
Alzheimer’s Disease: Current Perspectives and Advances in Physiological Modeling


Abstract
:1. Introduction
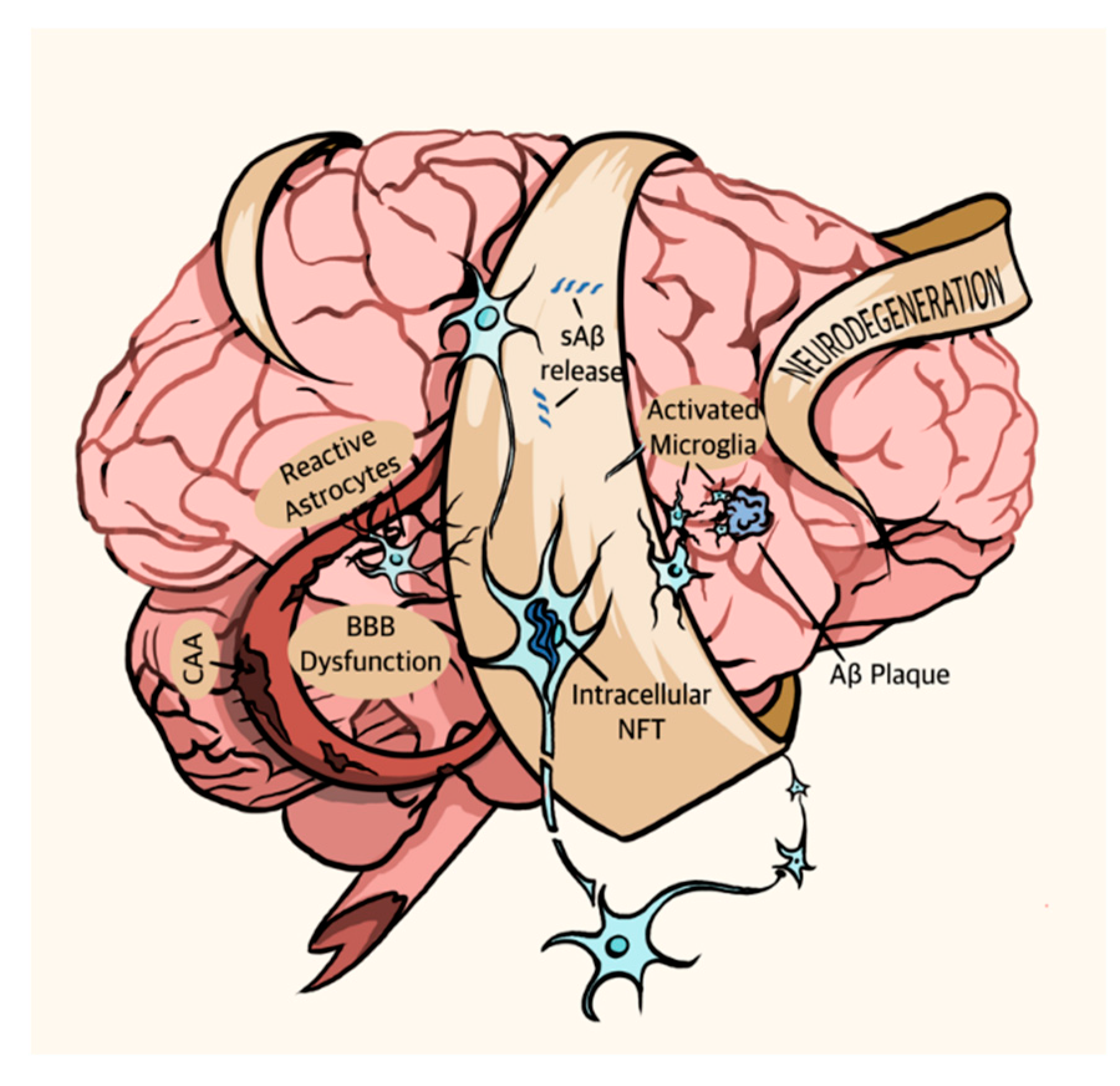
1.1. Alzheimer’s Disease Pathophysiology
1.1.1. Amyloid Beta Peptides (Aβ)
1.1.2. Tau
1.1.3. Familial and Sporadic Alzheimer’s Disease, and Other Symptoms
2. Modeling Alzheimer’s Disease
2.1. Computational Modelling
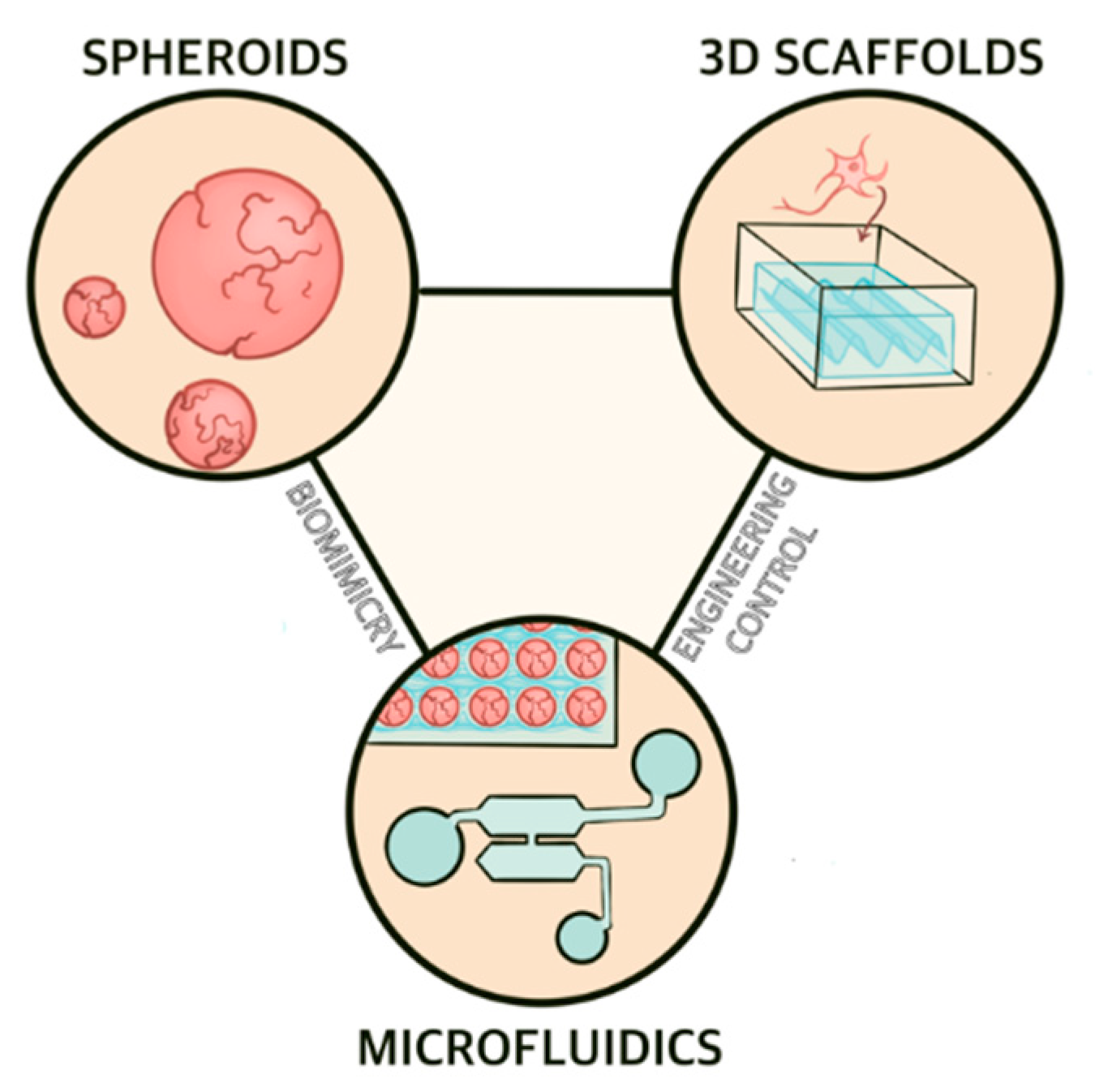
2.2. Experimental Models of Alzheimer’s Disease
2.2.1. Limitations of Transgenic Mouse Models
2.2.2. Microphysiological Modeling
2.2.3. Three Dimensional Scaffolds
2.2.4. Spheroids
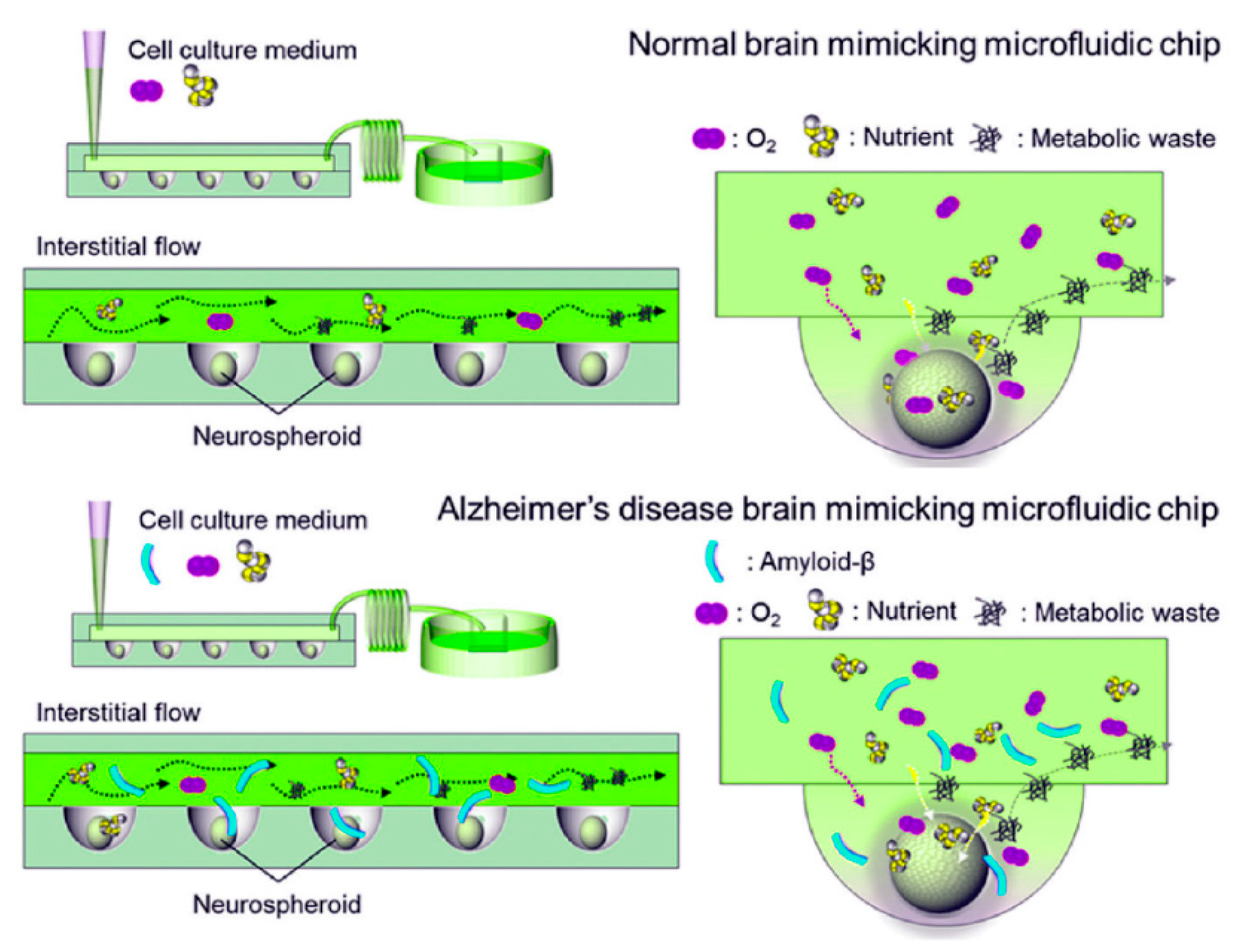
2.2.5. Microfluidics
3. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sengoku, R. Aging and Alzheimer’s Disease Pathology. Neuropathology 2020, 40, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Förstl, H.; Kurz, A. Clinical Features of Alzheimer’s Disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of Acetylcholinesterase Inhibitors in Alzheimer’s Disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, I.; Sorrentino, L.; Paoletti, A.; Marra, R.; Arbitrio, M. The State of The Art on Acetylcholinesterase Inhibitors in the Treatment of Alzheimer’s Disease. J. Cent. Nerv. Syst. Dis. 2021, 13, 11795735211029112. [Google Scholar] [CrossRef] [PubMed]
- Moreta, M.P.-G.; Burgos-Alonso, N.; Torrecilla, M.; Marco-Contelles, J.; Bruzos-Cidón, C. Efficacy of Acetylcholinesterase Inhibitors on Cognitive Function in Alzheimer’s Disease. Review of Reviews. Biomedicines 2021, 9, 1689. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Yan, R. A Close Look at BACE1 inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263. [Google Scholar] [CrossRef]
- Mullard, A. Landmark Alzheimer’s Drug Approval Confounds Research Community. Nature 2021, 594, 309–310. [Google Scholar] [CrossRef]
- Walsh, S.; Merrick, R.; Milne, R.; Brayne, C. Aducanumab for Alzheimer’s Disease? BMJ 2021, 374, n1682. [Google Scholar] [CrossRef]
- Cummings, J.; Salloway, S. Aducanumab: Appropriate Use Recommendations. J. Prev. Alzheimer’s Dis. 2021, 8, 398–410. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Banerjee, D. A Primer on the Evolution of Aducanumab: The First Antibody Approved for Treatment of Alzheimer’s Disease. J. Alzheimers Dis. JAD 2021, 83, 1537–1552. [Google Scholar] [CrossRef] [PubMed]
- Inacio, P. FDA OKs Phase I Trial of Nasal Spray Immunotherapy Protollin. Alzhiemer’s Today. 2021. Available online: https://alzheimersnewstoday.com/2021/08/24/fda-oks-alzheimers-trial-testing-protollin-nasal-spray-immunotherapy/ (accessed on 3 December 2021).
- Huang, L.-K.; Chao, S.-P.; Hu, C.-J. Clinical Trials of New Drugs for Alzheimer Disease. J. Biomed. Sci. 2020, 27, 18. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Bateman, R.J.; Munsell, L.Y.; Morris, J.C.; Swarm, R.; Yarasheski, K.E.; Holtzman, D.M. Quantifying CNS Protein Production and Clearance Rates in Humans Using in Vivo Stable Isotope Labeling, Immunoprecipitation, and Tandem Mass Spectrometry. Nat. Med. 2006, 12, 856–861. [Google Scholar] [CrossRef]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid β-Peptide Is Produced by Cultured Cells during Normal Metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Gu, L.; Guo, Z. Alzheimer’s Aβ42 and Aβ40 Peptides Form Interlaced Amyloid Fibrils. J. Neurochem. 2013, 126, 305–311. [Google Scholar] [CrossRef]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Bierer, L.M.; Hof, P.R.; Purohit, D.P.; Carlin, L.; Schmeidler, J.; Davis, K.L.; Perl, D.P. Neocortical Neurofibrillary Tangles Correlate with Dementia Severity in Alzheimer’s Disease. Arch. Neurol. 1995, 52, 81–88. [Google Scholar] [CrossRef]
- Neuropil Threads-MeSH-NCBI. Available online: https://www.ncbi.nlm.nih.gov/mesh?Db=mesh&Cmd=DetailsSearch&Term=%22Neuropil+Threads%22%5BMeSH+Terms%5D (accessed on 4 September 2021).
- Ash, E.L. 27-Dementia. In On Call Neurology, 3rd ed.; Marshall, R.S., Mayer, S.A., Eds.; On Call Series; Saunders-Elsevier-Health Sciences Division: Philadelphia, PA, USA, 2007; pp. 401–417. ISBN 978-1-4160-2375-3. [Google Scholar]
- Chávez-Gutiérrez, L.; Szaruga, M. Mechanisms of Neurodegeneration—Insights from Familial Alzheimer’s Disease. Semin. Cell Dev. Biol. 2020, 105, 75–85. [Google Scholar] [CrossRef]
- Kummer, M.P.; Heneka, M.T. Truncated and Modified Amyloid-Beta Species. Alzheimers Res. Ther. 2014, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Spillantini, M.G. A Century of Alzheimer’s Disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two Transmembrane Aspartates in Presenilin-1 Required for Presenilin Endoproteolysis and γ-Secretase Activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The Mechanism of γ-Secretase Dysfunction in Familial Alzheimer Disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–186. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-β Plaques Enhance Alzheimer’s Brain Tau-Seeded Pathologies by Facilitating Neuritic Plaque Tau Aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and Tau—A Toxic Pas de Deux in Alzheimer’s Disease. Nat. Rev. Neurosci. 2011, 12, 67–72. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Imbimbo, B.P.; Ippati, S.; Watling, M. Should Drug Discovery Scientists Still Embrace the Amyloid Hypothesis for Alzheimer’s Disease or Should They Be Looking Elsewhere? Expert Opin. Drug Discov. 2020, 15, 1241–1251. [Google Scholar] [CrossRef] [PubMed]
- Caselli, R.J.; Knopman, D.S.; Bu, G. An Agnostic Reevaluation of the Amyloid Cascade Hypothesis of Alzheimer’s Disease Pathogenesis: The Role of APP Homeostasis. Alzheimers Dement. 2020, 16, 1582–1590. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Tolar, M.; Abushakra, S.; Sabbagh, M. The Path Forward in Alzheimer’s Disease Therapeutics: Reevaluating the Amyloid Cascade Hypothesis. Alzheimers Dement. 2020, 16, 1553–1560. [Google Scholar] [CrossRef]
- Frisoni, G.B.; Altomare, D.; Thal, D.R.; Ribaldi, F.; van der Kant, R.; Ossenkoppele, R.; Blennow, K.; Cummings, J.; van Duijn, C.; Nilsson, P.M.; et al. The Probabilistic Model of Alzheimer Disease: The Amyloid Hypothesis Revised. Nat. Rev. Neurosci. 2022, 23, 53–66. [Google Scholar] [CrossRef]
- Musiek, E.S.; Gomez-Isla, T.; Holtzman, D.M. Aducanumab for Alzheimer Disease: The Amyloid Hypothesis Moves from Bench to Bedside. J. Clin. Investig. 2021, 131, e154889. [Google Scholar] [CrossRef]
- Canevelli, M.; Bruno, G.; Cesari, M. The Sterile Controversy on the Amyloid Cascade Hypothesis. Neurosci. Biobehav. Rev. 2017, 83, 472–473. [Google Scholar] [CrossRef]
- Hunter, S.; Brayne, C. Amyloid in the Ageing Brain: New Frameworks and Perspectives. Aging Brain 2021, 1, 100008. [Google Scholar] [CrossRef]
- Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer Disease: Risk, Mechanisms and Therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Husain, M.A.; Laurent, B.; Plourde, M. APOE and Alzheimer’s Disease: From Lipid Transport to Physiopathology and Therapeutics. Front. Neurosci. 2021, 15, 85. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Grammas, P. Neurovascular Dysfunction, Inflammation and Endothelial Activation: Implications for the Pathogenesis of Alzheimer’s Disease. J. Neuroinflam. 2011, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, J.M.; Kwan, J.; Malek-Ahmadi, M.; Maarouf, C.L.; Kokjohn, T.A.; Belden, C.; Sabbagh, M.N.; Beach, T.G.; Roher, A.E. Morphological and Pathological Evolution of the Brain Microcirculation in Aging and Alzheimer’s Disease. PLoS ONE 2012, 7, e36893. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolós, M.; Perea, J.R.; Avila, J. Alzheimer’s Disease as an Inflammatory Disease. Biomol. Concepts 2017, 8, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Iturria-Medina, Y.; Sotero, R.C.; Toussaint, P.J.; Evans, A.C.; Alzheimer’s Disease Neuroimaging Initiative. Epidemic Spreading Model to Characterize Misfolded Proteins Propagation in Aging and Associated Neurodegenerative Disorders. PLOS Comput. Biol. 2014, 10, e1003956. [Google Scholar] [CrossRef] [Green Version]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Liu, X.; Jiang, R.; Yan, X.; Ling, Z. Roles and Mechanisms of Gut Microbiota in Patients With Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 650047. [Google Scholar] [CrossRef]
- Harris, S.A.; Harris, E.A. Herpes Simplex Virus Type 1 and Other Pathogens Are Key Causative Factors in Sporadic Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 319–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, K.; Kar, S.; Das, R.N. Chapter 5—Computational Chemistry. In Understanding the Basics of QSAR for Applications in Pharmaceutical Sciences and Risk Assessment; Roy, K., Kar, S., Das, R.N., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 151–189. ISBN 978-0-12-801505-6. [Google Scholar]
- Leonard, C.; Phillips, C.; McCarty, J. Insight Into Seeded Tau Fibril Growth From Molecular Dynamics Simulation of the Alzheimer’s Disease Protofibril Core. Front. Mol. Biosci. 2021, 8, 109. [Google Scholar] [CrossRef]
- Wan, J.; Gong, Y.; Xu, Z.; Dong, X.; Wei, G.; Zhang, Q. Molecular Dynamics Simulations Reveal the Destabilization Mechanism of Alzheimer’s Disease-Related Tau R3-R4 Protofilament by Norepinephrine. Biophys. Chem. 2021, 271, 106541. [Google Scholar] [CrossRef]
- Fan, H.-M.; Gu, R.-X.; Wang, Y.-J.; Pi, Y.-L.; Zhang, Y.-H.; Xu, Q.; Wei, D.-Q. Destabilization of Alzheimer’s Aβ42 Protofibrils with a Novel Drug Candidate Wgx-50 by Molecular Dynamics Simulations. J. Phys. Chem. B 2015, 119, 11196–11202. [Google Scholar] [CrossRef]
- Petrella, J.R.; Hao, W.; Rao, A.; Doraiswamy, P.M. Computational Causal Modeling of the Dynamic Biomarker Cascade in Alzheimer’s Disease. Comput. Math. Methods Med. 2019, 2019, e6216530. [Google Scholar] [CrossRef] [Green Version]
- Caligiore, D.; Silvetti, M.; D’Amelio, M.; Puglisi-Allegra, S.; Baldassarre, G. Computational Modeling of Cate cholamines Dysfunction in Alzheimer’s Disease at Pre-Plaque Stage. J. Alzheimers Dis. 2020, 77, 275–290. [Google Scholar] [CrossRef] [PubMed]
- Hasselmo, M.E. A computational model of the progression of Alzheimer’s disease. MD Comput. 1997, 14, 181–191. [Google Scholar]
- Hassan, M.; Abbas, Q.; Seo, S.-Y.; Shahzadi, S.; Al Ashwal, H.; Zaki, N.; Iqbal, Z.; Moustafa, A.A. Computational Modeling and Biomarker Studies of Pharmacological Treatment of Alzheimer’s Disease (Review). Mol. Med. Rep. 2018, 18, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Brodland, G.W. How Computational Models Can Help Unlock Biological Systems. Semin. Cell Dev. Biol. 2015, 47–48, 62–73. [Google Scholar] [CrossRef] [Green Version]
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 2019, 10, 362. [Google Scholar] [CrossRef]
- Cipriani, G.; Dolciotti, C.; Picchi, L.; Bonuccelli, U. Alzheimer and His Disease: A Brief History. Neurol. Sci. 2011, 32, 275–279. [Google Scholar] [CrossRef]
- Elder, G.A.; Sosa, M.A.G.; Gasperi, R.D. Transgenic Mouse Models of Alzheimer’s Disease. Mt. Sinai J. Med. J. Transl. Pers. Med. 2010, 77, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP Mouse Models for Alzheimer’s Disease Preclinical Studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single App Knock-in Mouse Models of Alzheimer’s Disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Hamanaka, H.; Katoh-Fukui, Y.; Suzuki, K.; Kobayashi, M.; Suzuki, R.; Motegi, Y.; Nakahara, Y.; Takeshita, A.; Kawai, M.; Ishiguro, K.; et al. Altered Cholesterol Metabolism in Human Apolipoprotein E4 Knock-in Mice. Hum. Mol. Genet. 2000, 9, 353–361. [Google Scholar] [CrossRef]
- Gong, J.-S.; Kobayashi, M.; Hayashi, H.; Zou, K.; Sawamura, N.; Fujita, S.C.; Yanagisawa, K.; Michikawa, M. Apolipoprotein E (ApoE) Isoform-Dependent Lipid Release from Astrocytes Prepared from Human ApoE3 and ApoE4 Knock-in Mice. J. Biol. Chem. 2002, 277, 29919–29926. [Google Scholar] [CrossRef] [Green Version]
- Tesseur, I.; Van Dorpe, J.; Spittaels, K.; Van den Haute, C.; Moechars, D.; Van Leuven, F. Expression of Human Apolipoprotein E4 in Neurons Causes Hyperphosphorylation of Protein Tau in the Brains of Transgenic Mice. Am. J. Pathol. 2000, 156, 951–964. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, J.; Paradis, A.-L.; Boucher, M.; Andrieu, L.; Barnéoud, P.; Rondi-Reig, L. Flexibility as a Marker of Early Cognitive Decline in Humanized Apolipoprotein E Ε4 (ApoE4) Mice. Neurobiol. Aging 2021, 102, 129–138. [Google Scholar] [CrossRef]
- Ma, Q.-L.; Zhu, C.; Morselli, M.; Su, T.; Pelligrini, M.; Lu, Z.; Jones, M.; Denver, P.; Castro, D.; Gu, X.; et al. The Novel Omega-6 Fatty Acid Docosapentaenoic Acid Positively Modulates Brain Innate Immune Response for Resolving Neuroinflammation at Early and Late Stages of Humanized APOE-Based Alzheimer’s Disease Models. Front. Immunol. 2020, 11, 2364. [Google Scholar] [CrossRef]
- Huynh, T.-P.V.; Wang, C.; Tran, A.C.; Tabor, G.T.; Mahan, T.E.; Francis, C.M.; Finn, M.B.; Spellman, R.; Manis, M.; Tanzi, R.E.; et al. Lack of Hepatic ApoE Does Not Influence Early Aβ Deposition: Observations from a New APOE Knock-in Model. Mol. Neurodegener. 2019, 14, 37. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, C.T.; Maldonado Weng, J.; LaDu, M.J. Alzheimer’s Disease Pathology in APOE Transgenic Mouse Models: The Who, What, When, Where, Why, and How. Neurobiol. Dis. 2020, 139, 104811. [Google Scholar] [CrossRef]
- Jucker, M. The Benefits and Limitations of Animal Models for Translational Research in Neurodegenerative Diseases. Nat. Med. 2010, 16, 1210–1214. [Google Scholar] [CrossRef]
- Jessen, F.; Spottke, A.; Boecker, H.; Brosseron, F.; Buerger, K.; Catak, C.; Fliessbach, K.; Franke, C.; Fuentes, M.; Heneka, M.T.; et al. Design and First Baseline Data of the DZNE Multicenter Observational Study on Predementia Alzheimer’s Disease (DELCODE). Alzheimers Res. Ther. 2018, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.M.; Golde, T.E.; Lagier-Tourenne, C. Animal Models of Neurodegenerative Diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s Disease Drug-Development Pipeline: Few Candidates, Frequent Failures. Alzheimers Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cummings, J. Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin. Transl. Sci. 2018, 11, 147–152. [Google Scholar] [CrossRef]
- Morgan, D.; Diamond, D.M.; Gottschall, P.E.; Ugen, K.E.; Dickey, C.; Hardy, J.; Duff, K.; Jantzen, P.; DiCarlo, G.; Wilcock, D.; et al. Aβ Peptide Vaccination Prevents Memory Loss in an Animal Model of Alzheimer’s Disease. Nature 2000, 408, 982–985. [Google Scholar] [CrossRef]
- Nicoll, J.A.R.; Buckland, G.R.; Harrison, C.H.; Page, A.; Harris, S.; Love, S.; Neal, J.W.; Holmes, C.; Boche, D. Persistent Neuropathological Effects 14 Years Following Amyloid-β Immunization in Alzheimer’s Disease. Brain 2019, 142, 2113–2126. [Google Scholar] [CrossRef] [Green Version]
- Cirit, M.; Stokes, C.L. Maximizing the Impact of Microphysiological Systems with in Vitro—In Vivo Translation. Lab Chip 2018, 18, 1831–1837. [Google Scholar] [CrossRef] [Green Version]
- Damiati, S.; Kompella, U.B.; Damiati, S.A.; Kodzius, R. Microfluidic Devices for Drug Delivery Systems and Drug Screening. Genes 2018, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Stett, A.; Egert, U.; Guenther, E.; Hofmann, F.; Meyer, T.; Nisch, W.; Haemmerle, H. Biological Application of Microelectrode Arrays in Drug Discovery and Basic Research. Anal. Bioanal. Chem. 2003, 377, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Strickland, J.D.; Lefew, W.R.; Crooks, J.; Hall, D.; Ortenzio, J.N.; Dreher, K.; Shafer, T.J. In Vitro Screening of Metal Oxide Nanoparticles for Effects on Neural Function Using Cortical Networks on Microelectrode Arrays. Nanotoxicology 2016, 10, 619–628. [Google Scholar] [CrossRef]
- Obien, M.E.J.; Deligkaris, K.; Bullmann, T.; Bakkum, D.J.; Frey, U. Revealing Neuronal Function through Microelectrode Array Recordings. Front. Neurosci. 2015, 8, 423. [Google Scholar] [CrossRef] [Green Version]
- Penney, J.; Ralvenius, W.T.; Tsai, L.-H. Modeling Alzheimer’s Disease with IPSC-Derived Brain Cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef] [Green Version]
- Ko, K.R.; Frampton, J.P. Developments in 3D Neural Cell Culture Models: The Future of Neurotherapeutics Testing? Expert Rev. Neurother. 2016, 16, 739–741. [Google Scholar] [CrossRef] [Green Version]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Téglási, A.; Bock, I.; Giudice, M.L.; Táncos, Z.; Molnár, K.; László, L.; et al. Neurons Derived from Sporadic Alzheimer’s Disease IPSCs Reveal Elevated TAU Hyperphosphorylation, Increased Amyloid Levels, and GSK3B Activation. Alzheimers Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Irons, H.R.; Cullen, D.K.; Shapiro, N.P.; Lambert, N.A.; Lee, R.H.; LaPlaca, M.C. Three-Dimensional Neural Constructs: A Novel Platform for Neurophysiological Investigation. J. Neural Eng. 2008, 5, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Laplaca, M.; Vernekar, V.; Shoemaker, J.; Cullen, D.K.; Coulter, W. Three-Dimensional Neuronal Cultures. In Methods Bioeng. 3D Tissue Eng.; Chapter 11; Berthiaume, F., Morgan, J., Eds.; Artech House Publishers: Boston, MA, USA, 2010; Volume 11, pp. 187–204. ISBN 978-1-59693-458-0. [Google Scholar]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A Three-Dimensional Human Neural Cell Culture Model of Alzheimer’s Disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Kwak, S.S.; Washicosky, K.J.; Brand, E.; von Maydell, D.; Aronson, J.; Kim, S.; Capen, D.E.; Cetinbas, M.; Sadreyev, R.; Ning, S.; et al. Amyloid-Β42/40 Ratio Drives Tau Pathology in 3D Human Neural Cell Culture Models of Alzheimer’s Disease. Nat. Commun. 2020, 11, 1377. [Google Scholar] [CrossRef] [PubMed]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.-T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.-H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [Green Version]
- Kapr, J.; Petersilie, L.; Distler, T.; Lauria, I.; Bendt, F.; Sauter, C.M.; Boccaccini, A.R.; Rose, C.R.; Fritsche, E. Human Induced Pluripotent Stem Cell-Derived Neural Progenitor Cells Produce Distinct Neural 3D In Vitro Models Depending on Alginate/Gellan Gum/Laminin Hydrogel Blend Properties. Adv. Healthc. Mater. 2021, 10, e2100131. [Google Scholar] [CrossRef]
- Ranjan, V.D.; Qiu, L.; Lee, J.W.-L.; Chen, X.; Jang, S.E.; Chai, C.; Lim, K.-L.; Tan, E.-K.; Zhang, Y.; Huang, W.M.; et al. A Microfiber Scaffold-Based 3D in Vitro Human Neuronal Culture Model of Alzheimer’s Disease. Biomater. Sci. 2020, 8, 4861–4874. [Google Scholar] [CrossRef]
- Cui, G.-H.; Shao, S.-J.; Yang, J.-J.; Liu, J.-R.; Guo, H.-D. Designer Self-Assemble Peptides Maximize the Therapeutic Benefits of Neural Stem Cell Transplantation for Alzheimer’s Disease via Enhancing Neuron Differentiation and Paracrine Action. Mol. Neurobiol. 2016, 53, 1108–1123. [Google Scholar] [CrossRef] [Green Version]
- DeQuach, J.A.; Yuan, S.H.; Goldstein, L.S.B.; Christman, K.L. Decellularized Porcine Brain Matrix for Cell Culture and Tissue Engineering Scaffolds. Tissue Eng. Part A 2011, 17, 2583–2592. [Google Scholar] [CrossRef] [Green Version]
- Simsa, R.; Rothenbücher, T.; Gürbüz, H.; Ghosheh, N.; Emneus, J.; Jenndahl, L.; Kaplan, D.L.; Bergh, N.; Serrano, A.M.; Fogelstrand, P. Brain Organoid Formation on Decellularized Porcine Brain ECM Hydrogels. PLoS ONE 2021, 16, e0245685. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Pekkanen-Mattila, M.; Shahsavani, M.; Falk, A.; Teixeira, A.I.; Herland, A. A 3D Alzheimer’s Disease Culture Model and the Induction of P21-Activated Kinase Mediated Sensing in IPSC Derived Neurons. Biomaterials 2014, 35, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Bagrov, D.; Gazizova, Y.; Podgorsky, V.; Udovichenko, I.; Danilkovich, A.; Prusakov, K.; Klinov, D. Morphology and Aggregation of RADA-16-I Peptide Studied by AFM, NMR and Molecular Dynamics Simulations. Pept. Sci. 2016, 106, 72–81. [Google Scholar] [CrossRef]
- Dzamba, D.; Harantova, L.; Butenko, O.; Anderova, M. Glial Cells—The Key Elements of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 894–911. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The Role of Astroglia in Alzheimer’s Disease: Pathophysiology and Clinical Implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The Role of Innate Immune Responses and Neuroinflammation in Amyloid Accumulation and Progression of Alzheimer’s Disease. Immunol. Cell Biol. 2020, 98, 28–41. [Google Scholar] [CrossRef]
- Papadimitriou, C.; Celikkaya, H.; Cosacak, M.I.; Mashkaryan, V.; Bray, L.; Bhattarai, P.; Brandt, K.; Hollak, H.; Chen, X.; He, S.; et al. 3D Culture Method for Alzheimer’s Disease Modeling Reveals Interleukin-4 Rescues Aβ42-Induced Loss of Human Neural Stem Cell Plasticity. Dev. Cell 2018, 46, 85–101.e8. [Google Scholar] [CrossRef] [Green Version]
- Cairns, D.M.; Rouleau, N.; Parker, R.N.; Walsh, K.G.; Gehrke, L.; Kaplan, D.L. A 3D Human Brain–like Tissue Model of Herpes-Induced Alzheimer’s Disease. Sci. Adv. 2020, 6, eaay8828. [Google Scholar] [CrossRef]
- Simpson, L.W.; Szeto, G.L.; Boukari, H.; Good, T.A.; Leach, J.B. Impact of Four Common Hydrogels on Amyloid-β (Aβ) Aggregation and Cytotoxicity: Implications for 3D Models of Alzheimer’s Disease. ACS Omega 2020, 5, 20250–20260. [Google Scholar] [CrossRef]
- Castillo, G.M.; Lukito, W.; Peskind, E.; Raskind, M.; Kirschner, D.A.; Yee, A.G.; Snow, A.D. Laminin Inhibition of β-Amyloid Protein (Aβ) Fibrillogenesis and Identification of an Aβ Binding Site Localized to the Globular Domain Repeats on the Laminin a Chain. J. Neurosci. Res. 2000, 62, 451–462. [Google Scholar] [CrossRef]
- Smith, I.; Silveirinha, V.; Stein, J.L.; de la Torre-Ubieta, L.; Farrimond, J.A.; Williamson, E.M.; Whalley, B.J. Human Neural Stem Cell-Derived Cultures in Three-Dimensional Substrates Form Spontaneously Functional Neuronal Networks. J. Tissue Eng. Regen. Med. 2017, 11, 1022–1033. [Google Scholar] [CrossRef]
- Poli, D.; Pastore, V.P.; Massobrio, P. Functional Connectivity in in Vitro Neuronal Assemblies. Front. Neural Circuits 2015, 9, 57. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.; Jeong, S.; Lee, J.-H.; Sun, W.; Choi, N.; Cho, I.-J. 3D High-Density Microelectrode Array with Optical Stimulation and Drug Delivery for Investigating Neural Circuit Dynamics. Nat. Commun. 2021, 12, 492. [Google Scholar] [CrossRef]
- Chang, C.-W.; Chiou, J.-C. Development of a Three Dimensional Neural Sensing Device by a Stacking Method. Sensors 2010, 10, 4238–4252. [Google Scholar] [CrossRef]
- De Souza, N. Organoids. Nat. Methods 2018, 15, 23. [Google Scholar] [CrossRef]
- Yin, J.; VanDongen, A.M. Enhanced Neuronal Activity and Asynchronous Calcium Transients Revealed in a 3D Organoid Model of Alzheimer’s Disease. ACS Biomater. Sci. Eng. 2021, 7, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Arber, C.; Toombs, J.; Lovejoy, C.; Ryan, N.S.; Paterson, R.W.; Willumsen, N.; Gkanatsiou, E.; Portelius, E.; Blennow, K.; Heslegrave, A.; et al. Familial Alzheimer’s Disease Patient-Derived Neurons Reveal Distinct Mutation-Specific Effects on Amyloid Beta. Mol. Psychiatry 2020, 25, 2919–2931. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-K.; Sanchez, C.V.; Chen, M.; Morin, P.J.; Wells, J.M.; Hanlon, E.B.; Xia, W. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0163072. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.-L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J.; et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human IPSC-Derived Brain Cell Types. Neuron 2018, 98, 1141–1154.e7. [Google Scholar] [CrossRef] [Green Version]
- Bi, F.-C.; Yang, X.-H.; Cheng, X.-Y.; Deng, W.-B.; Guo, X.-L.; Yang, H.; Wang, Y.; Li, J.; Yao, Y. Optimization of Cerebral Organoids: A More Qualified Model for Alzheimer’s Disease Research. Transl. Neurodegener. 2021, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Generation of Cerebral Organoids from Human Pluripotent Stem Cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Rothenbücher, T.S.P.; Gürbüz, H.; Pereira, M.P.; Heiskanen, A.; Emneus, J.; Martinez-Serrano, A. Next Generation Human Brain Models: Engineered Flat Brain Organoids Featuring Gyrification. Biofabrication 2021, 13, 011001. [Google Scholar] [CrossRef]
- Ao, Z.; Cai, H.; Wu, Z.; Song, S.; Karahan, H.; Kim, B.; Lu, H.-C.; Kim, J.; Mackie, K.; Guo, F. Tubular human brain organoids to model microglia-mediated neuroinflammation. Lab Chip. 2021, 21, 2751–2762. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic Organs-on-Chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Li, Y.; Li, D.; Zhao, P.; Nandakumar, K.; Wang, L.; Song, Y. Microfluidics-Based Systems in Diagnosis of Alzheimer’s Disease and Biomimetic Modeling. Micromachines 2020, 11, 787. [Google Scholar] [CrossRef]
- Choi, Y.J.; Chae, S.; Kim, J.H.; Barald, K.F.; Park, J.Y.; Lee, S.-H. Neurotoxic Amyloid Beta Oligomeric Assemblies Recreated in Microfluidic Platform with Interstitial Level of Slow Flow. Sci. Rep. 2013, 3, 1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, B.; Jeong, G.S.; Hyun, J.K.; Lee, C.J.; Lee, S.-H. Three-Dimensional Brain-on-a-Chip with an Interstitial Level of Flow and Its Application as an in Vitro Model of Alzheimer’s Disease. Lab Chip 2014, 15, 141–150. [Google Scholar] [CrossRef]
- Cho, H.; Hashimoto, T.; Wong, E.; Hori, Y.; Wood, L.B.; Zhao, L.; Haigis, K.M.; Hyman, B.T.; Irimia, D. Microfluidic Chemotaxis Platform for Differentiating the Roles of Soluble and Bound Amyloid-β on Microglial Accumulation. Sci. Rep. 2013, 3, 1823. [Google Scholar] [CrossRef] [Green Version]
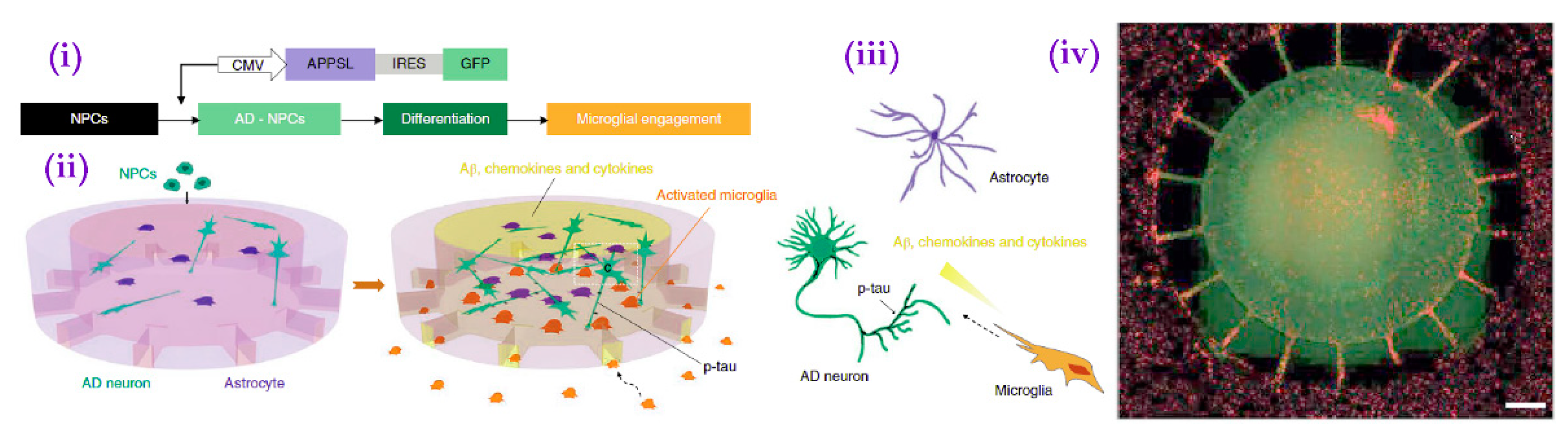
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D Human Triculture System Modeling Neurodegeneration and Neuroinflammation in Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Kilinc, D.; Vreulx, A.-C.; Mendes, T.; Flaig, A.; Marques-Coelho, D.; Verschoore, M.; Demiautte, F.; Amouyel, P.; Neuro-CEB Brain Bank; Eysert, F.; et al. Pyk2 Overexpression in Postsynaptic Neurons Blocks Amyloid Β1–42-Induced Synaptotoxicity in Microfluidic Co-Cultures. Brain Commun. 2020, 2, fcaa139. [Google Scholar] [CrossRef]
- Katsikoudi, A.; Ficulle, E.; Cavallini, A.; Sharman, G.; Guyot, A.; Zagnoni, M.; Eastwood, B.J.; Hutton, M.; Bose, S. Quantitative Propagation of Assembled Human Tau from Alzheimer’s Disease Brain in Microfluidic Neuronal Cultures. J. Biol. Chem. 2020, 295, 13079–13093. [Google Scholar] [CrossRef]
- Shin, Y.; Choi, S.H.; Kim, E.; Bylykbashi, E.; Kim, J.A.; Chung, S.; Kim, D.Y.; Kamm, R.D.; Tanzi, R.E. Blood–Brain Barrier Dysfunction in a 3D In Vitro Model of Alzheimer’s Disease. Adv. Sci. 2019, 6, 1900962. [Google Scholar] [CrossRef] [Green Version]
- Erdener, Ş.E.; Dalkara, T. Small Vessels Are a Big Problem in Neurodegeneration and Neuroprotection. Front. Neurol. 2019, 10, 889. [Google Scholar] [CrossRef]
- Jorfi, M.; D’Avanzo, C.; Tanzi, R.E.; Kim, D.Y.; Irimia, D. Human Neurospheroid Arrays for In Vitro Studies of Alzheimer’s Disease. Sci. Rep. 2018, 8, 2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Ao, Z.; Hu, L.; Moon, Y.; Wu, Z.; Lu, H.-C.; Kim, J.; Guo, F. Acoustofluidic Assembly of 3D Neurospheroids to Model Alzheimer’s Disease. Analyst 2020, 145, 6243–6253. [Google Scholar] [CrossRef]
- Cho, A.N.; Jin, Y.; An, Y.; Kim, J.; Choi, Y.S.; Lee, J.S.; Kim, J.; Choi, W.Y.; Koo, D.J.; Yu, W.; et al. Microfluidic device with brain extracellular matrix promotes structural and functional maturation of human brain organoids. Nat. Commun. 2021, 12, 4730. [Google Scholar] [CrossRef]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Bräuninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human Cerebral Organoids Recapitulate Gene Expression Programs of Fetal Neocortex Development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Technology Used | Cell Type and Treatment | Results | Year |
|---|---|---|---|---|
| Choi et al. | Matrigel as 3D scaffold | Commercial hNPCs overexpressing FAD-mutated APP and PSEN-1 | Creation of fAD-mutated hNPC line with the following characteristics: Increased overall AB levels at 6 weeks, inc. AB 42:40 ratio in some mutated cell lines. Both AB and tau pathology, including insoluble extracellular AB deposits and intracellular tau aggregates. Inhibition of AB production → dec. tau pathology GSK-3 regulated AB-mediated tau phosphorylation | 2014 |
| Raja et al. | Spheroid | fAD patient-derived iPSCs (multiple cell lines) | Creation of AD patient-derived cerebral organoid with the following characteristics: Spontaneous AB accumulation and aggregation, subsequent spontaneous p-tau accumulation Endosome abnormalities. Treatment with B- and y-secretase inhibitors subsequently reduced AB deposition, then p-tau accumulation | 2016 |
| Lee et. al. | Spheroid (see Raja et al.) | sAD patient-derived iPSCs (multiple cell lines) | BACE1 and y-secretase inhibitors reduced AB levels in some, but not all, patient-derived cell lines Reduced efficacy of inhibitors in spheroids compared to 2D culture, likely a result of decreased diffusion | 2016 |
| Lin et al. | Spheroid co-culture | Isogenic AOPE4/APOE3 iPSCs differentiated into neurons, astrocytes & microglia. APOE4 iPSCs were generated by editing APOE3 iPSCs using CRISPR/Cas9 gene editing. | APOE4 organoids displayed heightened AD phenotypes compared to APOE3 organoids at 6 months | 2018 |
| Park et al. | Multi-chambered microfluidic triculture system | Commercial hNPCs overexpressing fAD-mutated APP and differentiated to neurons and astrocytes (see Choi et al.). Repeated with commercial iPSCs. | fAD neurons and astrocytes in the central chamber induced activation and migration of microglia added to the peripheral chambers toward the central chamber and mimicked AD pathologies including AB aggregation, p-tau accumulation, and neuroinflammation. | 2018 |
| Shin et al. | 5-chambered PDMS Microfluidic BBB-on-a-chip | Commercial hNPCs overexpressing fAD-mutated APP and APP/PSEN1 (see Choi et al.) Commercial brain endothelial cells | Increased bEC monolayer permeability, decreased expression of tight junction proteins, and vascular endothelial AB deposition upon co-culture with fAD-expressing cells | 2019 |
| Cairns et al. | Engineered multi-sectional 3D scaffold infected with HSV-1 | Human-induced neural stem cells generated from foreskin fibroblasts through direct reprogramming (bypasses the pluripotent state) | Generation of AB and p-tau positive plaques, reactive astrocytes, and neuroinflammation, as well as loss of network functionality | 2020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Josephine Boder, E.; Banerjee, I.A. Alzheimer’s Disease: Current Perspectives and Advances in Physiological Modeling. Bioengineering 2021, 8, 211. https://doi.org/10.3390/bioengineering8120211
Josephine Boder E, Banerjee IA. Alzheimer’s Disease: Current Perspectives and Advances in Physiological Modeling. Bioengineering. 2021; 8(12):211. https://doi.org/10.3390/bioengineering8120211
Chicago/Turabian StyleJosephine Boder, E., and Ipsita A. Banerjee. 2021. "Alzheimer’s Disease: Current Perspectives and Advances in Physiological Modeling" Bioengineering 8, no. 12: 211. https://doi.org/10.3390/bioengineering8120211
APA StyleJosephine Boder, E., & Banerjee, I. A. (2021). Alzheimer’s Disease: Current Perspectives and Advances in Physiological Modeling. Bioengineering, 8(12), 211. https://doi.org/10.3390/bioengineering8120211