang, H.-X.; Cao, Y.-L. Temperature sensitive cathode materials for safer lithium-ion batteries. Energy Environ. Sci. 2011, 4, 2845–2848. [Google Scholar] [CrossRef]
- Zhang, J.C.; Zheng, X.; Chen, M.; Yang, X.Y.; Cao, W.L. Synthesis and application of solar cells of poly(3-decylthiophene)/N/titanium dioxide hybrid. eXPRESS Polym. Lett. 2011, 5, 401–408. [Google Scholar]
- Senthilkumar, B.; Thenamirtham, P.; Selvan, R. Structural and electrochemical properties of polythiophene. Appl. Suf. Sci. 2011, 257, 9063–9067. [Google Scholar] [CrossRef]
- Yam, C.M.; Deluge, M.; Tang, D.; Kumar, A.; Cai, C. Preparation, characterization, resistance to protein adsorption, and specific avidin-biotin binding of poly(amidoamine) dendrimers, functionalized with oligo(ethylene glycol) on gold. J. Colloid Interface Sci. 2006, 296, 118–130. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Olubi, O.; London, L.; Sannigrahi, B.; Nagappan, P.; Williams, M.; Khan, I.M. Fabrication of Bioactive Surfaces by Functionalization of Electroactive and Surface-Active Block Copolymers. Bioengineering 2014, 1, 134-153. https://doi.org/10.3390/bioengineering1030134
Olubi O, London L, Sannigrahi B, Nagappan P, Williams M, Khan IM. Fabrication of Bioactive Surfaces by Functionalization of Electroactive and Surface-Active Block Copolymers. Bioengineering. 2014; 1(3):134-153. https://doi.org/10.3390/bioengineering1030134
Chicago/Turabian StyleOlubi, Omotunde, Laurisa London, Biswajit Sannigrahi, Peri Nagappan, Michael Williams, and Ishrat M. Khan. 2014. "Fabrication of Bioactive Surfaces by Functionalization of Electroactive and Surface-Active Block Copolymers" Bioengineering 1, no. 3: 134-153. https://doi.org/10.3390/bioengineering1030134
APA StyleOlubi, O., London, L., Sannigrahi, B., Nagappan, P., Williams, M., & Khan, I. M. (2014). Fabrication of Bioactive Surfaces by Functionalization of Electroactive and Surface-Active Block Copolymers. Bioengineering, 1(3), 134-153. https://doi.org/10.3390/bioengineering1030134