
Advanced Glycation End-Products in Skeletal Muscle Aging


 ,
, 

Abstract
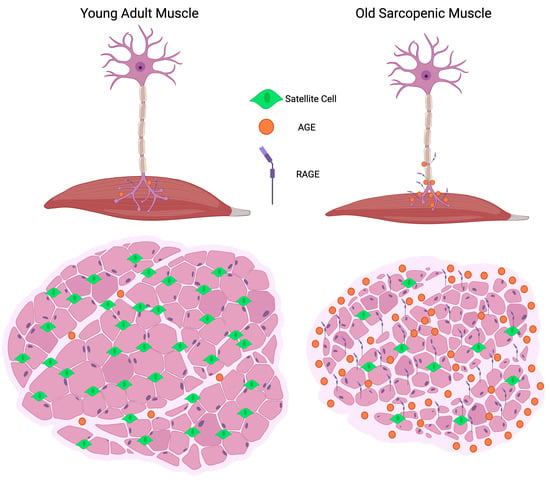
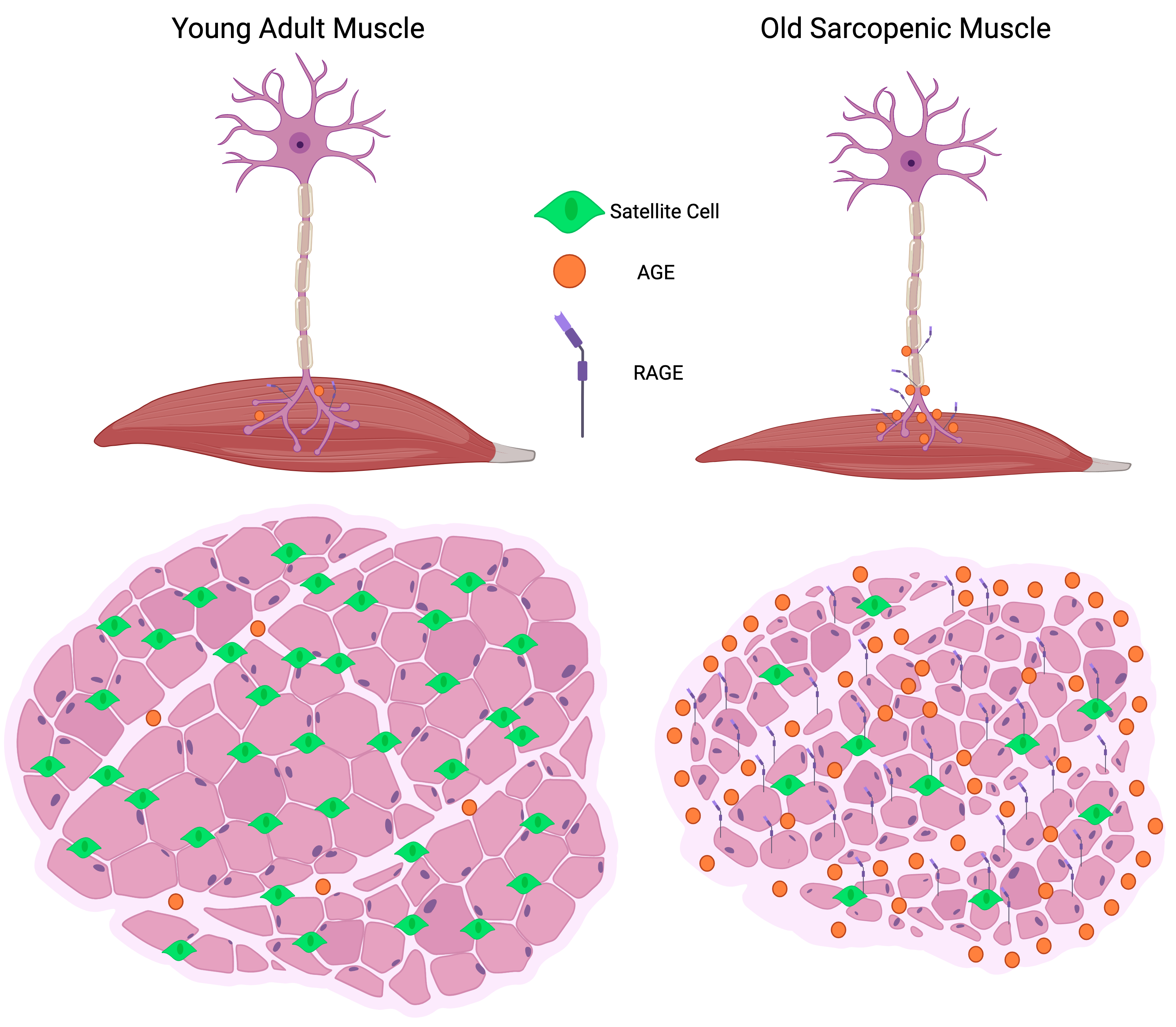
:
{kind=link}
{kind=link}
{kind=link}
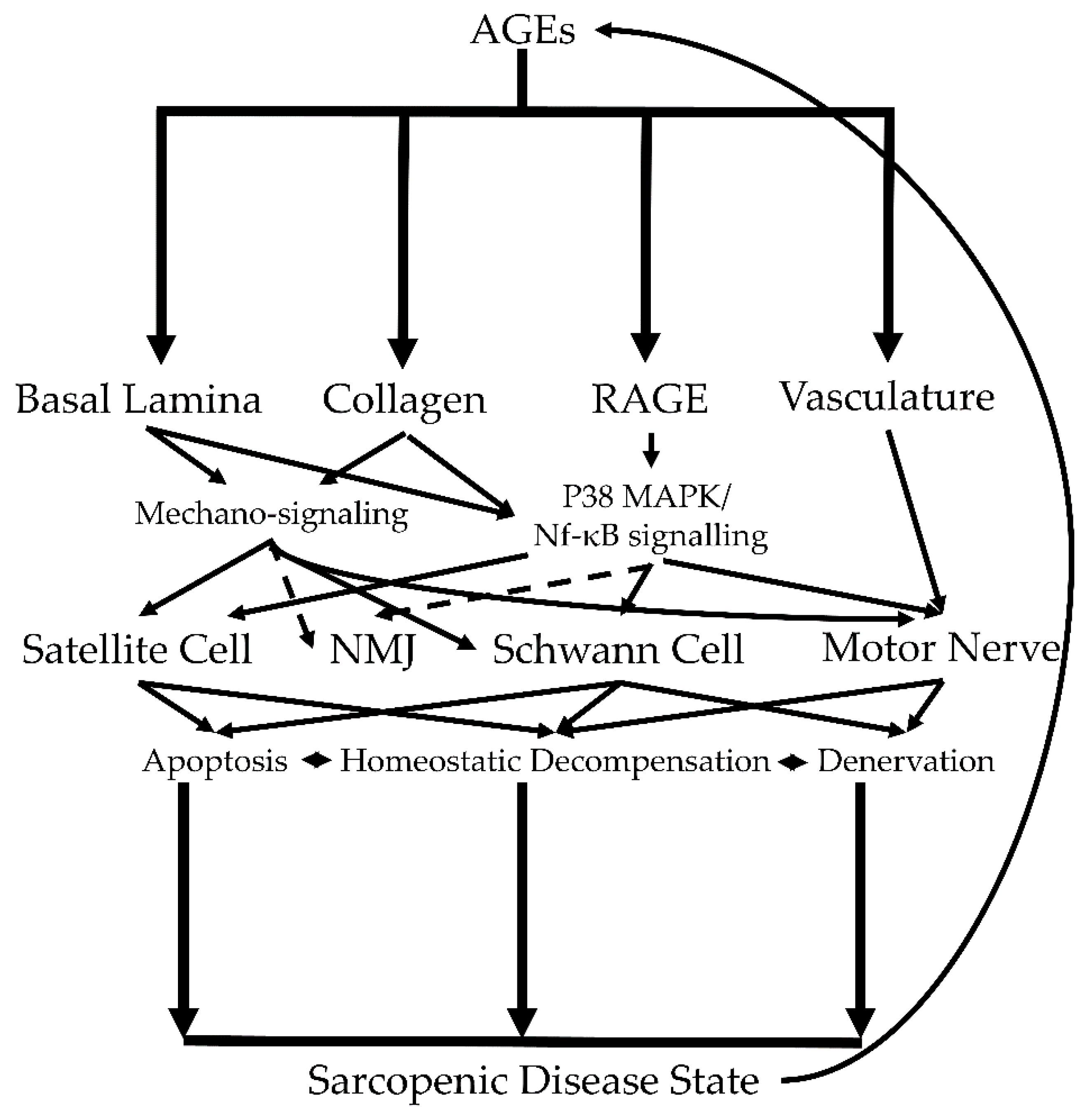{kind=link}
1. Introduction
2. Skeletal Muscle and the Aging Extracellular Matrix
2.1. ECM Composition Is Unique to the Muscle Fiber Type
2.2. Satellite Cells and Aging
2.3. The AGE-RAGE Axis in Skeletal Muscle Aging
2.4. Aging of Skeletal Muscle Collagen
2.5. Basal Lamina Aging
2.6. ECM Aging Is Muscle-Specific
3. Peripheral Nerve Involvement in Aging and Atrophy
3.1. Overview and Neuromuscular Junction Anatomy
3.2. Endplate Fragmentation and Aging
3.3. Insights from Diabetic Neuropathy, AGEs, and RAGE
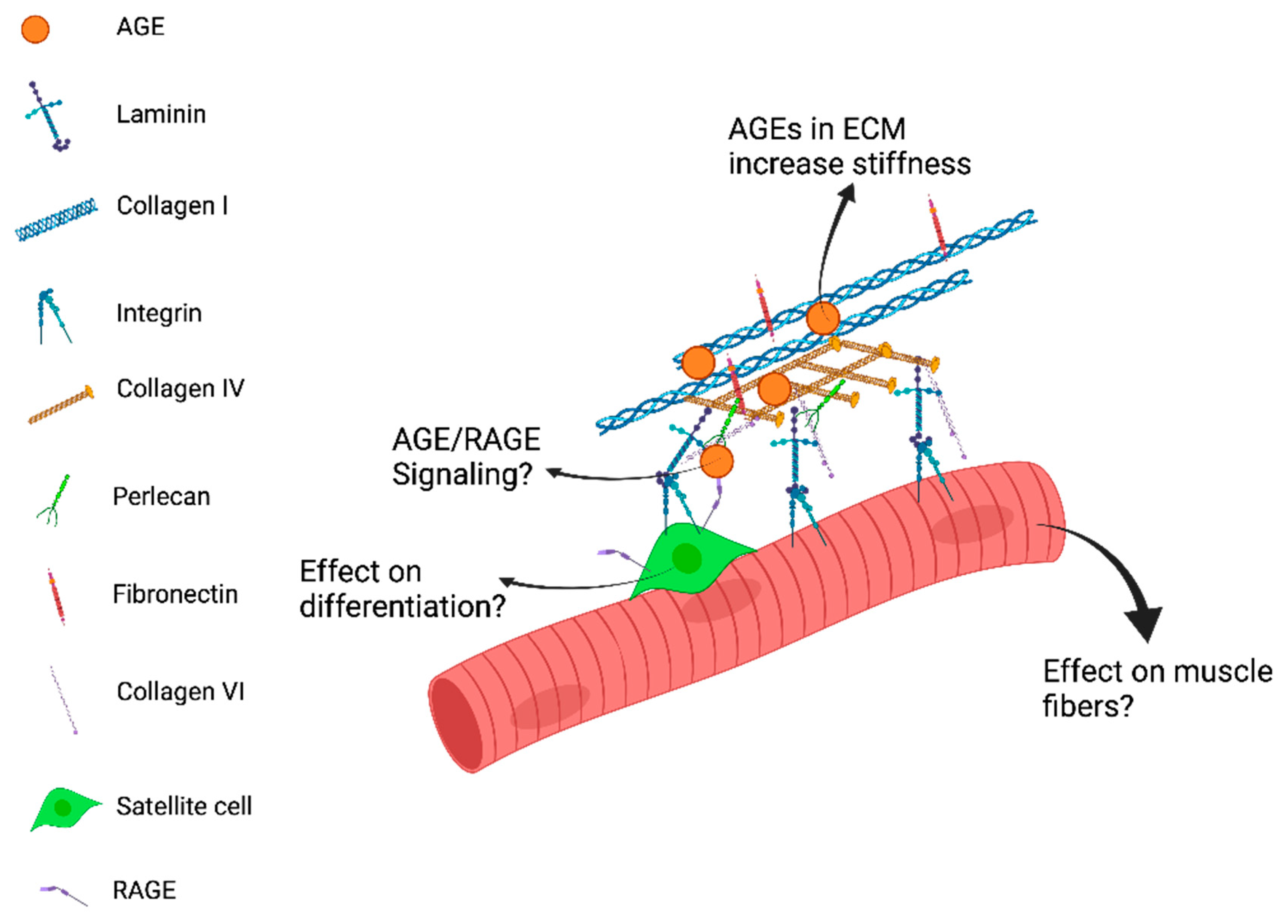
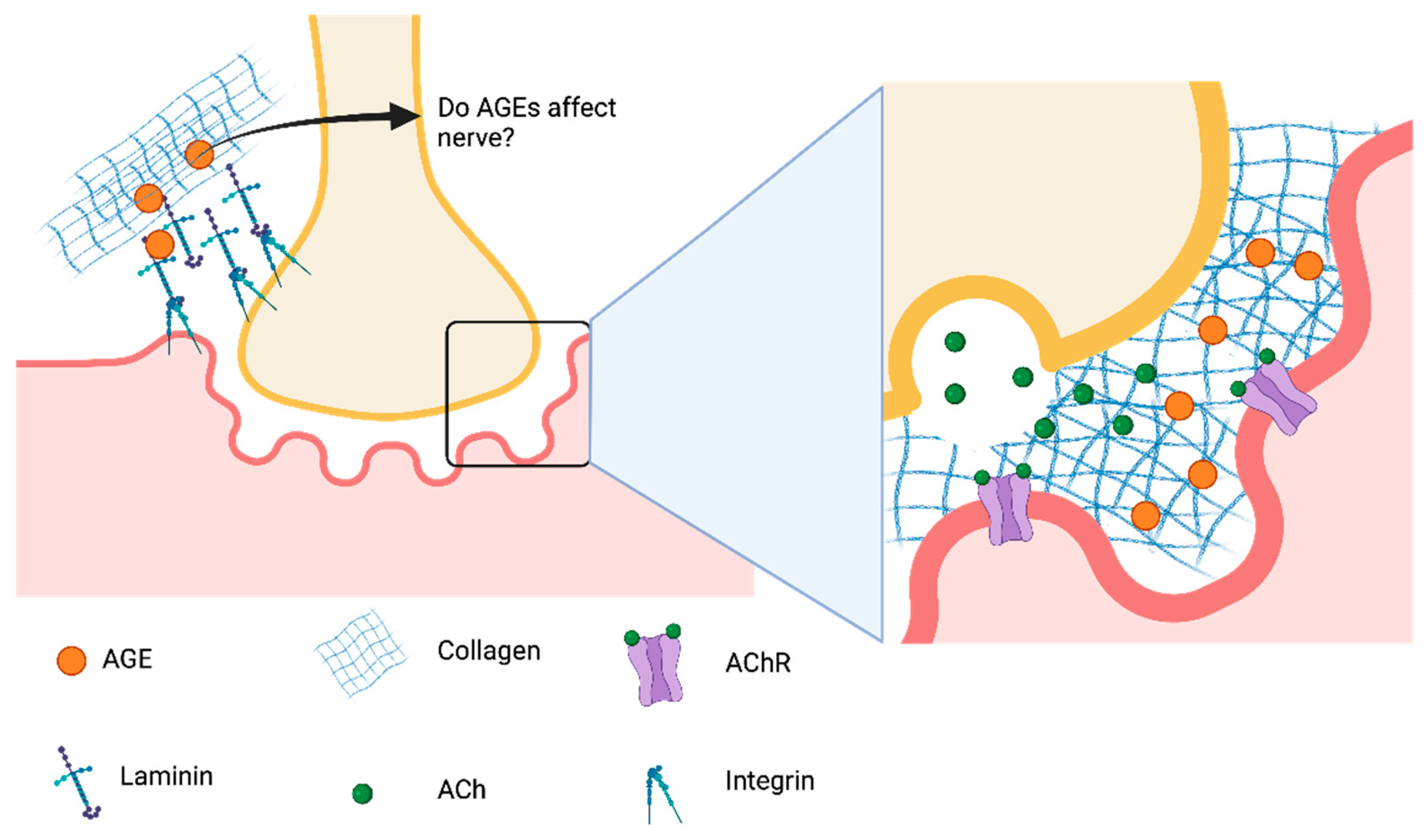
4. Future Directions
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vollset, S.E.; Goren, E.; Yuan, C.W.; Cao, J.; Smith, A.E.; Hsiao, T.; Bisignano, C.; Azhar, G.S.; Castro, E.; Chalek, J.; et al. Fertility, Mortality, Migration, and Population Scenarios for 195 Countries and Territories from 2017 to 2100: A Forecasting Analysis for the Global Burden of Disease Study. Lancet 2020, 396, 1285–1306. [Google Scholar] [CrossRef]
- Frontera, W.R.; Miljkovic, N.; Lim, J.-Y.; Miljkovic, I. Aging of Skeletal Muscle Fibers. Ann. Rehabil. Med. Rev. Artic. Ann. Rehabil. Med. 2015, 39, 155–162. [Google Scholar] [CrossRef]
- Doherty, T.J. Invited Review: Aging and Sarcopenia. J. Appl. Physiol. 2003, 95, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.L.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef] [PubMed]
- Walston, J.D. Sarcopenia in Older Adults. Curr. Opin. Rheumatol. 2012, 24, 623–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, W. On the relationship between strength of grip and certain mental and sensory characters. Biometrika 1924, 16, 299–327. [Google Scholar] [CrossRef]
- Berg, B.N. Muscular Dystrophy in Aging Rats. J. Gerontol. 1956, 11, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Siparsky, P.N.; Kirkendall, D.T.; Garrett, W.E., Jr. Muscle Changes in Aging: Understanding Sarcopenia. Sports Health 2014, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Maillard, L.-C. Condensation Des Acides Amines En Presence de La Glycerine; Cycloglycylglycine et Polypeptides. C. R. Hebd. Seances Acad. Sci. 1911, 153, 1078–1080. [Google Scholar]
- Kawamura, S. Seventy Years of the Maillard Reaction. In The Maillard Reaction in Foods and Nutrition; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1983; Volume 215, pp. 1–3. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Yabu, A.; Nakamura, H. Advanced Glycation End Products in Musculoskeletal System and Disorders. Methods 2020. [Google Scholar] [CrossRef]
- Svensson, R.B.; Smith, S.T.; Moyer, P.J.; Magnusson, S.P. Effects of Maturation and Advanced Glycation on Tensile Mechanics of Collagen Fibrils from Rat Tail and Achilles Tendons. Acta Biomater. 2018, 70, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Gillies, A.R.; Lieber, R.L. Structure and Function of the Skeletal Muscle Extracellular Matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef] [Green Version]
- Unoki, H.; Yamagishi, S. Advanced Glycation End Products and Insulin Resistance. Curr. Pharm. Des. 2008, 14, 987–989. [Google Scholar] [CrossRef]
- Ahmad, K.; Lee, E.J.; Moon, J.S.; Park, S.-Y.; Choi, I. Multifaceted Interweaving Between Extracellular Matrix, Insulin Resistance, and Skeletal Muscle. Cells 2018, 7, 148. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, K.; Shaikh, S.; Lee, E.J.; Lee, Y.-H.; Choi, I. Consequences of Dicarbonyl Stress on Skeletal Muscle Proteins in Type 2 Diabetes. Curr. Protein Pept. Sci. 2019, 21, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Seynnes, O.; Schiaffino, S.; Blottner, D.; Csapo, R.; Gumpenberger, M.; Wessner, B. Skeletal Muscle Extracellular Matrix—What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front. Physiol 2020, 11, 253. [Google Scholar] [CrossRef] [Green Version]
- Chazaud, B. Inflammation during Skeletal Muscle Regeneration and Tissue Remodeling: Application to Exercise-Induced Muscle Damage Management. Immunol. Cell Biol. 2016, 94, 140–145. [Google Scholar] [CrossRef]
- Etienne, J.; Liu, C.; Skinner, C.M.; Conboy, M.J.; Conboy, I.M. Skeletal Muscle as an Experimental Model of Choice to Study Tissue Aging and Rejuvenation. Skelet. Muscle 2020, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kragstrup, T.W.; Kjaer, M.; Mackey, A.L.; Wenzel Kragstrup, T. Structural, Biochemical, Cellular, and Functional Changes in Skeletal Muscle Extracellular Matrix with Aging. Scand. J. Med. Sci. Sports 2011, 21, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite Cells and the Muscle Stem Cell Niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [Green Version]
- Bentzinger, C.F.; Wang, Y.X.; von Maltzahn, J.; Soleimani, V.D.; Yin, H.; Rudnicki, M.A. Fibronectin Regulates Wnt7a Signaling and Satellite Cell Expansion. Cell Stem Cell 2013, 12, 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzinger, C.F.; von Maltzahn, J.; Rudnicki, M.A. Extrinsic Regulation of Satellite Cell Specification. Stem Cell Res. Ther. 2010, 1, 27. [Google Scholar] [CrossRef] [Green Version]
- Rayagiri, S.S.; Ranaldi, D.; Raven, A.; Mohamad Azhar, N.I.F.; Lefebvre, O.; Zammit, P.S.; Borycki, A.-G. Basal Lamina Remodeling at the Skeletal Muscle Stem Cell Niche Mediates Stem Cell Self-Renewal. Nat. Commun. 2018, 9, 1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, R.W.D. Morphology of Perimysial and Endomysial Connective Tissue in Skeletal Muscle. Tissue Cell 1981, 13, 681–690. [Google Scholar] [CrossRef]
- Gao, Y.; Waas, A.M.; Faulkner, J.A.; Kostrominova, T.Y.; Wineman, A.S. Micromechanical Modeling of the Epimysium of the Skeletal Muscles. J. Biomech. 2008, 41, 1–10. [Google Scholar] [CrossRef]
- Purslow, P.P.; Trotter, J.A. The Morphology and Mechanical Properties of Endomysium in Series-Fibred Muscles: Variations with Muscle Length. J. Muscle Res. Cell Motil. 1994, 15, 299–308. [Google Scholar] [CrossRef]
- Bowman, W. On the Minute Structure and Movements of Voluntary Muscle. R. J. R. Taylor 1840, 3, 457–501. [Google Scholar]
- Hu, P.; Geles, K.G.; Paik, J.H.; DePinho, R.A.; Tjian, R. Codependent Activators Direct Myoblast-Specific MyoD Transcription. Dev. Cell 2008, 15, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Mauro, A.; Sten-Knudsen, O. The role of the sarcolemma in muscle physiology. Acta Med. Scand. 1952, 142, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, V.; Suominen, H.; Heikkinen, E. Collagen of Slow Twitch and Fast Twitch Muscle Fibres in Different Types of Rat Skeletal Muscle. Eur. J. Appl. Physiol. Occup. Physiol. 1984, 52, 235–242. [Google Scholar] [CrossRef]
- Sanes, J.R. The Basement Membrane/Basal Lamina of Skeletal Muscle. J. Biol. Chem. 2003, 278, 12601–12604. [Google Scholar] [CrossRef] [Green Version]
- Grzelkowska-Kowalczyk, K. The Importance of Extracellular Matrix in Skeletal Muscle Development and Function. Compos. Funct. Extracell. Matrix Hum. Body 2016. [Google Scholar] [CrossRef] [Green Version]
- Mauro, A. Satellite Cell of Skeletal Muscle Fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef]
- Aziz, A.; Sebastian, S.; Dilworth, J.J. The Origin and Fate of Muscle Satellite Cells. Stem Cell Rev. Rep. 2012, 8, 609–622. [Google Scholar] [CrossRef] [PubMed]
- Mayer, U. Integrins: Redundant or Important Players in Skeletal Muscle? J. Biol. Chem. 2003, 278, 14587–14590. [Google Scholar] [CrossRef] [Green Version]
- Hantaü, D.; Gautron, J.; Labat-Robert, J. Immunolocalization of Fibronectin and Other Macromolecules of the Intercellular Matrix in the Striated Muscle Fiber of the Adult Rat. Top. Catal. 1983, 3, 381–391. [Google Scholar] [CrossRef]
- Florian Bentzinger, C.; Wang, Y.X.; Rudnicki, M.A. Building Muscle: Molecular Regulation of Myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4, a008342. [Google Scholar] [CrossRef]
- Kuang, S.; Gillespie, M.A.; Rudnicki, M.A. Niche Regulation of Muscle Satellite Cell Self-Renewal and Differentiation. Cell Stem Cell 2008, 2, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, S.; Kuroda, K.; Le Grand, F.; Rudnicki, M.A. Asymmetric Self-Renewal and Commitment of Satellite Stem Cells in Muscle. Cell 2007, 129, 999–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.B. Myogenic Programs of Mouse Muscle Cell Lines: Expression of Myosin Heavy Chain Isoforms, MyoD1, and Myogenin. J. Cell Biol. 1990, 111, 1149–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehrle, U.; Düsterhöft, S.; Pette, D. Effects of Chronic Electrical Stimulation on Myosin Heavy Chain Expression in Satellite Cell Cultures Derived from Rat Muscles of Different Fiber-Type Composition. Differentiation 1994, 58, 37–46. [Google Scholar] [CrossRef]
- Stern-Straeter, J.; Bonaterra, G.A.; Kassner, S.S.; Zügel, S.; Hörmann, K.; Kinscherf, R.; Goessler, U.R. Characterization of Human Myoblast Differentiation for Tissue-Engineering Purposes by Quantitative Gene Expression Analysis. J. Tissue Eng. Regen. Med. 2011, 5, e197–e206. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.M. Denervation and the Aging of Skeletal Muscle. Basic Appl. Myol. 2004, 14, 135–139. [Google Scholar]
- Dedkov, E.I.; Kostrominova, T.Y.; Borisov, A.B.; Carlson, B.M. MyoD and Myogenin Protein Expression in Skeletal Muscles of Senile Rats. Cell Tissue Res 2003, 311, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Musarò, A.; Cusella De Angelis, M.G.; Germani, A.; Ciccarelli, C.; Molinaro, M.; Zani, B.M. Enhanced Expression of Myogenic Regulatory Genes in Aging Skeletal Muscle. Exp. Cell Res. 1995, 221, 241–248. [Google Scholar] [CrossRef]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.E.; Elliston, K.; Stern, D.; Shaw, A. Cloning and Expression of a Cell Surface Receptor for Advanced Glycosylation End Products of Proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Riuzzi, F.; Sorci, G.; Sagheddu, R.; Chiappalupi, S.; Salvadori, L.; Donato, R. RAGE in the Pathophysiology of Skeletal Muscle. J. Cachexia Sarcopenia Muscle 2018, 9, 1213–1234. [Google Scholar] [CrossRef] [Green Version]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “RAGE” in Lung Disease: The Receptor for Advanced Glycation Endproducts (RAGE) Is a Major Mediator of Pulmonary Inflammatory Responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef] [PubMed]
- D’Agati, V.; Schmidt, A.M. RAGE and the Pathogenesis of Chronic Kidney Disease. Nat. Rev. Nephrol. 2010, 6, 352–360. [Google Scholar] [CrossRef]
- Yan, S.F.; Yan, S.D.; Ramasamy, R.; Schmidt, A.M. Tempering the Wrath of RAGE: An Emerging Therapeutic Strategy against Diabetic Complications, Neurodegeneration, and Inflammation. Ann. Med. 2009, 41, 408–422. [Google Scholar] [CrossRef] [Green Version]
- Riuzzi, F.; Sorci, G.; Sagheddu, R.; Donato, R. HMGB1-RAGE Regulates Muscle Satellite Cell Homeostasis through P38-MAPK- and Myogenindependent Repression of Pax7 Transcription. J. Cell Sci. 2012, 125, 1440–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagheddu, R.; Chiappalupi, S.; Salvadori, L.; Riuzzi, F.; Donato, R.; Sorci, G. Targeting RAGE as a Potential Therapeutic Approach to Duchenne Muscular Dystrophy. Hum. Mol. Genet. 2018, 27, 3734–3746. [Google Scholar] [CrossRef]
- Chiappalupi, S.; Sorci, G.; Vukasinovic, A.; Salvadori, L.; Sagheddu, R.; Coletti, D.; Renga, G.; Romani, L.; Donato, R.; Riuzzi, F. Targeting RAGE Prevents Muscle Wasting and Prolongs Survival in Cancer Cachexia. J. Cachexia. Sarcopenia Muscle 2020, 11, 929–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beccafico, S.; Riuzzi, F.; Puglielli, C.; Mancinelli, R.; Fulle, S.; Sorci, G.; Donato, R. Human Muscle Satellite Cells Show Age-Related Differential Expression of S100B Protein and RAGE. Age 2011, 33, 523–541. [Google Scholar] [CrossRef] [Green Version]
- Sorci, G.; Riuzzi, F.; Arcuri, C.; Giambanco, I.; Donato, R. Amphoterin Stimulates Myogenesis and Counteracts the Antimyogenic Factors Basic Fibroblast Growth Factor and S100B via RAGE Binding Downloaded From. Mol. Cell. Biol. 2004, 24, 4880–4894. [Google Scholar] [CrossRef] [Green Version]
- Rauvala, H.; Rouhiainen, A. RAGE as a Receptor of HMGB1 (Amphoterin): Roles in Health and Disease. Curr. Mol. Med. 2007, 7, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Riuzzi, F.; Sorci, G.; Beccafico, S.; Donato, R. S100B Engages RAGE or BFGF/FGFR1 in Myoblasts Depending on Its Own Concentration and Myoblast Density. Implications for Muscle Regeneration. PLoS ONE 2012, 7, e28700. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.-E.; Chiu, Y.-L.; Kao, T.-W.; Chen, W.-L. Elevated Level of the Soluble Receptor for Advanced Glycation End-Products Involved in Sarcopenia: An Observational Study. BMC Geriatr. 2021, 21, 531. [Google Scholar] [CrossRef]
- Peng, Y.; Kim, J.-M.; Park, H.-S.; Yang, A.; Islam, C.; Lakatta, E.G.; Lin, L. AGE-RAGE Signal Generates a Specific NF-ΚB RelA “Barcode” That Directs Collagen I Expression. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bernet, J.D.; Doles, J.D.; Hall, J.K.; Kelly Tanaka, K.; Carter, T.A.; Olwin, B.B. P38 MAPK Signaling Underlies a Cell-Autonomous Loss of Stem Cell Self-Renewal in Skeletal Muscle of Aged Mice. Nat. Med. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Verzár, F. Aging of the Collagen Fiber. Int. Rev. Connect. Tissue Res. 1964, 2, 243–300. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929. [Google Scholar] [CrossRef] [Green Version]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Collagen: The Fibrous Proteins of the Matrix. In Molecular Cell Biology, 4th ed.; W. H. Freeman: NewYork, NY, USA, 2000. [Google Scholar]
- Haus, J.M.; Carrithers, J.A.; Trappe, S.W.; Trappe, T.A. Collagen, Cross-Linking, and Advanced Glycation End Products in Aging Human Skeletal Muscle. J. Appl. Physiol. 2007, 103, 2068–2076. [Google Scholar] [CrossRef]
- Schaub, M.C. The Ageing of Collagen in the Striated Muscle. Gerontology 1963, 8, 16–35. [Google Scholar] [CrossRef]
- Goldspink, G.; Fernandes, K.; Williams, P.E.; Wells, D.J. Age-Related Changes in Collagen Gene Expression in the Muscles of Mdx Dystrophic and Normal Mice. Neuromuscul. Disord. 1994, 4, 183–191. [Google Scholar] [CrossRef]
- Haseeb, M.A.; Patnaik, B.K. Age-Related Changes in Collagenous and Noncollagenous Proteins of Skeletal Muscle of a Short-Lived Species of Reptile. Gerontology 1978, 24, 343–347. [Google Scholar] [CrossRef]
- Hindle, A.G.; Horning, M.; Mellish, J.-A.E.; Lawler, J.M. Diving into Old Age: Muscular Senescence in a Large-Bodied, Long-Lived Mammal, the Weddell Seal (Leptonychotes Weddellii). J. Exp. Biol. 2009, 212, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmichael, D.J.; Lawrie, R.A. Bovine Collagen. I. Changes in Collagen Solubility with Animal Age. Int. J. Food Sci. Technol. 1967, 2, 299–311. [Google Scholar] [CrossRef]
- Dickerson, J.W.T.; Widdowson, E.M. Chemical Changes in Skeletal Muscle during Development. Biochem. J. 1960, 74, 247. [Google Scholar] [CrossRef] [Green Version]
- Lacraz, G.; Rouleau, A.-J.; Couture, V.; Söllrald, T.; Drouin, G.; Veillette, N.; Grandbois, M.; Grenier, G. Increased Stiffness in Aged Skeletal Muscle Impairs Muscle Progenitor Cell Proliferative Activity. PLoS ONE 2015, 10, e0136217. [Google Scholar] [CrossRef] [Green Version]
- Ducomps, C.; Mauriege, P.; Darche, B.; Combes, S.; Lebas, F.; Doutreloux, J.P. Effects of Jump Training on Passive Mechanical Stress and Stiffness in Rabbit Skeletal Muscle: Role of Collagen. Acta Physiol. Scand. 2003, 178, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Verzár, F. The Stages and Consequences of Ageing of Collagen. Gerontology 1969, 15, 233–239. [Google Scholar] [CrossRef]
- Herchenhan, A.; Uhlenbrock, F.; Eliasson, P.; Weis, M.; Eyre, D.; Kadler, K.E.; Magnusson, S.P.; Kjaer, M. Lysyl Oxidase Activity Is Required for Ordered Collagen Fibrillogenesis by Tendon Cells. J. Biol. Chem. 2015, 290, 16440–16450. [Google Scholar] [CrossRef] [Green Version]
- Heinemeier, K.M.; Schjerling, P.; Heinemeier, J.; Magnusson, S.P.; Kjaer, M. Lack of Tissue Renewal in Human Adult Achilles Tendon Is Revealed by Nuclear Bomb 14C. FASEB J. 2013, 27, 2074. [Google Scholar] [CrossRef] [Green Version]
- Elliott, B.; Youl Moon, H.; Levinger, I.; Reggiani, C.; Naro, F.; Venturelli, M.; Monaco, L.; Toniolo, L.; Muti, E.; Milanese, C.; et al. Skeletal Muscle Fiber Size and Gene Expression in the Oldest-Old With Differing Degrees of Mobility. Front. Physiol. 2019, 10, 313. [Google Scholar] [CrossRef] [Green Version]
- Wessner, B.; Liebensteiner, M.; Nachbauer, W.; Csapo, R. Age-Specific Response of Skeletal Muscle Extracellular Matrix to Acute Resistance Exercise: A Pilot Study. Eur. J. Sport Sci. 2018, 19, 354–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.R.; Hammers, D.W.; Sweeney, H.L.; Barton, E.R. Increased Collagen Cross-linking Is a Signature of Dystrophin-deficient Muscle. Muscle Nerve 2016, 54, 71. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.M.; Abbott, C.; Peelor, F.F.; Lopes, E.B.P.; Griffin, T.M.; Miller, B.F. Determining Resistance to Protein Turnover in Aged Skeletal Muscle Collagen Using a Novel Stable Isotope Timecourse Approach. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Snow, L.M.; Fugere, N.A.; Thompson, L.V. Advanced Glycation End-Product Accumulation and Associated Protein Modification in Type II Skeletal Muscle with Aging. J. Gerontol. A. Biol. Sci. Med. Sci. 2007, 62, 1204–1210. [Google Scholar] [CrossRef] [Green Version]
- Olson, L.C.; Nguyen, T.M.; Heise, R.L.; Boyan, B.D.; Schwartz, Z.; McClure, M.J. Advanced Glycation End Products Are Retained in Decellularized Muscle Matrix Derived from Aged Skeletal Muscle. Int. J. Mol. Sci. 2021, 22, 8832. [Google Scholar] [CrossRef]
- Gulati, A.K.; Reddi, A.H.; Zalewski, A.A. Distribution of Fibronectin in Normal and Regenerating Skeletal Muscle. Anat. Rec. 1982, 204, 175–183. [Google Scholar] [CrossRef]
- Lukjanenko, L.; Jung, M.J.; Hegde, N.; Perruisseau-Carrier, C.; Migliavacca, E.; Rozo, M.; Karaz, S.; Jacot, G.; Schmidt, M.; Li, L.; et al. Loss of Fibronectin from the Aged Stem Cell Niche Affects the Regenerative Capacity of Skeletal Muscle in Mice. Nat. Med. 2016, 22, 897. [Google Scholar] [CrossRef] [Green Version]
- Kovanen, V.; Suominen, H.; Risteli, J.; Risteli, L. Type IV Collagen and Laminin in Slow and Fast Skeletal Muscle in Rats—Effects of Age and Life-Time Endurance Training. Top. Catal. 1988, 8, 145–153. [Google Scholar] [CrossRef]
- Stearns-Reider, K.M.; D’Amore, A.; Beezhold, K.; Rothrauff, B.; Cavalli, L.; Wagner, W.R.; Vorp, D.A.; Tsamis, A.; Shinde, S.; Zhang, C.; et al. Aging of the Skeletal Muscle Extracellular Matrix Drives a Stem Cell Fibrogenic Conversion. Aging Cell 2017, 16, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Kiss, A.A.; Somlyai-Popovics, N.; Kiss, M.; Boldogkői, Z.; Csiszár, K.; Mink, M. Type IV Collagen Is Essential for Proper Function of Integrin-Mediated Adhesion in Drosophila Muscle Fibers. Int. J. Mol. Sci. 2019, 20, 5124. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, Y.; Ikegami, K.; Sujino, M.; Koinuma, S.; Nagano, M.; Oi, Y.; Onishi, T.; Sugiyo, S.; Takeda, I.; Kaji, H.; et al. Effects of Aging on Basement Membrane of the Soleus Muscle during Recovery Following Disuse Atrophy in Rats. Exp. Gerontol. 2017, 98, 153–161. [Google Scholar] [CrossRef]
- Bartling, B.; Desole, M.; Rohrbach, S.; Silber, R.E.; Simm, A. Age-Associated Changes of Extracellular Matrix Collagen Impair Lung Cancer Cell Migration. FASEB J. 2009, 23, 1510–1520. [Google Scholar] [CrossRef]
- Tarsio, J.F.; Reger, L.A.; Furcht, L.T. Decreased Interaction of Fibronectin, Type IV Collagen, and Heparin Due to Nonenzymatic Glycation. Implications for Diabetes Mellitus. Biochemistry 1987, 26, 1014–1020. [Google Scholar] [CrossRef]
- Pastino, A.K.; Greco, T.M.; Mathias, R.A.; Cristea, I.M.; Schwarzbauer, J.E. Stimulatory Effects of Advanced Glycation Endproducts (AGEs) on Fibronectin Matrix Assembly. Matrix Biol. 2017, 59, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Ohlendieck, K. Proteomic Profiling of Fast-To-Slow Muscle Transitions during Aging. Front. Physiol. 2011, 2, 105. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, S.D.; McCormick, R.J.; Vadlamudi, R.K.; Thomas, D.P. Age and Training Alter Collagen Characteristics in Fast- and Slow-Twitch Rat Limb Muscle. J. Appl. Physiol. 1993, 75, 1670–1674. [Google Scholar] [CrossRef]
- Nilwik, R.; Snijders, T.; Leenders, M.; Groen, B.B.; van Kranenburg, J.; Verdijk, L.B.; van Loon, L.J. The Decline in Skeletal Muscle Mass with Aging Is Mainly Attributed to a Reduction in Type II Muscle Fiber Size. Exp. Gerontol. 2013, 48, 492–498. [Google Scholar] [CrossRef]
- Larsson, L.; Edström, L. Effects of Age on Enzyme-Histochemical Fibre Spectra and Contractile Properties of Fast- and Slow-Twitch Skeletal Muscles in the Rat. J. Neurol. Sci. 1986, 76, 69–89. [Google Scholar] [CrossRef]
- Kirkendall, D.T.; Garrett, W.E. The Effects of Aging and Training on Skeletal Muscle. Am. J. Sports Med. 1998, 26, 598–602. [Google Scholar] [CrossRef]
- Larsson, L.; Karlsson, J. Isometric and Dynamic Endurance as a Function of Age and Skeletal Muscle Characteristics. Acta Physiol. Scand. 1978, 104, 129–136. [Google Scholar] [CrossRef]
- Barany, M.; Close, R. The Transformation of Myosin in Cross-Innervated Rat Muscles. J. Physiol. 1971, 213, 455–474. [Google Scholar] [CrossRef] [Green Version]
- Evans, W.J.; Lexell, J. Human Aging, Muscle Mass, and Fiber Type Composition. J. Gerontol. Ser. A 1995, 50, 11–16. [Google Scholar] [CrossRef]
- Gibson, M.C.; Schultz, E. The Distribution of Satellite Cells and Their Relationship to Specific Fiber Types in Soleus and Extensor Digitorum Longus Muscles. Anat. Rec. 1982, 202, 329–337. [Google Scholar] [CrossRef]
- Schmalbruch, H.; Hellhammer, U. The Number of Nuclei in Adult Rat Muscles with Special Reference to Satellite Cells. Anat. Rec. 1977, 189, 169–175. [Google Scholar] [CrossRef]
- Snow, M.H. A Quantitative Ultrastructural Analysis of Satellite Cells in Denervated Fast and Slow Muscles of the Mouse. Anat. Rec. 1983, 207, 593–604. [Google Scholar] [CrossRef]
- Mackey, A.L.; Kjaer, M.; Charifi, N.; Henriksson, J.; Bojsen-Moller, J.; Holm, L.; Kadi, F. Assessment of Satellite Cell Number and Activity Status in Human Skeletal Muscle Biopsies. Muscle Nerve 2009, 40, 455–465. [Google Scholar] [CrossRef]
- Okada, S.; Nonaka, I.; Chou, S.M. Muscle Fiber Type Differentiation and Satellite Cell Populations in Normally Grown and Neonatally Denervated Muscles in the Rat. Acta Neuropathol. 1984, 65, 90–98. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; von Maltzahn, J.; Dumont, N.A.; Stark, D.A.; Wang, Y.X.; Nhan, K.; Frenette, J.; Cornelison, D.D.W.; Rudnicki, M.A. Wnt7a Stimulates Myogenic Stem Cell Motility and Engraftment Resulting in Improved Muscle Strength. J. Cell Biol. 2014, 205, 97–111. [Google Scholar] [CrossRef]
- Verdijk, L.B.; Snijders, T.; Drost, M.; Delhaas, T.; Kadi, F.; van Loon, L.J.C. Satellite Cells in Human Skeletal Muscle; from Birth to Old Age. Age 2014, 36, 545. [Google Scholar] [CrossRef] [Green Version]
- Verdijk, L.B.; Koopman, R.; Schaart, G.; Meijer, K.; Savelberg, H.H.; van Loon, L.J.C. Satellite Cell Content Is Specifically Reduced in Type II Skeletal Muscle Fibers in the Elderly. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E151–E157. [Google Scholar] [CrossRef] [Green Version]
- Horwath, O.; Moberg, M.; Larsen, F.J.; Philp, A.; Apró, W.; Ekblom, B. Influence of Sex and Fiber Type on the Satellite Cell Pool in Human Skeletal Muscle. Scand. J. Med. Sci. Sports 2021, 31, 303–312. [Google Scholar] [CrossRef]
- Deschenes, M.; Gaertner, J.; O’Reilly, S. The Effects of Sarcopenia on Muscles with Different Recruitment Patterns and Myofiber Profiles. Curr. Aging Sci. 2014, 6, 266–272. [Google Scholar] [CrossRef]
- Ramamurthy, B.; Larsson, L. Detection of an Aging-Related Increase in Advanced Glycation End Products in Fast- and Slow-Twitch Skeletal Muscles in the Rat. Biogerontology 2013, 14, 293–301. [Google Scholar] [CrossRef]
- Yang, X.; Arber, S.; William, C.; Li, L.; Tanabe, Y.; Jessell, T.M.; Birchmeier, C.; Burden, S.J. Patterning of Muscle Acetylcholine Receptor Gene Expression in the Absence of Motor Innervation. Neuron 2001, 30, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Sanes, J.R.; Lichtman, J.W. Induction, Assembly, Maturation and Maintenance of a Postsynaptic Apparatus. Nat. Rev. Neurosci. 2001, 2, 791–805. [Google Scholar] [CrossRef]
- Patton, B.L.; Miner, J.H.; Chiu, A.Y.; Sanes, J.R. Distribution and Function of Laminins in the Neuromuscular System of Developing, Adult, and Mutant Mice. J. Cell Biol. 1997, 139, 1507–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, R.S.; Nishimune, H. The role of laminins in the organization and function of neuromuscular junctions. Matrix Biol. 2017, 57–58, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.-M.; Yu, H.; Chen, Z.-L. Laminins in Peripheral Nerve Development and Muscular Dystrophy. Mol. Neurobiol. 2007, 35, 288–297. [Google Scholar] [CrossRef]
- Tricaud, N. Myelinating Schwann Cell Polarity and Mechanically-Driven Myelin Sheath Elongation. Front. Cell. Neurosci. 2018, 11, 414. [Google Scholar] [CrossRef] [Green Version]
- Gordon, T. Peripheral Nerve Regeneration and Muscle Reinnervation. Int. J. Mol. Sci. 2020, 21, 8652. [Google Scholar] [CrossRef] [PubMed]
- Willadt, S.; Nash, M.; Slater, C.R. Age-Related Fragmentation of the Motor Endplate Is Not Associated with Impaired Neuromuscular Transmission in the Mouse Diaphragm. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Bao, Z.; Cui, C.; Chow, S.K.-H.; Qin, L.; Wong, R.M.Y.; Cheung, W.-H. AChRs Degeneration at NMJ in Aging-Associated Sarcopenia–A Systematic Review. Front. Aging Neurosci. 2020, 12, 454. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal Muscle Atrophy and the E3 Ubiquitin Ligases MuRF1 and MAFbx/Atrogin-1. Am. J. Physiol.-Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.-Q.; Peng, J.; Wang, A.-H.; Luo, Z.-G. Tumor Necrosis Factor Alpha Mediates Neuromuscular Synapse Elimination. Cell Discov. 2020, 6, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Monaco, C.M.F.; Gingrich, M.A.; Hawke, T.J. Considering Type 1 Diabetes as a Form of Accelerated Muscle Aging. Exerc. Sport Sci. Rev. 2019, 47, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Mesinovic, J.; Zengin, A.; Courten, B.D.; Ebeling, P.R.; Scott, D. Sarcopenia and Type 2 Diabetes Mellitus: A Bidirectional Relationship. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 1057–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popescu, S.; Timar, B.; Baderca, F.; Simu, M.; Diaconu, L.; Velea, I.; Timar, R. Age as an Independent Factor for the Development of Neuropathy in Diabetic Patients. Clin. Interv. Aging 2016, 11, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.J.; Price, S.A.; Chilton, L.; Calcutt, N.A.; Tomlinson, D.R.; Verkhratsky, A.; Fernyhough, P. Insulin Prevents Depolarization of the Mitochondrial Inner Membrane in Sensory Neurons of Type 1 Diabetic Rats in the Presence of Sustained Hyperglycemia. Diabetes 2003, 52, 2129–2136. [Google Scholar] [CrossRef] [Green Version]
- Dobretsov, M.; Romanovsky, D.; Stimers, J.R. Early Diabetic Neuropathy: Triggers and Mechanisms. World J. Gastroenterol. 2007, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Brisset, M.; Nicolas, G. Peripheral Neuropathies and Aging. Geriatr. Psychol. Neuropsychiatr. Vieil. 2018, 16, 409–413. [Google Scholar] [CrossRef]
- Almurdhi, M.M.; Reeves, N.D.; Bowling, F.L.; Boulton, A.J.M.; Jeziorska, M.; Malik, R.A. Reduced Lower-Limb Muscle Strength and Volume in Patients With Type 2 Diabetes in Relation to Neuropathy, Intramuscular Fat, and Vitamin D Levels. Diabetes Care 2016, 39, 441–447. [Google Scholar] [CrossRef] [Green Version]
- Andersen, H.; Nielsen, S.; Mogensen, C.E.; Jakobsen, J. Muscle Strength in Type 2 Diabetes. Diabetes 2004, 53, 1543–1548. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Loenneke, J.P.; Thiebaud, R.S.; Fukunaga, T. Age-Related Site-Specific Muscle Wasting of Upper and Lower Extremities and Trunk in Japanese Men and Women. Age (Omaha) 2014, 36, 813. [Google Scholar] [CrossRef]
- Abe, T.; Sakamaki, M.; Yasuda, T.; Bemben, M.G.; Kondo, M.; Kawakami, Y.; Fukunaga, T. Age-Related, Site-Specific Muscle Loss in 1507 Japanese Men and Women Aged 20 to 95 Years. J. Sports Sci. Med. 2011, 10, 145. [Google Scholar] [PubMed]
- Mori, H.; Kuroda, A.; Araki, M.; Suzuki, R.; Taniguchi, S.; Tamaki, M.; Akehi, Y.; Matsuhisa, M. Advanced Glycation End-Products Are a Risk for Muscle Weakness in Japanese Patients with Type 1 Diabetes. J. Diabetes Investig. 2017, 8, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Kuroda, A.; Ishizu, M.; Ohishi, M.; Takashi, Y.; Otsuka, Y.; Taniguchi, S.; Tamaki, M.; Kurahashi, K.; Yoshida, S.; et al. Association of Accumulated Advanced Glycation End-Products with a High Prevalence of Sarcopenia and Dynapenia in Patients with Type 2 Diabetes. J. Diabetes Investig. 2019, 10, 1332–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piasecki, M.; Ireland, A.; Jones, D.A.; McPhee, J.S. Age-Dependent Motor Unit Remodelling in Human Limb Muscles. Biogerontology 2016, 17, 485. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.M.; Conwit, R.A.; Ferrucci, L.; Metter, E.J. Age-Associated Changes in Motor Unit Physiology: Observations From the Baltimore Longitudinal Study of Aging. Arch. Phys. Med. Rehabil. 2009, 90, 1237. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-Y.; Yang, R.-S.; Sheu, M.-L.; Chan, D.-C.; Yang, T.-H.; Tsai, K.-S.; Chiang, C.-K.; Liu, S.-H. Advanced Glycation End-Products Induce Skeletal Muscle Atrophy and Dysfunction in Diabetic Mice via a RAGE-Mediated, AMPK-down-Regulated, Akt Pathway. J. Pathol. 2016, 238, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced Glycation End Products and Diabetic Complications. Korean J. Physiol. Pharmacol. 2014, 18, 1. [Google Scholar] [CrossRef] [Green Version]
- Wilson, N.M.; Wright, D.E. Experimental Motor Neuropathy in Diabetes. Handb. Clin. Neurol. 2014, 126, 461–467. [Google Scholar] [CrossRef]
- Dyck, P.J.; Kratz, K.M.; Karnes, J.L.; Litchy, W.J.; Klein, R.; Pach, J.M.; Wilson, D.M.; O’Brien, P.C.; Melton, L.J. The Prevalence by Staged Severity of Various Types of Diabetic Neuropathy, Retinopathy, and Nephropathy in a Population-based Cohort. Neurology 1993, 43, 817. [Google Scholar] [CrossRef]
- Feldman, E.L.; Nave, K.A.; Jensen, T.S.; Bennett, D.L.H. New Horizons in Diabetic Neuropathy: Mechanisms, Bioenergetics, and Pain. Neuron 2017, 93, 1296–1313. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, K. Diabetes Mellitus-Related Dysfunction of the Motor System. Int. J. Mol. Sci. 2020, 21, 7485. [Google Scholar] [CrossRef] [PubMed]
- Ramji, N.; Toth, C.; Kennedy, J.; Zochodne, D.W. Does Diabetes Mellitus Target Motor Neurons? Neurobiol. Dis. 2007, 26, 301–311. [Google Scholar] [CrossRef]
- Estrada-Bonilla, Y.C.; Castro, P.A.; Luna, G.L.; Souza, A.B.; Santos, G.S.; Salvini, T.F.; Leal, A.M.; Russo, T.L. Reaching Task Performance Is Associated to Neuromuscular Junction Adaptations in Rats with Induced Diabetes Mellitus. Braz. J. Med. Biol. Res. 2020, 53. [Google Scholar] [CrossRef] [PubMed]
- Cameron, N.E.; Cotter, M.A.; Dines, K.; Love, A. Effects of Aminoguanidine on Peripheral Nerve Function and Polyol Pathway Metabolites in Streptozotocin-Diabetic Rats. Diabetologia 1992, 35, 946–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, N.E.; Gibson, T.M.; Nangle, M.R.; Cotter, M.A. Inhibitors of Advanced Glycation End Product Formation and Neurovascular Dysfunction in Experimental Diabetes. Ann. N. Y. Acad. Sci. 2005, 1043, 784–792. [Google Scholar] [CrossRef]
- Nishizawa, Y.; Wada, R.I.; Baba, M.; Takeuchi, M.; Hanyu-Itabashi, C.; Yagihashi, S. Neuropathy Induced by Exogenously Administered Advanced Glycation End-Products in Rats. J. Diabetes Investig. 2010, 1, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öztürk, G.; Şekeroğlu, M.R.; Erdoğan, E.; Öztürk, M. The Effect of Non-Enzymatic Glycation of Extracellular Matrix Proteins on Axonal Regeneration in Vitro. Acta Neuropathol. 2006, 112, 627–632. [Google Scholar] [CrossRef]
- Duran-Jimenez, B.; Dobler, D.; Moffatt, S.; Rabbani, N.; Streuli, C.H.; Thornalley, P.J.; Tomlinson, D.R.; Gardiner, N.J. Advanced Glycation End Products in Extracellular Matrix Proteins Contribute to the Failure of Sensory Nerve Regeneration in Diabetes. Diabetes 2009, 58, 2893–2903. [Google Scholar] [CrossRef] [Green Version]
- Gumy, L.F.; Bampton, E.T.W.; Tolkovsky, A.M. Hyperglycaemia Inhibits Schwann Cell Proliferation and Migration and Restricts Regeneration of Axons and Schwann Cells from Adult Murine DRG. Mol. Cell. Neurosci. 2008, 37, 298–311. [Google Scholar] [CrossRef]
- Sango, K.; Horie, H.; Saito, H.; Ajiki, K.; Tokashiki, A.; Takeshita, K.; Ishigatsubo, Y.; Kawano, H.; Ishikawa, Y. Diabetes Is Not a Potent Inducer of Neuronal Cell Death in Mouse Sensory Ganglia, but It Enhances Neurite Regeneration in Vitro. Life Sci. 2002, 71, 2351–2368. [Google Scholar] [CrossRef]
- Saito, H.; Sango, K.; Horie, H.; Ikeda, H.; Ishigatsubo, Y.; Ishikawa, Y.; Inoue, S. Enhanced Neural Regeneration from Transected Vagus Nerve Terminal in Diabetic Mice in Vitro. Neuroreport 1999, 10, 1025–1028. [Google Scholar] [CrossRef]
- Nishida, N.; Yamagishi, S.-I.; Mizukami, H.; Yagihashi, S. Impaired Nerve Fiber Regeneration in Axotomized Peripheral Nerves in Streptozotocin-Diabetic Rats. J. Diabetes Investig. 2013, 4, 533–539. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Terada, M.; Taniguchi, Y.; Sasaki, T.; Maeda, K.; Haneda, M.; Kashiwagi, A.; Kikkawa, R. Impaired Regeneration and No Amelioration with Aldose Reductase Inhibitor in Crushed Unmyelinated Nerve Fibers of Diabetic Rats. Neuroreport 1999, 10, 2405–2409. [Google Scholar] [CrossRef]
- Sango, K.; Mizukami, H.; Horie, H.; Yagihashi, S. Impaired Axonal Regeneration in Diabetes. Perspective on the Underlying Mechanism from In Vivo and In Vitro Experimental Studies. Front. Endocrinol. (Lausanne) 2017, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Eckersley, L. Role of the Schwann Cell in Diabetic Neuropathy. Int. Rev. Neurobiol. 2002, 50, 293–321. [Google Scholar] [CrossRef]
- Ota, K.; Nakamura, J.; Li, W.; Kozakae, M.; Watarai, A.; Nakamura, N.; Yasuda, Y.; Nakashima, E.; Naruse, K.; Watabe, K.; et al. Metformin Prevents Methylglyoxal-Induced Apoptosis of Mouse Schwann Cells. Biochem. Biophys. Res. Commun. 2007, 357, 270–275. [Google Scholar] [CrossRef]
- Xu, S.; Bao, W.; Men, X.; Liu, Y.; Sun, J.; Li, J.; Liu, H.; Cai, H.; Zhang, W.; Lou, J.; et al. Interleukin-10 Protects Schwann Cells against Advanced Glycation End Products-Induced Apoptosis via NF-ΚB Suppression. Exp. Clin. Endocrinol. Diabetes 2019, 128, 89–96. [Google Scholar] [CrossRef]
- Sekido, H.; Suzuki, T.; Jomori, T.; Takeuchi, M.; Yabe-Nishimura, C.; Yagihashi, S. Reduced Cell Replication and Induction of Apoptosis by Advanced Glycation End Products in Rat Schwann Cells. Biochem. Biophys. Res. Commun. 2004, 320, 241–248. [Google Scholar] [CrossRef]
- Fukunaga, M.; Miyata, S.; Liu, B.F.; Miyazaki, H.; Hirota, Y.; Higo, S.; Hamada, Y.; Ueyama, S.; Kasuga, M. Methylglyoxal Induces Apoptosis through Activation of P38 MAPK in Rat Schwann Cells. Biochem. Biophys. Res. Commun. 2004, 320, 689–695. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olson, L.C.; Redden, J.T.; Schwartz, Z.; Cohen, D.J.; McClure, M.J. Advanced Glycation End-Products in Skeletal Muscle Aging. Bioengineering 2021, 8, 168. https://doi.org/10.3390/bioengineering8110168
Olson LC, Redden JT, Schwartz Z, Cohen DJ, McClure MJ. Advanced Glycation End-Products in Skeletal Muscle Aging. Bioengineering. 2021; 8(11):168. https://doi.org/10.3390/bioengineering8110168
Chicago/Turabian StyleOlson, Lucas C., James T. Redden, Zvi Schwartz, David J. Cohen, and Michael J. McClure. 2021. "Advanced Glycation End-Products in Skeletal Muscle Aging" Bioengineering 8, no. 11: 168. https://doi.org/10.3390/bioengineering8110168
APA StyleOlson, L. C., Redden, J. T., Schwartz, Z., Cohen, D. J., & McClure, M. J. (2021). Advanced Glycation End-Products in Skeletal Muscle Aging. Bioengineering, 8(11), 168. https://doi.org/10.3390/bioengineering8110168