
Indirect Nuclear Magnetic Resonance (NMR) Spectroscopic Determination of Acrylamide in Coffee Using Partial Least Squares (PLS) Regression

 , , and
, , and 
Abstract
:1. Introduction
2. Results
2.1. Calibration of the PLS Model
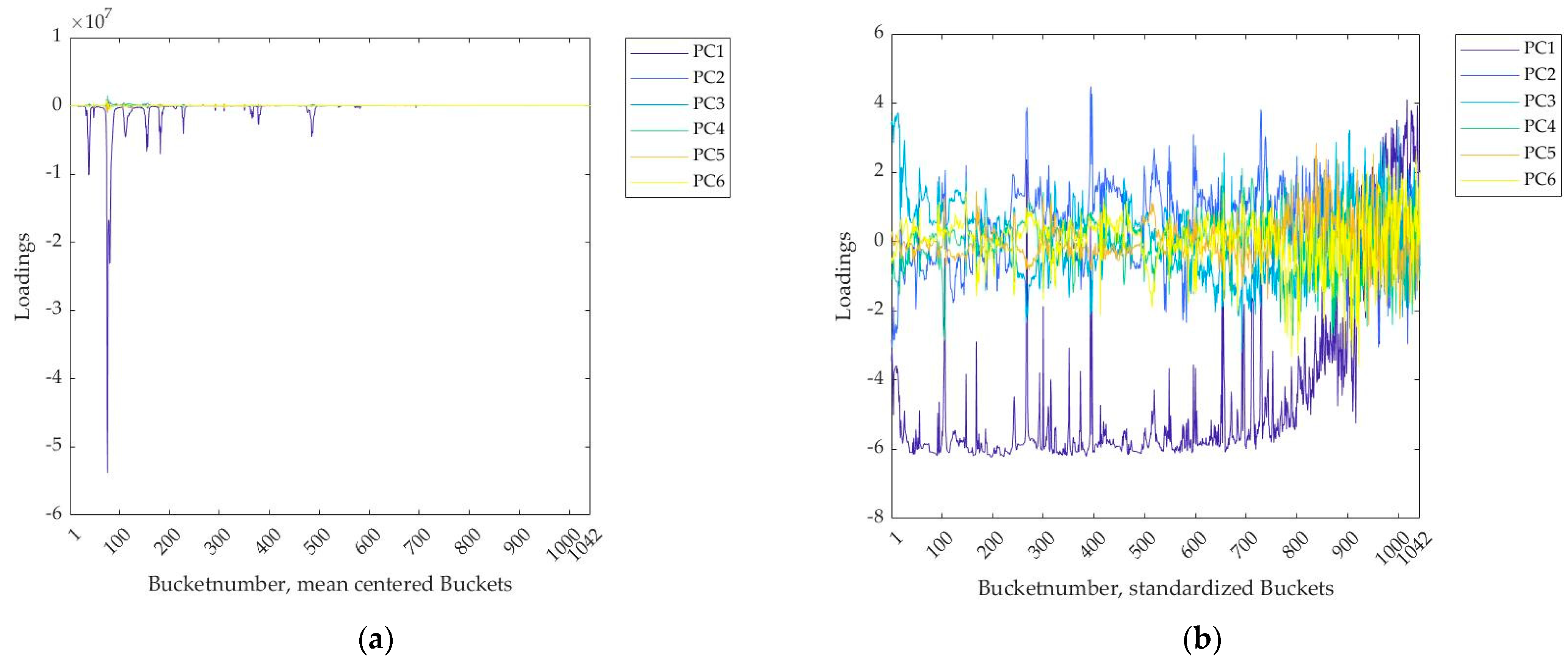
2.1.1. Data Preprocessing
2.1.2. Variable Selection
2.1.3. Spiking
2.2. Parameters of the PLS Model
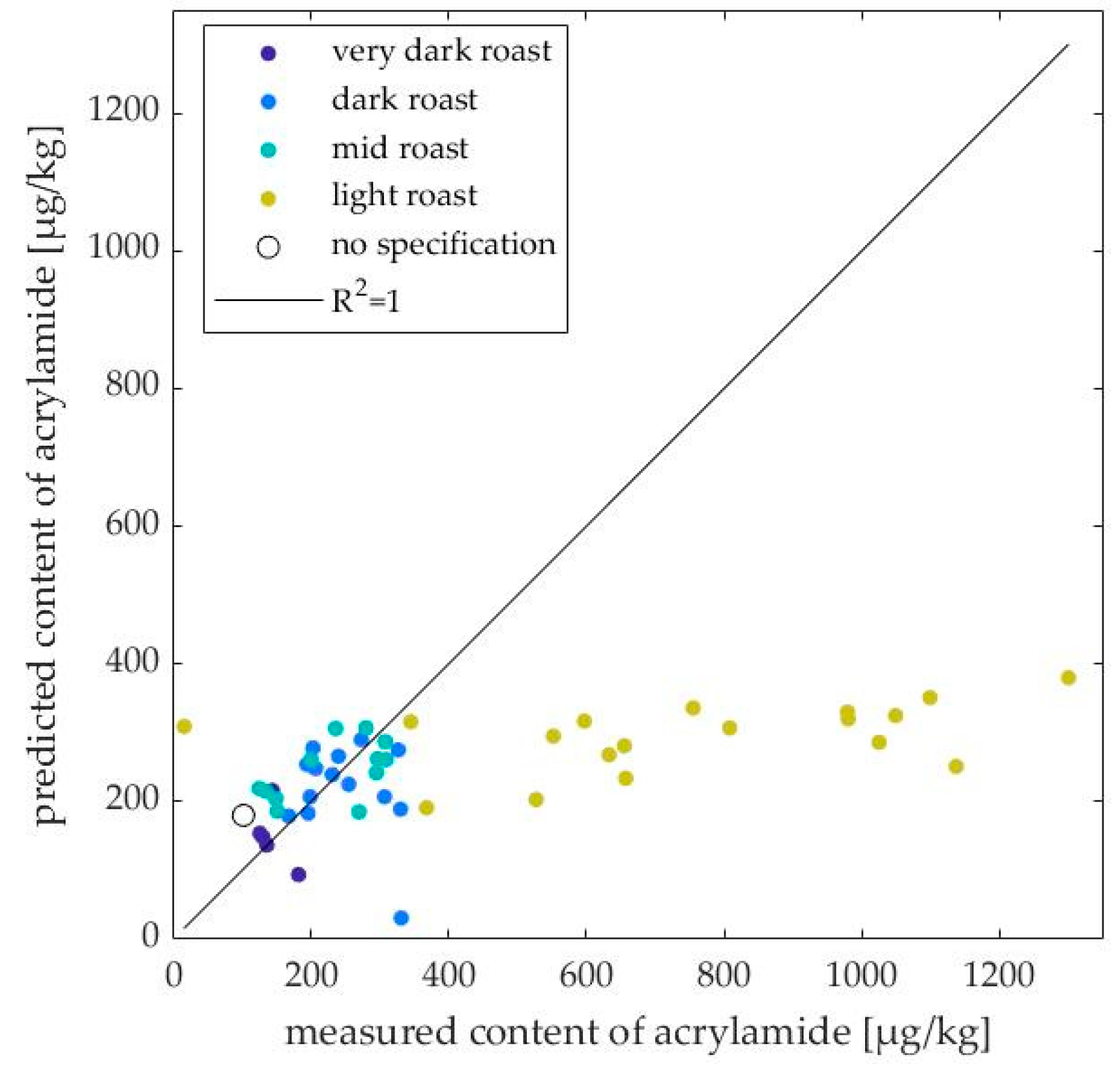
2.3. Application of the PLS Model to Unknown Data
3. Discussion
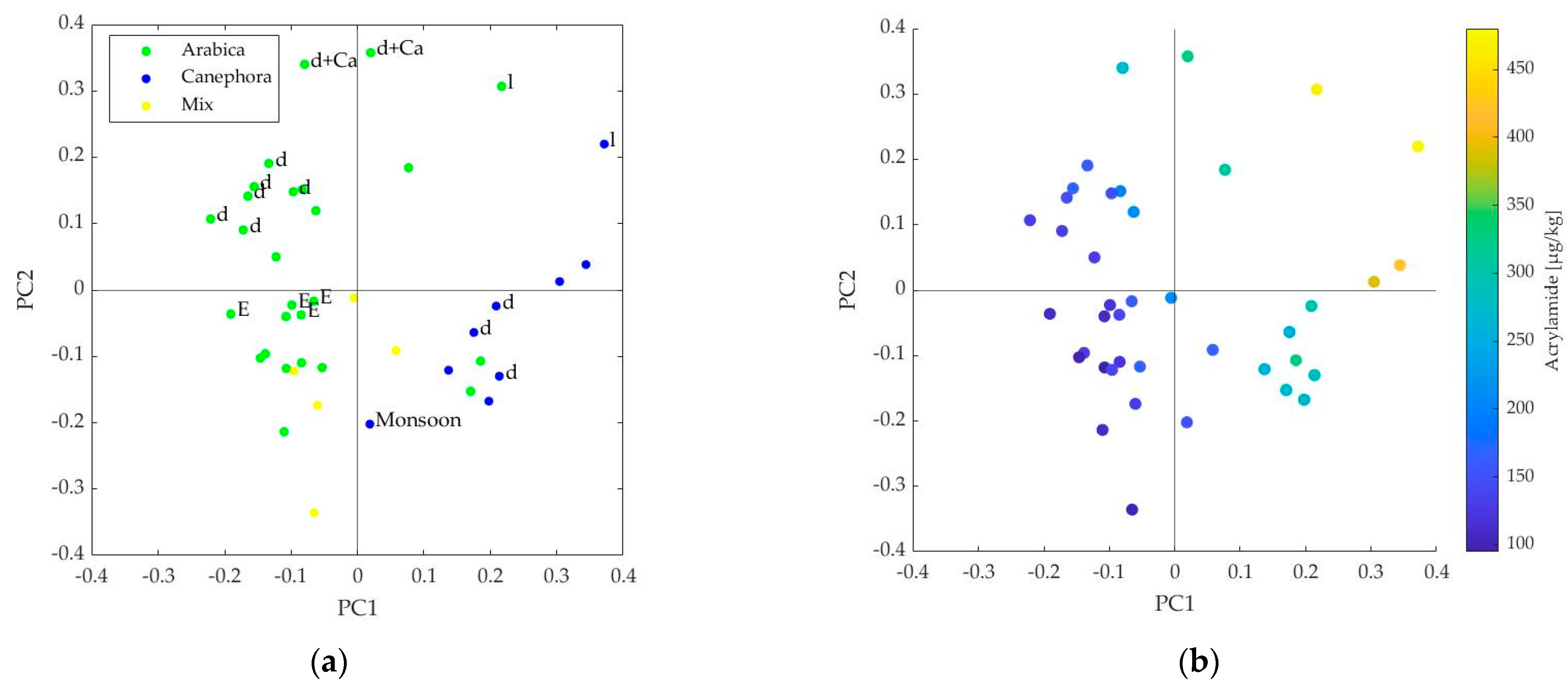
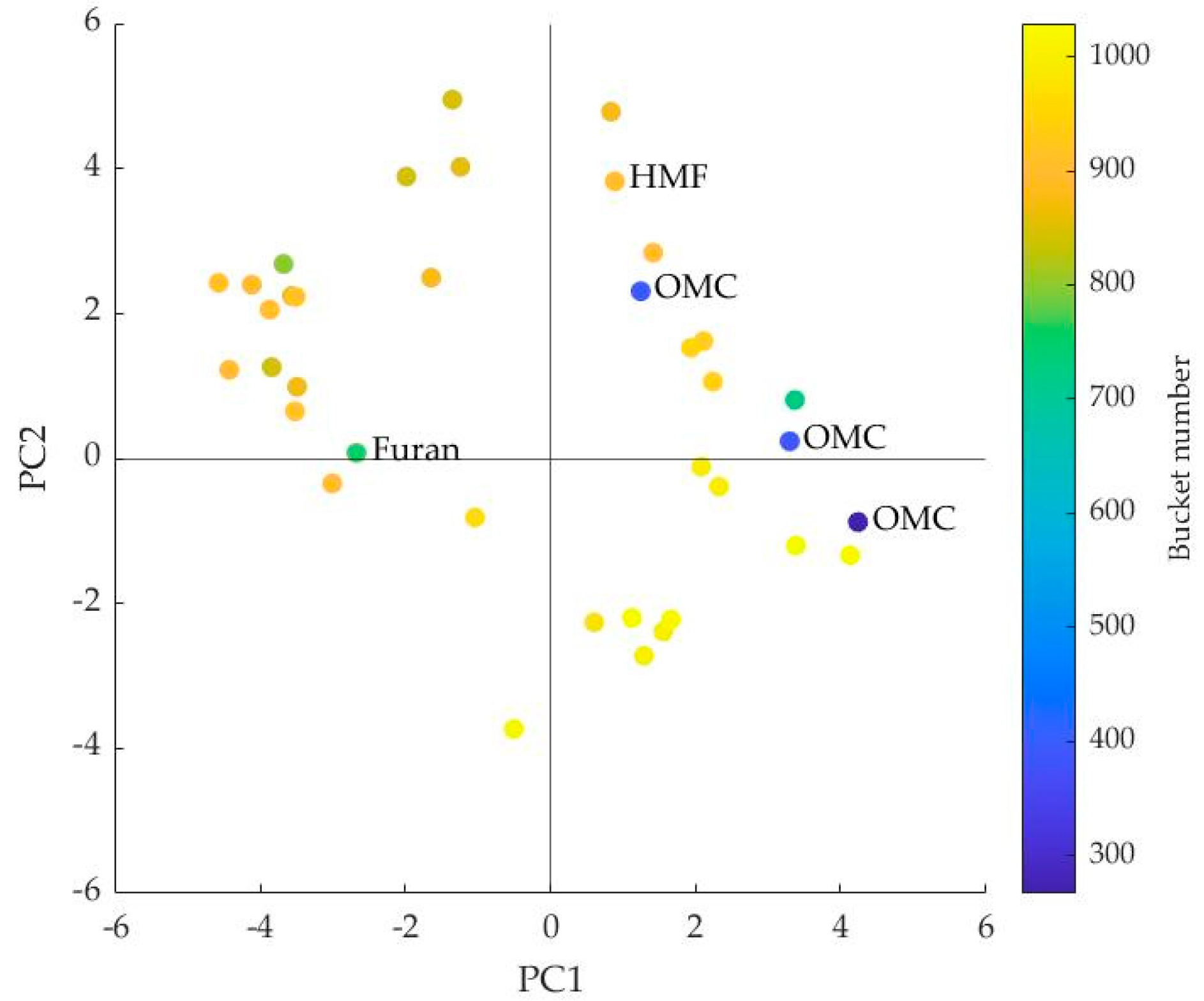
3.1. Interpretation of the PLS Components
3.2. Applicability of the PLS Model
3.3. Similar Applications in Literature
4. Materials and Methods
4.1. Samples
4.2. Materials
- NMR instrument: Ultrashield 400;
- Console: Avance III-400;
- Sample head: 5 mm PASEI 1H/D 13C;
- Sampler: SampleXpress;
- Sample head cooling: BCU05;
- TopSpin, Version 4.0.9.
4.3. Analytical Methodology
4.3.1. LC–MS/MS, Method 1, Calibration Set
4.3.2. LC–MS/MS, Method 2, Test Set
4.3.3. NMR Spectroscopy
4.3.4. Spiking Experiments
4.4. Multivariate Data Analysis
4.5. Validation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bucket Number | ppm | Signal | Compound |
|---|---|---|---|
| 267 | 3.16 | s | OMC |
| 393 | 4.42 | d, J = 12.55 Hz | OMC |
| 394 | 4.43 | ||
| 730 | 7.88 | m | |
| 752 | 8.1 | s | Furan |
| 797 | 8.55 | ||
| 842 | 9 | ||
| 844 | 9.02 | ||
| 847 | 9.05 | ||
| 859 | 9.17 | ||
| 860 | 9.18 | ||
| 873 | 9.31 | ||
| 877 | 9.35 | m | |
| 879 | 9.37 | t | |
| 894 | 9.52 | s | |
| 898 | 9.56 | ||
| 900 | 9.58 | ||
| 904 | 9.62 | s | HMF |
| 906 | 9.64 | ||
| 909 | 9.67 | m | |
| 911 | 9.69 | m | |
| 913 | 9.71 | ||
| 915 | 9.73 | ||
| 936 | 9.94 | m | |
| 943 | 10.01 | ||
| 952 | 10.1 | ||
| 966 | 10.24 | m | |
| 973 | 10.31 | ||
| 998 | 10.56 | ||
| 999 | 10.57 | ||
| 1002 | 10.6 | ||
| 1005 | 10.63 | ||
| 1018 | 10.76 | ||
| 1022 | 10.8 | ||
| 1025 | 10.83 | ||
| 1026 | 10.84 | ||
| 1029 | 10.87 |
| Sample Id. | Roaster Type | Roasting Degree | Coffee Type | Acrylamide (µg/kg) | |||
|---|---|---|---|---|---|---|---|
| Method 1 | Method 2a | Method 2b | PLS Result | ||||
| 467 | IR | light | A, Catuaí | 350 | 340 | 316 | |
| 468 | IR | mid | A, Catuaí | 238 | 235 | 306 | |
| 469 | IR | dark | A, Catuaí | 207 | 206 | 248 | |
| 470 | IR | mid | A, Catuaí | 153 | 146 | 204 | |
| 471 | IR | mid/dark | A, Catuaí | 125 | 125 | 219 | |
| 472 | IR | mid/dark | A, Catuaí | 134 | 136 | 215 | |
| 473 | IR | mid | A, Catuaí | 150 | 152 | 186 | |
| 474 | IR | light | A, Catuaí | 968 | 991 | 330 | |
| 475 | IR | light | A, Catuaí | 660 | 655 | 234 | |
| 476 | IR | mid | A, Catuaí | 297 | 296 | 262 | |
| 477 | IR | dark | A, Catuaí | 198 | 195 | 183 | |
| 478 | IR | light | A, Catuaí | 536 | 518 | 203 | |
| 479 | IR | mid | A, Catuaí | 248 | 331 | 287 | 261 |
| 480 | IR | light | A, Catuaí | 1020 | 1020 | 1079 | 325 |
| 481 | IR | light | A, Catuaí | 1430 | 1200 | 1399 | 380 |
| 482 | IR | dark | A, Catuaí | 224 | 347 | 306 | 275 |
| 483 | IR | dark | A, Catuaí | 206 | 280 | 230 | 225 |
| 484 | Drum | light | A, Catuaí | 344 | 393 | 191 | |
| 485 | Drum | mid | A, Catuaí | 274 | 266 | 185 | |
| 486 | Drum | dark | A, Catuaí | 301 | 361 | 31 | |
| 487 | Drum | mid | A, Catuaí | 227 | 294 | 296 | 242 |
| 488 | Drum | light | A, Catuaí | 471 | 593 | 601 | 317 |
| 489 | Drum | light | A, Catuaí | 588 | 757 | 859 | 307 |
| 490 | Drum | dark | A, Catuaí | 250 | 319 | 295 | 207 |
| 491 | Drum | dark | A, Catuaí | 244 | 326 | 333 | 189 |
| 492 | Drum | A, Catuaí | 102 | 103 | 180 | ||
| 493 | IR | dark | R | 224 | 257 | 266 | |
| 494 | IR | mid | R | 325 | 290 | 287 | |
| 495 | IR | light | R | 960 | 1089 | 286 | |
| 496 | IR | dark | A, Malabar | 245 | 218 | 239 | |
| 497 | IR | mid | A, Malabar | 254 | 305 | 307 | |
| 498 | IR | light | A, Malabar | 583 | 683 | 268 | |
| 499 | IR | very dark | R | 151 | 121 | 137 | |
| 500 | IR | very dark | R | 130 | 131 | 149 | |
| 501 | IR | very dark | A, Malabar | 177 | 187 | 94 | |
| 502 | IR | very dark | A, Malabar | 121 | 131 | 154 | |
| 503 | IR | dark | Mix 20% R | 167 | 169 | 179 | |
| 504 | IR | dark | Mix 40% R | 197 | 192 | 254 | |
| 505 | IR | dark | Mix 60% R | 211 | 195 | 278 | |
| 506 | IR | dark | Mix 80% R | 264 | 282 | 290 | |
| 507 | IR | dark | R | 199 | 199 | 207 | |
| 508 | IR | light | Mix 20% R | 527 | 576 | 295 | |
| 509 | IR | light | Mix 40% R | 603 | 706 | 281 | |
| 510 | IR | light | Mix 60% R | 1051 | 910 | 320 | |
| 511 | IR | light | Mix 80% R | 1111 | 1164 | 251 | |
| 512 | IR | light | R | 792 | 719 | 336 | |
| 513 | IR | 6.5min | A, Catuaí | 15 | 17 | 309 | |
| 514 | IR | 13 min | A, Catuaí | 1099 | 1099 | 351 | |
| 515 | IR | 19.5 min | A, Catuaí | 201 | 200 | 261 | |
| 516 | IR | 26 min | A, Catuaí | 146 | 142 | 216 | |
References
- International Agency for Research on Cancer (IARC). Agents Classified by the IARC Monographs, Volumes 1–129. 2020. Available online: https://monographs.iarc.fr/list-of-classifications (accessed on 10 February 2021).
- Bergmark, E. Hemoglobin Adducts of Acrylamide and Acrylonitrile in Laboratory Workers, Smokers and Nonsmokers. Chem. Res. Toxicol. 1997, 10, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Tareke, E.; Rydberg, P.; Karlsson, P.; Eriksson, S.; Törnqvist, M. Acrylamide: A Cooking Carcinogen? Chem. Res. Toxicol. 2000, 13, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Tareke, E.; Rydberg, P.; Karlsson, P.; Eriksson, S.; Törnqvist, M. Analysis of Acrylamide, a Carcinogen Formed in Heated Foodstuffs. J. Agric. Food Chem. 2002, 50, 4998–5006. [Google Scholar] [CrossRef] [PubMed]
- Stadler, R.H.; Robert, F.; Riediker, S.; Varga, N.; Davidek, T.; Devaud, S.; Goldmann, T.; Hau, J.; Blank, I. In-Depth Mechanistic Study on the Formation of Acrylamide and Other Vinylogous Compounds by the Maillard Reaction. J. Agric. Food Chem. 2004, 52, 5550–5558. [Google Scholar] [CrossRef] [PubMed]
- Weisshaar, R.; Gutsche, B. Formation of acrylamide in heated potato products—Model experiments pointing to asparagine as precursor. Deut. Lebensm. Rundsch. 2002, 98, 397–400. [Google Scholar]
- European Commission. Commission Regulation (EU) 2017/2158 of 20 November 2017 Establishing Mitigation Measures and Benchmark Levels for the Reduction of the Presence of Acrylamide in Food. Off. J. Europ. Union 2017, L324, 24–44. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R2158&from=DE (accessed on 10 February 2021).
- Lachenmeier, D.W.; Schwarz, S.; Teipel, J.; Hegmanns, M.; Kuballa, T.; Walch, S.G.; Breitling-Utzmann, C.M. Potential Antagonistic Effects of Acrylamide Mitigation during Coffee Roasting on Furfuryl Alcohol, Furan and 5-Hydroxymethylfurfural. Toxics 2019, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- CEN/TC 275. EN 16618:2015. Food Analysis—Determination of Acrylamide in Food by Liquid Chromatography Tandem Mass Spectrometry (LC-ESI-MS/MS); European Committee for Standardization (CEN): Brussels, Belgium, 2015. [Google Scholar]
- Kessler, W. Multivariate Datenanalyse für die Pharma-, Bio- und Prozessanalytik: Ein Lehrbuch, 1st ed.; WILEY-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Monakhova, Y.B.; Ruge, W.; Kuballa, T.; Ilse, M.; Winkelmann, O.; Diehl, B.; Thomas, F.; Lachenmeier, D.W. Rapid approach to identify the presence of Arabica and Robusta species in coffee using 1H NMR spectroscopy. Food Chem. 2015, 182, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Defernez, M.; Wren, E.; Watson, A.D.; Gunning, Y.; Colquhoun, I.J.; Le Gall, G.; Williamson, D.; Kemsley, E.K. Low-field 1H NMR spectroscopy for distinguishing between arabica and robusta ground roast coffees. Food Chem. 2017, 216, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lachenmeier, D.W.; Teipel, J.; Scharinger, A.; Kuballa, A.; Walch, S.G.; Grosch, F.; Bunzel, M.; Okaru, A.O.; Schwarz, S. Fully Automated Identification of Coffee Species and Simultaneous Quantification of Furfuryl Alcohol Using NMR Spectroscopy. J. AOAC Int. 2020, 103, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Okaru, A.O.; Scharinger, A.; Rajcic de Rezende, T.; Teipel, J.; Kuballa, T.; Walch, S.G.; Lachenmeier, D.W. Validation of a Quantitative Proton Nuclear Magnetic Resonance Spectroscopic Screening Method for Coffee Quality and Authenticity (NMR Coffee Screener). Foods 2020, 9, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, M.; Toffanin, R.; Palo, D.D.; Zatti, L.; Segre, A. High-resolution 1H NMR investigation of coffee. J. Sci. Food Agric. 1999, 79, 869–878. [Google Scholar] [CrossRef]
- Shmueli, G.; Ray, S.; Velasquez Estrada, J.M.; Chatla, S.B. The elephant in the room: Predictive performance of PLS models. J. Bus. Res. 2016, 69, 4552–4564. [Google Scholar] [CrossRef]
- Moon, J.K.; Shibamoto, T. Role of Roasting Conditions in the Profile of Volatile Flavor Chemicals Formed from Coffee Beans. J. Agric. Food Chem. 2009, 57, 5823–5831. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Furihata, K.; Koda, M.; Hu, F.; Miyakawa, T.; Tanokura, M. Roasting Process of Coffee Beans as Studied by Nuclear Magnetic Resonance: Time Course of Changes in Composition. J. Agric. Food Chem. 2012, 60, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Tchibo GmbH, Kaffee-Akademie. Available online: https://www.tchibo.de/kaffee-akademie-c400103027.html (accessed on 10 February 2021).
- Consonni, R.; Cagliani, L.R.; Cogliati, C. NMR based geographical characterization of roasted coffee. Talanta 2012, 88, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Febvay, L.; Hamon, E.; Recht, R.; Andres, N.; Vincent, M.; Aoudé-Werner, D.; This, H. Identification of markers of thermal processing (“roasting”) in aqueous extracts of Coffea arabica L. seeds through NMR fingerprinting and chemometrics. Magn. Reson. Chem. 2019, 57, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Ruenz, M.; Bakuradze, T.; Eisenbrand, G.; Richling, E. Monitoring urinary mercapturic acids as biomarkers of human dietary exposure to acrylamide in combination with acrylamide uptake assessment based on duplicate diets. Arch. Toxicol. 2016, 90, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, T.; Sæbø, S.; Liland, K.H. Comparison of variable selection methods in partial least squares regression. J. Chemom. 2020, 34, 100. [Google Scholar] [CrossRef] [Green Version]
- Rajalahti Kvalheim, T.A. Mass Spectral Profiling and Multivariate Analysis for Detection of Biomarker Signatures: Application to Multiple Sclerosis; University Bergen: Bergen, Norway, 2010. [Google Scholar]
- Chong, I.G.; Jun, C.H. Performance of some variable selection methods when multicollinearity is present. Chemom. Int. Lab. Syst. 2005, 78, 103–112. [Google Scholar] [CrossRef]
- Eigenvector, Genetic Algorithms for Variable Selection. Available online: http://wiki.eigenvector.com/index.php?title=GeneticAlgorithmsforVariableSelection (accessed on 10 February 2021).
- Eigenvector, rPLS. Available online: http://wiki.eigenvector.com/index.php?title=Rpls_2018 (accessed on 10 February 2021).
- Eigenvector, Interval PLS (IPLS) for Variable Selection. Available online: http://wiki.eigenvector.com/index.php?title=Interval_PLS_(IPLS)_for_Variable_Selection (accessed on 10 February 2021).
- Rinnan, Å.; Andersson, M.; Ridder, C.; Engelsen, S.B. Recursive weighted partial least squares (rPLS): An efficient variable selection method using PLS. J. Chemom. 2014, 28, 439–447. [Google Scholar] [CrossRef]
- Eigenvector, Using Cross-Validation. Available online: https://wiki.eigenvector.com/index.php?title=Using_Cross-Validation (accessed on 10 February 2021).




| Method | Specification | Number of Selected Buckets | RMSECV (µg/kg) |
|---|---|---|---|
| GA | Interval size 5 | 170 | 38 |
| Forward iPLS | Interval size 1 | 23 | 18 |
| rPLS | Specified, Level 6 | 36 | 28 |
| suggested | 19 | 35 | |
| surveyed | 32 | 19 | |
| sRatio | Exclusion of last 45% | 315 | 48 |
| VIP | Exclusion of last 20% | 140 | 36 |
| RMSEP (µg/kg) | |
|---|---|
| Roasting degree | |
| Light (n = 18) | 546 |
| Medium (n = 10) | 52 |
| Medium/dark (n = 2) | 87 |
| Dark (n = 14) | 98 |
| Very dark (n = 5) | 53 |
| Type | |
| C. arabica, Catuaí (n = 19) | 97 |
| C. arabica, Malabar (n = 4) | 48 |
| C. canephora (n = 5) | 17 |
| Roaster | |
| IR (n = 26) | 49 |
| Drum (n = 6) | 151 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rief, V.; Felske, C.; Scharinger, A.; Krumbügel, K.; Stegmüller, S.; Breitling-Utzmann, C.M.; Richling, E.; Walch, S.G.; Lachenmeier, D.W. Indirect Nuclear Magnetic Resonance (NMR) Spectroscopic Determination of Acrylamide in Coffee Using Partial Least Squares (PLS) Regression. Beverages 2021, 7, 31. https://doi.org/10.3390/beverages7020031
Rief V, Felske C, Scharinger A, Krumbügel K, Stegmüller S, Breitling-Utzmann CM, Richling E, Walch SG, Lachenmeier DW. Indirect Nuclear Magnetic Resonance (NMR) Spectroscopic Determination of Acrylamide in Coffee Using Partial Least Squares (PLS) Regression. Beverages. 2021; 7(2):31. https://doi.org/10.3390/beverages7020031
Chicago/Turabian StyleRief, Vera, Christina Felske, Andreas Scharinger, Katrin Krumbügel, Simone Stegmüller, Carmen M. Breitling-Utzmann, Elke Richling, Stephan G. Walch, and Dirk W. Lachenmeier. 2021. "Indirect Nuclear Magnetic Resonance (NMR) Spectroscopic Determination of Acrylamide in Coffee Using Partial Least Squares (PLS) Regression" Beverages 7, no. 2: 31. https://doi.org/10.3390/beverages7020031
APA StyleRief, V., Felske, C., Scharinger, A., Krumbügel, K., Stegmüller, S., Breitling-Utzmann, C. M., Richling, E., Walch, S. G., & Lachenmeier, D. W. (2021). Indirect Nuclear Magnetic Resonance (NMR) Spectroscopic Determination of Acrylamide in Coffee Using Partial Least Squares (PLS) Regression. Beverages, 7(2), 31. https://doi.org/10.3390/beverages7020031