
Hypothermic Preservation of Adipose-Derived Mesenchymal Stromal Cells as a Viable Solution for the Storage and Distribution of Cell Therapy Products


 ,
,  , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
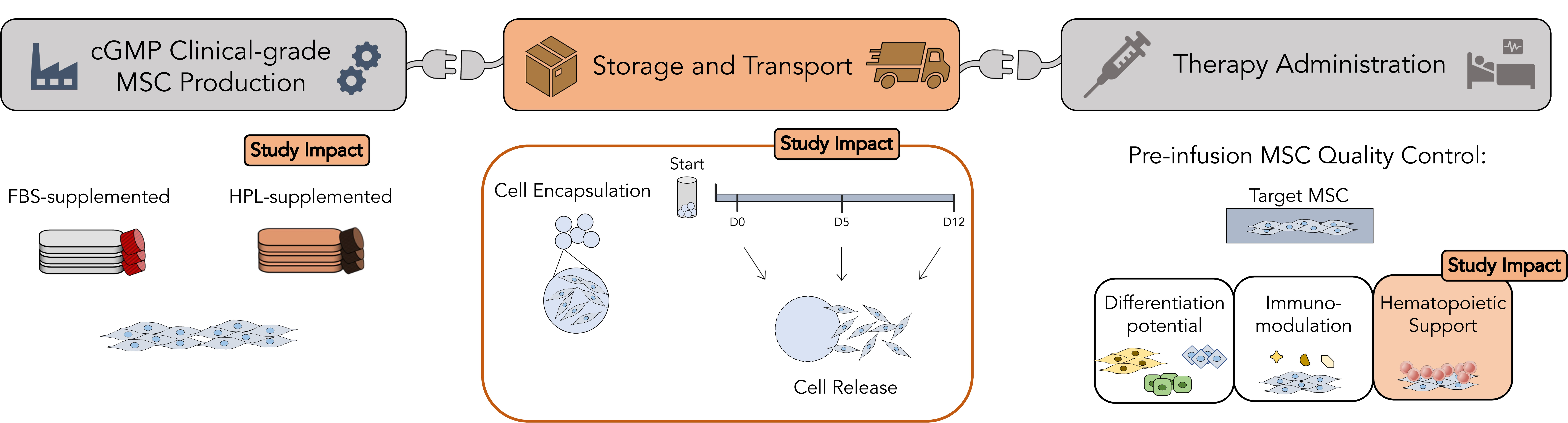
1. Introduction
2. Materials and Methods
2.1. Human Tissues
2.2. MSC(AT) Expansion
2.3. MSC(AT) Encapsulation
2.4. Glucose and Lactate Profiles
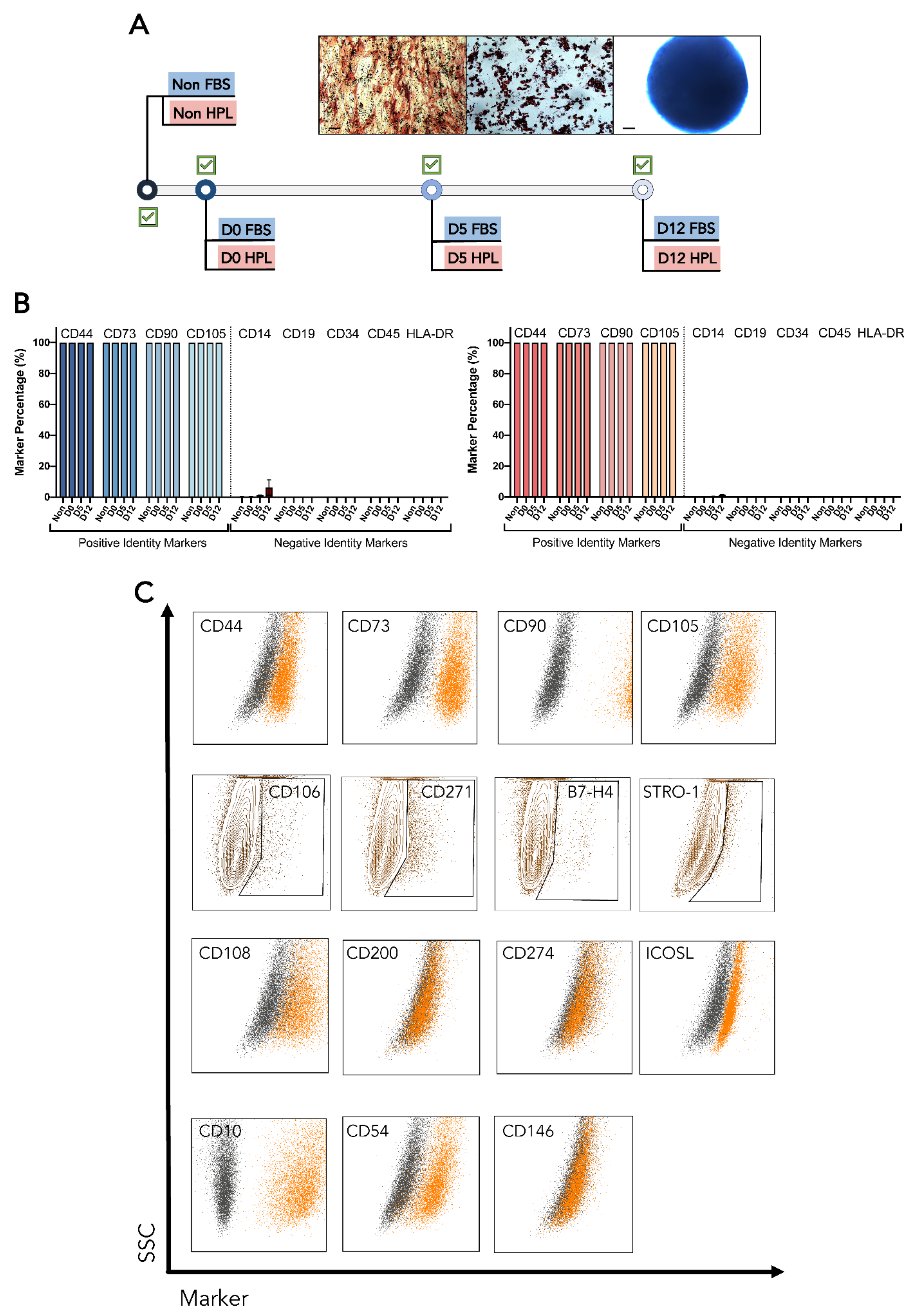
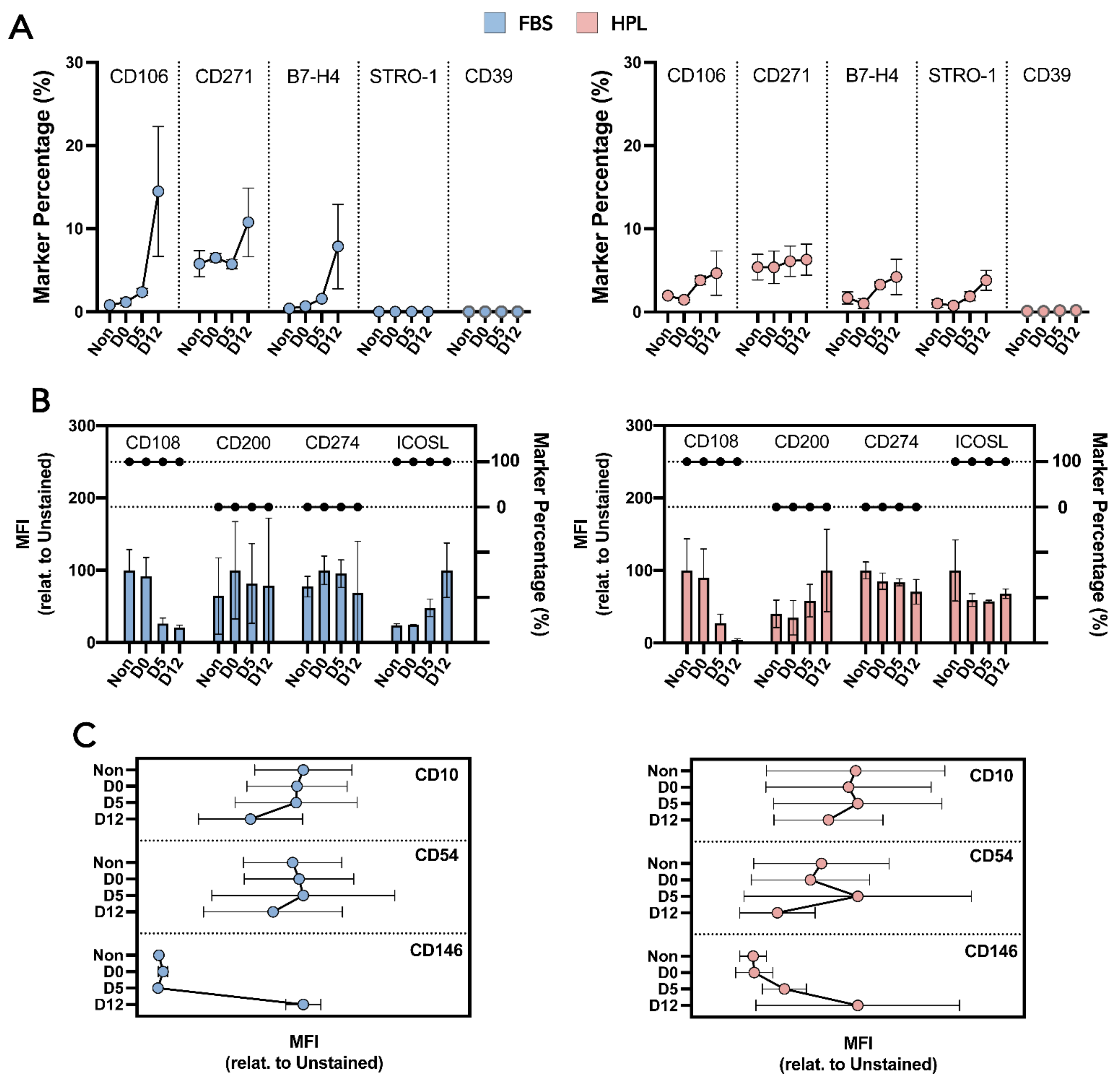
2.5. MSC(AT) Immunophenotype
2.6. MSC(AT) Multilineage Differentiation
2.6.1. Adipogenic Differentiation
2.6.2. Osteogenic Differentiation
2.6.3. Chondrogenic Differentiation
2.7. Hematopoietic Support Assay
2.7.1. UCB Mononuclear Cell (MNC(CB)) Isolation
2.7.2. Generation of a Cryopreserved CD34+ Pool from MNC(CB)
2.7.3. MSC(AT) Feeder Layer Preparation
2.7.4. Ex Vivo Expansion of HSPC
2.7.5. Proliferation Assay
2.7.6. In Vitro Clonogenic Assay
2.7.7. HSPC Immunophenotype
2.8. Statistical Analysis
3. Results
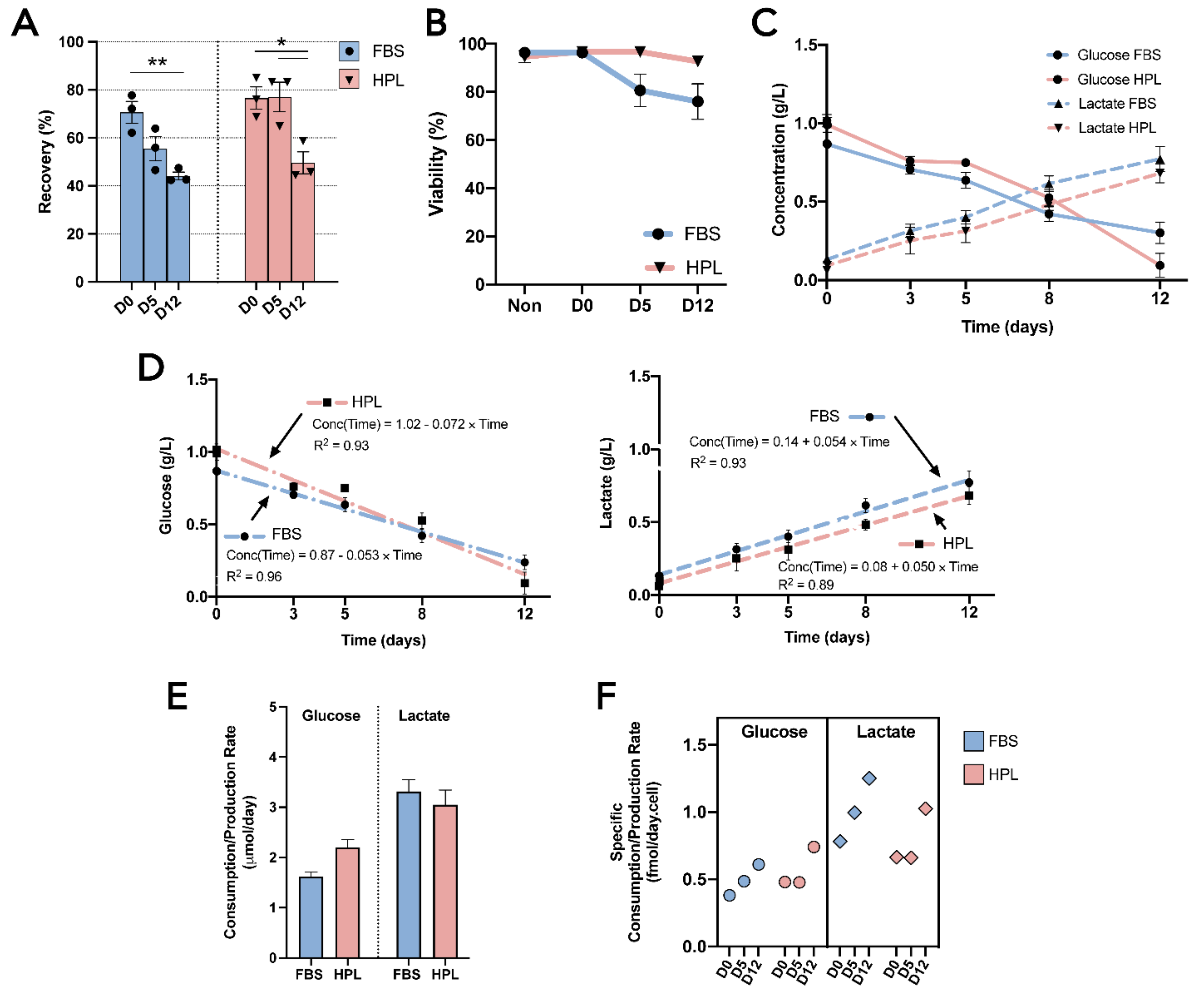
3.1. MSC(AT) Were Successfully Encapsulated and Able to Withstand Hypothermic Temperatures for up to 12 Days
3.2. Encapsulated MSC(AT) Demonstrated an Active Metabolism Regardless of the Expansion Medium
3.3. Upon Encapsulation, Released MSC(AT) Maintained Their Identity, Immunosuppressive Potential and Clonogenic Capabilities as Well as Their Differentiation Potential
3.4. Encapsulation Time Did Not Impact the Hematopoietic Support Capacity of MSC(AT)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aijaz, A.; Li, M.; Smith, D.; Khong, D.; LeBlon, C.; Fenton, O.S.; Olabisi, R.M.; Libutti, S.; Tischfield, J.; Maus, M.V.; et al. Biomanufacturing for clinically advanced cell therapies. Nat. Biomed. Eng. 2018, 2, 362–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.L.; Janes, M.E.; Kumbhojkar, N.; Kapate, N.; Clegg, J.R.; Prakash, S.; Heavey, M.K.; Zhao, Z.; Anselmo, A.C.; Mitragotri, S. Cell therapies in the clinic. Bioeng. Transl. Med. 2021, 6, e10214. [Google Scholar] [CrossRef] [PubMed]
- Hoogduijn, M.J.; Lombardo, E. Mesenchymal Stromal Cells Anno 2019: Dawn of the Therapeutic Era? Concise Review. Stem Cells Transl. Med. 2019, 8, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Kabat, M.; Bobkov, I.; Kumar, S.; Grumet, M. Trends in mesenchymal stem cell clinical trials 2004-2018: Is efficacy optimal in a narrow dose range? Stem Cells Transl. Med. 2020, 9, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Committee for Medicinal Products for Human Use. Alofisel Assessment Report; CHMP: London, UK, 2017. [Google Scholar]
- Galipeau, J.; Sensébé, L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celikkan, F.T.; Mungan, C.; Sucu, M.; Ulus, A.T.; Cinar, O.; Ili, E.G.; Can, A. Optimizing the transport and storage conditions of current Good Manufacturing Practice—Grade human umbilical cord mesenchymal stromal cells for transplantation (HUC-HEART Trial). Cytotherapy 2019, 21, 64–75. [Google Scholar] [CrossRef] [PubMed]
- de Almeida Fuzeta, M.; de Matos Branco, A.D.; Fernandes-Platzgummer, A.; da Silva, C.L.; Cabral, J.M.S. Addressing the Manufacturing Challenges of Cell-Based Therapies. In Advances in Biochemical Engineering/Biotechnology; Springer: Cham, Switzerland, 2019; Volume 171, pp. 225–278. ISBN 978-3-030-40463-5. [Google Scholar]
- Chilima, T.D.P.; Moncaubeig, F.; Farid, S.S. Impact of allogeneic stem cell manufacturing decisions on cost of goods, process robustness and reimbursement. Biochem. Eng. J. 2018, 137, 132–151. [Google Scholar] [CrossRef]
- Lipsitz, Y.Y.; Milligan, W.D.; Fitzpatrick, I.; Stalmeijer, E.; Farid, S.S.; Tan, K.Y.; Smith, D.; Perry, R.; Carmen, J.; Chen, A.; et al. A roadmap for cost-of-goods planning to guide economic production of cell therapy products. Cytotherapy 2017, 19, 1383–1391. [Google Scholar] [CrossRef]
- Ten Ham, R.M.T.; Hövels, A.M.; Hoekman, J.; Frederix, G.W.J.; Leufkens, H.G.M.; Klungel, O.H.; Jedema, I.; Veld, S.A.J.; Nikolic, T.; Van Pel, M.; et al. What does cell therapy manufacturing cost? A framework and methodology to facilitate academic and other small-scale cell therapy manufacturing costings. Cytotherapy 2020, 22, 388–397. [Google Scholar] [CrossRef]
- Swioklo, S.; Connon, C.J. Keeping cells in their place: The future of stem cell encapsulation. Expert Opin. Biol. Ther. 2016, 16, 1181–1183. [Google Scholar] [CrossRef]
- Ścieżyńska, A.; Soszyńska, M.; Szpak, P.; Krześniak, N.; Malejczyk, J.; Kalaszczyńska, I. Influence of Hypothermic Storage Fluids on Mesenchymal Stem Cell Stability: A Comprehensive Review and Personal Experience. Cells 2021, 10, 1043. [Google Scholar] [CrossRef]
- Meneghel, J.; Kilbride, P.; Morris, G.J. Cryopreservation as a Key Element in the Successful Delivery of Cell-Based Therapies—A Review. Front. Med. 2020, 7, 592242. [Google Scholar] [CrossRef]
- Cottle, C.; Porter, A.P.; Lipat, A.; Turner-Lyles, C.; Nguyen, J.; Moll, G.; Chinnadurai, R. Impact of Cryopreservation and Freeze-Thawing on Therapeutic Properties of Mesenchymal Stromal/Stem Cells and Other Common Cellular Therapeutics. Curr. Stem Cell Rep. 2022, 8, 72–92. [Google Scholar] [CrossRef]
- Pakzad, M.; Hassani, S.N.; Abbasi, F.; Hajizadeh-Saffar, E.; Taghiyar, L.; Fallah, N.; Haghparast, N.; Samadian, A.; Ganjibakhsh, M.; Dominici, M.; et al. A Roadmap for the Production of a GMP-Compatible Cell Bank of Allogeneic Bone Marrow-Derived Clonal Mesenchymal Stromal Cells for Cell Therapy Applications. Stem Cell Rev. Rep. 2022, 18, 2279–2295. [Google Scholar] [CrossRef]
- Moll, G.; Geißler, S.; Catar, R.; Ignatowicz, L.; Hoogduijn, M.J.; Strunk, D.; Bieback, K.; Ringdén, O. Cryopreserved or Fresh Mesenchymal Stromal Cells: Only a Matter of Taste or Key to Unleash the Full Clinical Potential of MSC Therapy? In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2016; Volume 951, pp. 77–98. ISBN 9783319454573. [Google Scholar]
- Bissoyi, A.; Kumar, A.; Rizvanov, A.A.; Nesmelov, A.; Gusev, O.; Patra, P.K.; Bit, A. Recent Advances and Future Direction in Lyophilisation and Desiccation of Mesenchymal Stem Cells. Stem Cells Int. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Dhall, S.; Sathyamoorthy, M.; Kuang, J.Q.; Hoffman, T.; Moorman, M.; Lerch, A.; Jacob, V.; Sinclair, S.M.; Danilkovitch, A. Properties of viable lyopreserved amnion are equivalent to viable cryopreserved amnion with the convenience of ambient storage. PLoS ONE 2018, 13, e0204060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltzer, J.; Aletti, M.; Frescaline, N.; Busson, E.; Lataillade, J.-J.; Martinaud, C. Mesenchymal Stromal Cells Based Therapy in Systemic Sclerosis: Rational and Challenges. Front. Immunol. 2018, 9, 2013. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, Y.; Chudickova, M.; Vackova, I.; Groh, T.; Kosnarova, E.; Cejkova, J.; Turnovcova, K.; Petrenko, A.; Sykova, E.; Kubinova, S. Clinically Relevant Solution for the Hypothermic Storage and Transportation of Human Multipotent Mesenchymal Stromal Cells. Stem Cells Int. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Freitas-Ribeiro, S.; Carvalho, A.F.; Costa, M.; Cerqueira, M.T.; Marques, A.P.; Reis, R.L.; Pirraco, R.P. Strategies for the hypothermic preservation of cell sheets of human adipose stem cells. PLoS ONE 2019, 14, e0222597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swioklo, S.; Constantinescu, A.; Connon, C.J. Alginate-Encapsulation for the Improved Hypothermic Preservation of Human Adipose-Derived Stem Cells. Stem Cells Transl. Med. 2016, 5, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaibaji, O.; Swioklo, S.; Shortt, A.; Figueiredo, F.C.; Connon, C.J. Hypothermically stored adipose-derived mesenchymal stromal cell alginate bandages facilitate use of paracrine molecules for corneal wound healing. Int. J. Mol. Sci. 2020, 21, 5849. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, F.; Andrade, P.Z.; Boura, J.S.; Abecasis, M.M.; Da Silva, C.L.; Cabral, J.M.S. Ex vivo expansion of human mesenchymal stem cells: A more effective cell proliferation kinetics and metabolism under hypoxia. J. Cell Physiol. 2010, 223, 27–35. [Google Scholar] [CrossRef]
- Moreira, F.; Mizukami, A.; de Souza, L.E.B.; Cabral, J.M.S.; da Silva, C.L.; Covas, D.T.; Swiech, K. Successful Use of Human AB Serum to Support the Expansion of Adipose Tissue-Derived Mesenchymal Stem/Stromal Cell in a Microcarrier-Based Platform. Front. Bioeng. Biotechnol. 2020, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Branco, A.; Bucar, S.; Moura-Sampaio, J.; Lilaia, C.; Cabral, J.M.S.; Fernandes-Platzgummer, A.; Lobato da Silva, C. Tailored Cytokine Optimization for ex vivo Culture Platforms Targeting the Expansion of Human Hematopoietic Stem/Progenitor Cells. Front. Bioeng. Biotechnol. 2020, 8, 573282. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Conget, P.A.; Minguell, J.J. Phenotypical and functional properties of human bone marrow mesenchymal progenitor cells. J. Cell Physiol. 1999, 181, 67–73. [Google Scholar] [CrossRef]
- Bucar, S.; de Matos Branco, A.D.; Mata, M.F.; Milhano, J.C.; Caramalho, Í.; Cabral, J.M.S.; Fernandes-Platzgummer, A.; da Silva, C.L. Influence of the mesenchymal stromal cell source on the hematopoietic supportive capacity of umbilical cord blood-derived CD34+-enriched cells. Stem Cell Res. Ther. 2021, 12, 399. [Google Scholar] [CrossRef] [PubMed]
- Shahryari, A.; Jazi, M.S.; Mohammadi, S.; Nikoo, H.R.; Nazari, Z.; Hosseini, E.S.; Burtscher, I.; Mowla, S.J.; Lickert, H. Development and clinical translation of approved gene therapy products for genetic disorders. Front. Genet. 2019, 10, 868. [Google Scholar] [CrossRef] [Green Version]
- Quinn, C.; Young, C.; Thomas, J.; Trusheim, M. Estimating the Clinical Pipeline of Cell and Gene Therapies and Their Potential Economic Impact on the US Healthcare System. Value Health 2019, 22, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahsoun, S.; Coopman, K.; Akam, E.C. The impact of cryopreservation on bone marrow-derived mesenchymal stem cells: A systematic review. J. Transl. Med. 2019, 17, 397. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, J. Concerns arising from MSC retrieval from cryostorage and effect on immune suppressive function and pharmaceutical usage in clinical trials. ISBT Sci. Ser. 2013, 8, 100–101. [Google Scholar] [CrossRef]
- Rahul, R.; Comeyne, L.; Agarwal, A. Distribution and Supply Chain Models in the Cell & Gene Therapy Landscape. Deloitte 2021, 1, 1–33. [Google Scholar]
- Bárcia, R.N.; Santos, J.M.; Teixeira, M.; Filipe, M.; Pereira, A.R.S.; Ministro, A.; Água-Doce, A.; Carvalheiro, M.; Gaspar, M.M.; Miranda, J.P.; et al. Umbilical cord tissue–derived mesenchymal stromal cells maintain immunomodulatory and angiogenic potencies after cryopreservation and subsequent thawing. Cytotherapy 2017, 19, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Lechanteur, C.; Briquet, A.; Bettonville, V.; Baudoux, E.; Beguin, Y. MSC manufacturing for academic clinical trials: From a clinical-grade to a full gmp-compliant process. Cells 2021, 10, 1320. [Google Scholar] [CrossRef]
- Lavrentieva, A.; Majore, I.; Kasper, C.; Hass, R. Effects of hypoxic culture conditions on umbilical cord-derived human mesenchymal stem cells. Cell Commun. Signal. 2010, 8, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanga, M.P.; Murasiewicz, H.; Pacek, A.W.; Nienow, A.W.; Coopman, K.; Hewitt, C.J. Expansion of bone marrow-derived human mesenchymal stem/stromal cells (hMSCs) using a two-phase liquid/liquid system. J. Chem. Technol. Biotechnol. 2017, 92, 1577–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmelo, J.G.; Fernandes-Platzgummer, A.; Diogo, M.M.; Silva, C.L.; Cabral, J.M.S. A xeno-free microcarrier-based stirred culture system for the scalable expansion of human mesenchymal stem/stromal cells isolated from bone marrow and adipose tissue. Biotechnol. J. 2015, 10, 1235–1247. [Google Scholar] [CrossRef]
- De Sousa Pinto, D.; Bandeiras, C.; de Almeida Fuzeta, M.; Rodrigues, C.A.V.; Jung, S.; Hashimura, Y.; Tseng, R.J.; Milligan, W.; Lee, B.; Ferreira, F.C.; et al. Scalable Manufacturing of Human Mesenchymal Stromal Cells in the Vertical-Wheel Bioreactor System: An Experimental and Economic Approach. Biotechnol. J. 2019, 14, 1800716. [Google Scholar] [CrossRef]
- Swioklo, S.; Ding, P.; Pacek, A.W.; Connon, C.J. Process parameters for the high-scale production of alginate-encapsulated stem cells for storage and distribution throughout the cell therapy supply chain. Process Biochem. 2017, 59, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cao, Y.; Zhao, G. Hypothermic Storage of Human Umbilical Vein Endothelial Cells and Their Hydrogel Constructs. Biopreserv. Biobank. 2020, 18, 305–310. [Google Scholar] [CrossRef]
- Damala, M.; Swioklo, S.; Koduri, M.A.; Mitragotri, N.S.; Basu, S.; Connon, C.J.; Singh, V. Encapsulation of human limbus-derived stromal/mesenchymal stem cells for biological preservation and transportation in extreme Indian conditions for clinical use. Sci. Rep. 2019, 9, 16950. [Google Scholar] [CrossRef]
- Guiotto, M.; Raffoul, W.; Hart, A.M.; Riehle, M.O.; Di Summa, P.G. Human platelet lysate to substitute fetal bovine serum in hMSC expansion for translational applications: A systematic review. J. Transl. Med. 2020, 18, 351. [Google Scholar] [CrossRef] [PubMed]
- Oikonomopoulos, A.; Van Deen, W.K.; Manansala, A.R.; Lacey, P.N.; Tomakili, T.A.; Ziman, A.; Hommes, D.W. Optimization of human mesenchymal stem cell manufacturing: The effects of animal/xeno-free media. Sci. Rep. 2015, 5, 16570. [Google Scholar] [CrossRef] [Green Version]
- Abdelrazik, H.; Spaggiari, G.M.; Chiossone, L.; Moretta, L. Mesenchymal stem cells expanded in human platelet lysate display a decreased inhibitory capacity on T- and NK-cell proliferation and function. Eur. J. Immunol. 2011, 41, 3281–3290. [Google Scholar] [CrossRef] [PubMed]
- Lipsitz, Y.Y.; Timmins, N.E.; Zandstra, P.W. Quality cell therapy manufacturing by design. Nat. Biotechnol. 2016, 34, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Levy, O.; Kuai, R.; Siren, E.M.J.; Bhere, D.; Milton, Y.; Nissar, N.; De Biasio, M.; Heinelt, M.; Reeve, B.; Abdi, R.; et al. Shattering barriers toward clinically meaningful MSC therapies. Sci. Adv. 2020, 6, eaba6884. [Google Scholar] [CrossRef]
- Galipeau, J.; Krampera, M.; Barrett, J.; Dazzi, F.; Deans, R.J.; DeBruijn, J.; Dominici, M.; Fibbe, W.E.; Gee, A.P.; Gimble, J.M.; et al. International Society for Cellular Therapy perspective on immune functional assays for mesenchymal stromal cells as potency release criterion for advanced phase clinical trials. Cytotherapy 2016, 18, 151–159. [Google Scholar] [CrossRef] [Green Version]
- Chinnadurai, R.; Rajan, D.; Qayed, M.; Arafat, D.; Garcia, M.; Liu, Y.; Kugathasan, S.; Anderson, L.J.; Gibson, G.; Galipeau, J. Potency Analysis of Mesenchymal Stromal Cells Using a Combinatorial Assay Matrix Approach. Cell Rep. 2018, 22, 2504–2517. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, A.C.; Igarashi, K.J.; Nakauchi, H. Haematopoietic stem cell self-renewal in vivo and ex vivo. Nat. Rev. Genet. 2020, 21, 541–554. [Google Scholar] [CrossRef]
- De Lima, M.; McNiece, I.; Robinson, S.N.; Munsell, M.; Eapen, M.; Horowitz, M.; Alousi, A.; Saliba, R.; McMannis, J.D.; Kaur, I.; et al. Cord-Blood Engraftment with Ex Vivo Mesenchymal-Cell Coculture. N. Engl. J. Med. 2012, 367, 2305–2315. [Google Scholar] [CrossRef] [Green Version]
- Crippa, S.; Santi, L.; Bosotti, R.; Porro, G.; Bernardo, M.E. Bone marrow-derived mesenchymal stromal cells: A novel target to optimize hematopoietic stem cell transplantation protocols in hematological malignancies and rare genetic disorders. J. Clin. Med. 2020, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- McNiece, I.K.; Harrington, J.; Turney, J.; Kellner, J.; Shpall, E.J. Ex vivo expansion of cord blood mononuclear cells on mesenchymal stem cells. Cytotherapy 2004, 6, 311–317. [Google Scholar] [CrossRef] [PubMed]
- da Silva, C.L.; Gonçalves, R.; Crapnell, K.B.; Cabral, J.M.S.; Zanjani, E.D.; Almeida-Porada, G. A human stromal-based serum-free culture system supports the ex vivo expansion/maintenance of bone marrow and cord blood hematopoietic stem/progenitor cells. Exp. Hematol. 2005, 33, 828–835. [Google Scholar] [CrossRef]
- Gonçalves, R.; da Silva, C.L.; Cabral, J.M.S.; Zanjani, E.D.; Almeida-Porada, G. A Stro-1+ human universal stromal feeder layer to expand/maintain human bone marrow hematopoietic stem/progenitor cells in a serum-free culture system. Exp. Hematol. 2006, 34, 1353–1359. [Google Scholar] [CrossRef]
- da Silva, C.L.; Gonçalves, R.; Porada, C.D.; Ascensão, J.L.; Zanjani, E.D.; Cabral, J.M.S.; Almeida-Porada, G. Differences amid bone marrow and cord blood hematopoietic stem/progenitor cell division kinetics. J. Cell Physiol. 2009, 220, 102–111. [Google Scholar] [CrossRef] [Green Version]
- da Silva, C.L.; Gonçalves, R.; dos Santos, F.; Andrade, P.Z.; Almeida-Porada, G.; Cabral, J.M.S. Dynamic cell-cell interactions between cord blood haematopoietic progenitors and the cellular niche are essential for the expansion of CD34+, CD34+CD38− and early lymphoid CD7+ cells. J. Tissue Eng. Regen. Med. 2010, 4, 149–158. [Google Scholar] [CrossRef]
- Andrade, P.Z.; dos Santos, F.; Almeida-Porada, G.; da Silva, C.L.; Cabral, J.M.S. Systematic delineation of optimal cytokine concentrations to expand hematopoietic stem/progenitor cells in co-culture with mesenchymal stem cells. Mol. Biosyst. 2010, 6, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Andrade, P.Z.; da Silva, C.L.; dos Santos, F.; Almeida-Porada, G.; Cabral, J.M.S. Initial CD34 + cell-enrichment of cord blood determines hematopoietic stem/progenitor cell yield upon ex vivo expansion. J. Cell Biochem. 2011, 112, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Andrade, P.Z.; de Soure, A.M.; dos Santos, F.; Paiva, A.; Cabral, J.M.S.; da Silva, C.L. Ex vivo expansion of cord blood haematopoietic stem/progenitor cells under physiological oxygen tensions: Clear-cut effects on cell proliferation, differentiation and metabolism. J. Tissue Eng. Regen. Med. 2015, 9, 1172–1181. [Google Scholar] [CrossRef] [PubMed]
- Nebel, S.; Lux, M.; Kuth, S.; Bider, F.; Dietrich, W.; Egger, D.; Boccaccini, A.R.; Kasper, C. Alginate Core–Shell Capsules for 3D Cultivation of Adipose-Derived Mesenchymal Stem Cells. Bioengineering 2022, 9, 66. [Google Scholar] [CrossRef]
- Costa, M.H.G.; McDevitt, T.C.; Cabral, J.M.S.; da Silva, C.L.; Ferreira, F.C. Tridimensional configurations of human mesenchymal stem/stromal cells to enhance cell paracrine potential towards wound healing processes. J. Biotechnol. 2017, 262, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Fernandez, T.; Tierney, E.G.; Cunniffe, G.M.; O’Brien, F.J.; Kelly, D.J. Gene Delivery of TGF-β3 and BMP2 in an MSC-Laden Alginate Hydrogel for Articular Cartilage and Endochondral Bone Tissue Engineering. Tissue Eng. Part A 2016, 22, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.I.; Moroni, L.; Barrias, C.C. Modulating Alginate Hydrogels for Improved Biological Performance as Cellular 3D Microenvironments. Front. Bioeng. Biotechnol. 2020, 8, 665. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.; Shea, L.D. Advances in islet encapsulation technologies. Nat. Rev. Drug Discov. 2017, 16, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Mao, A.S.; Özkale, B.; Shah, N.J.; Vining, K.H.; Descombes, T.; Zhang, L.; Tringides, C.M.; Wong, S.W.; Shin, J.W.; Scadden, D.T.; et al. Programmable microencapsulation for enhanced mesenchymal stem cell persistence and immunomodulation. Proc. Natl. Acad. Sci. USA 2019, 116, 15392–15397. [Google Scholar] [CrossRef] [Green Version]
- Levit, R.D.; Landázuri, N.; Phelps, E.A.; Brown, M.E.; García, A.J.; Davis, M.E.; Joseph, G.; Long, R.; Safley, S.A.; Suever, J.D.; et al. Cellular Encapsulation Enhances Cardiac Repair. J. Am. Heart Assoc. 2013, 2, e000367. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Branco, A.; Tiago, A.L.; Laranjeira, P.; Carreira, M.C.; Milhano, J.C.; dos Santos, F.; Cabral, J.M.S.; Paiva, A.; da Silva, C.L.; Fernandes-Platzgummer, A. Hypothermic Preservation of Adipose-Derived Mesenchymal Stromal Cells as a Viable Solution for the Storage and Distribution of Cell Therapy Products. Bioengineering 2022, 9, 805. https://doi.org/10.3390/bioengineering9120805
Branco A, Tiago AL, Laranjeira P, Carreira MC, Milhano JC, dos Santos F, Cabral JMS, Paiva A, da Silva CL, Fernandes-Platzgummer A. Hypothermic Preservation of Adipose-Derived Mesenchymal Stromal Cells as a Viable Solution for the Storage and Distribution of Cell Therapy Products. Bioengineering. 2022; 9(12):805. https://doi.org/10.3390/bioengineering9120805
Chicago/Turabian StyleBranco, André, Ana L. Tiago, Paula Laranjeira, Maria C. Carreira, João C. Milhano, Francisco dos Santos, Joaquim M. S. Cabral, Artur Paiva, Cláudia L. da Silva, and Ana Fernandes-Platzgummer. 2022. "Hypothermic Preservation of Adipose-Derived Mesenchymal Stromal Cells as a Viable Solution for the Storage and Distribution of Cell Therapy Products" Bioengineering 9, no. 12: 805. https://doi.org/10.3390/bioengineering9120805
APA StyleBranco, A., Tiago, A. L., Laranjeira, P., Carreira, M. C., Milhano, J. C., dos Santos, F., Cabral, J. M. S., Paiva, A., da Silva, C. L., & Fernandes-Platzgummer, A. (2022). Hypothermic Preservation of Adipose-Derived Mesenchymal Stromal Cells as a Viable Solution for the Storage and Distribution of Cell Therapy Products. Bioengineering, 9(12), 805. https://doi.org/10.3390/bioengineering9120805