
A Comparative View on Molecular Alterations and Potential Therapeutic Strategies for Canine Oral Melanoma


Abstract
:1. Introduction
2. Methods Available to Study Molecular Alterations in COM
2.1. DNA Analysis
2.2. RNA Analysis
2.3. Immunohistochemistry
2.4. Cell Lines
3. Mutational Landscape in Canine Oral Melanoma
3.1. Chromosomal Rearrangements
3.2. Copy Number Aberrations
3.3. Recurrent Single Nucleotide Variants
3.4. Notable Genes Lacking Recurrent Aberrations in COM
4. Epigenetic Alterations in Canine Oral Melanoma
5. MicroRNAs
5.1. MicroRNA-203
5.2. MicroRNA-205
5.3. MicroRNA-145
5.4. Circulating and Exosomal MicroRNAs as Biomarkers in Plasma
5.5. Hypoxia-Induced MicroRNAs
6. Cellular Pathways and Processes Deregulated in Canine Oral Melanoma
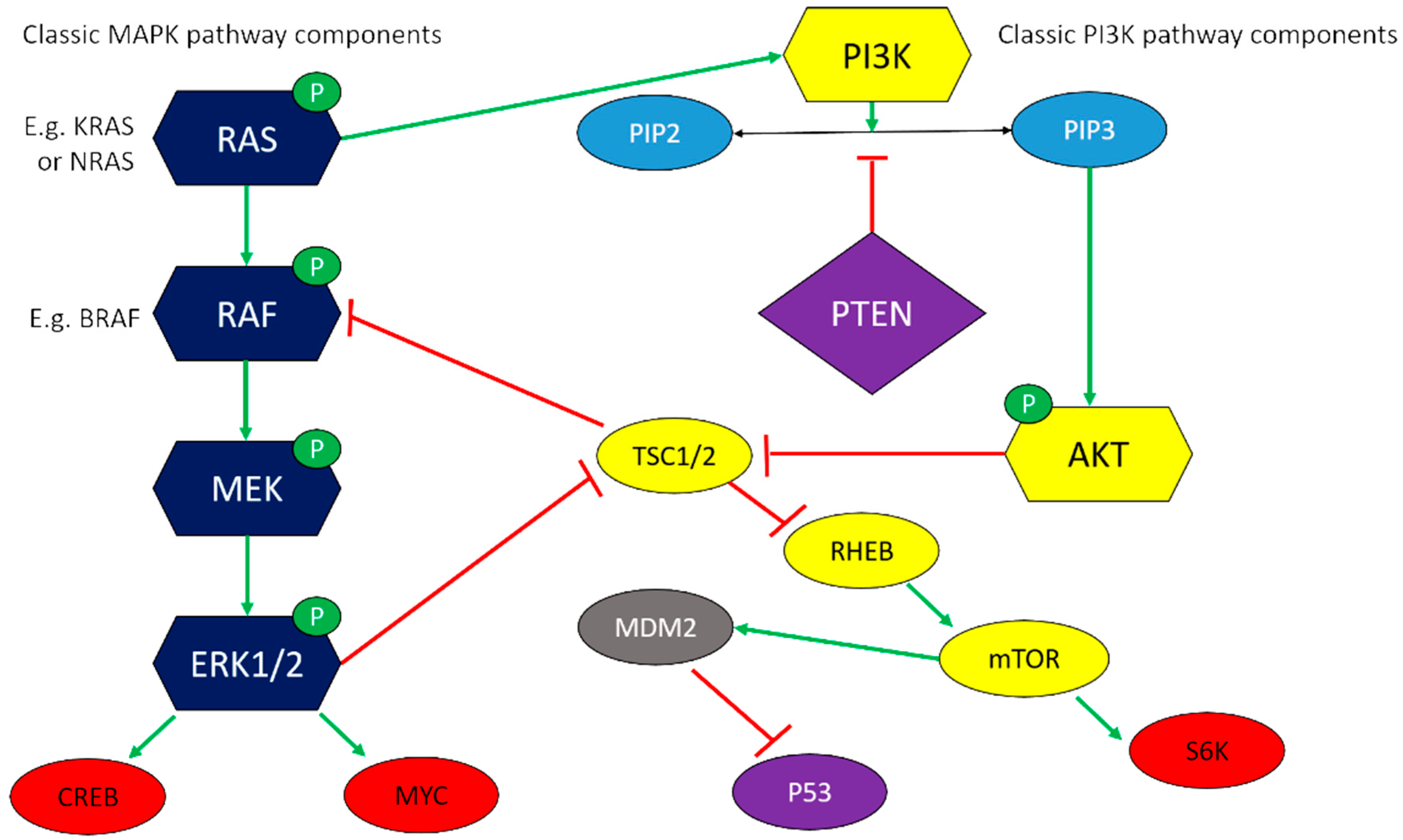
6.1. Pro-Survival Mitogenic Pathways
6.2. Loss of Cell Cycle Control and DNA Damage Response
6.3. Molecular Deregulation Promoting Metastasis
6.3.1. Cadherin Switching
6.3.2. Changes in Cell Motility
6.3.3. Angiogenesis
6.4. Deregulation of the Immune Microenvironment
6.4.1. Cyclooxygenase-2
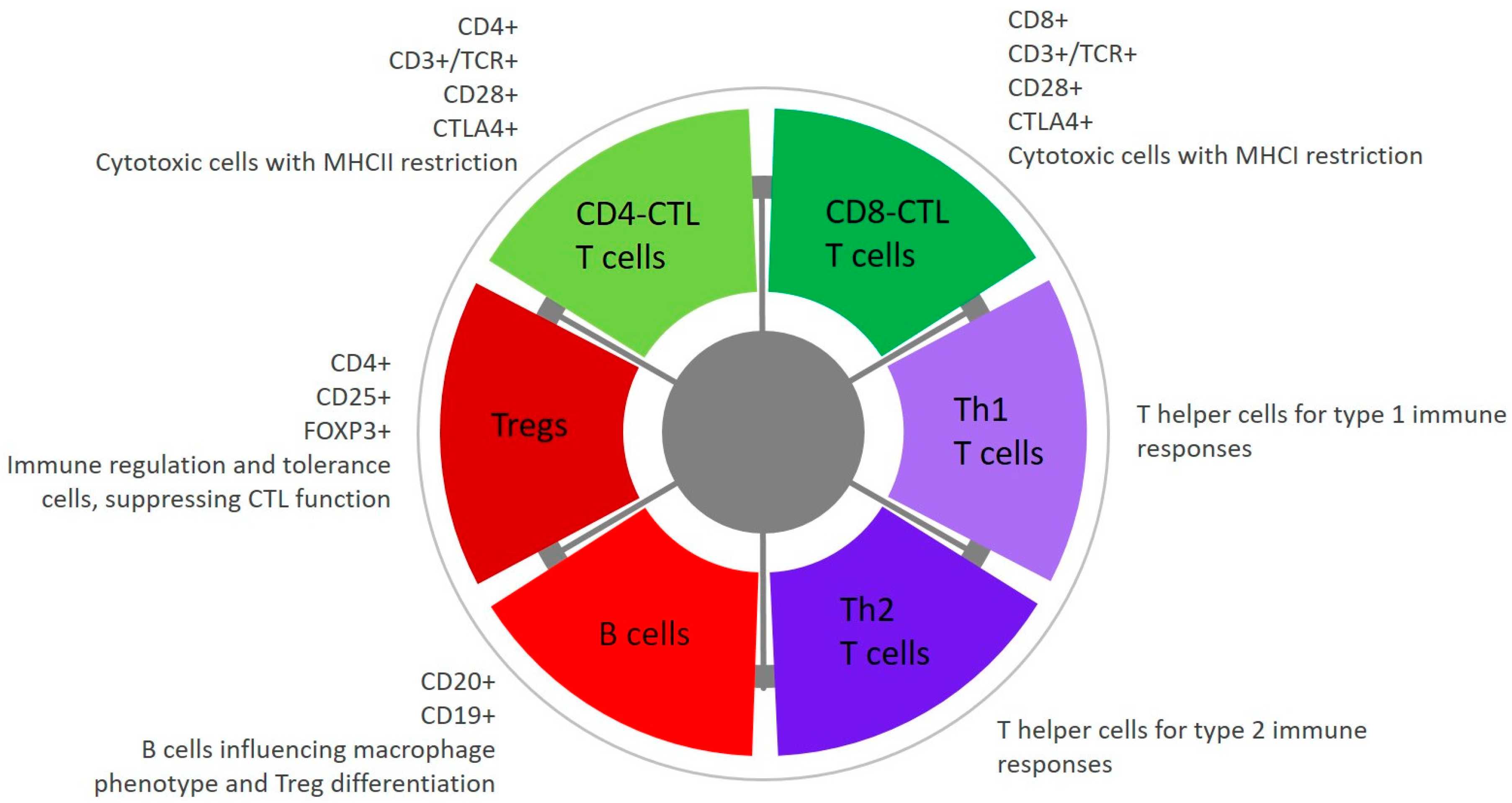
6.4.2. Tumour Infiltrating Lymphocytes and Mechanisms of Immune Tolerance
7. Potential Therapeutic Targets and Future Directions
7.1. Targeting Deregulated Signalling Pathways
7.2. Reinstating Apoptosis
7.3. Targeting the Immune Response
7.3.1. Immune Checkpoint Inhibitors
7.3.2. Vaccination Strategies
7.4. Harnessing Cancer-Associated Antigens
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, S.H.; Goldschmidt, M.H.; McManus, P.M. A comparative review of melanocytic neoplasms. Vet. Pathol. 2002, 39, 651–678. [Google Scholar] [CrossRef]
- Gillard, M.; Cadieu, E.; De Brito, C.; Abadie, J.; Vergier, B.; Devauchelle, P.; Degorce, F.; Dréano, S.; Primot, A.; Dorso, L.; et al. Naturally occurring melanomas in dogs as models for non-UV pathways of human melanomas. Pigment Cell Melanoma Res. 2014, 27, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, W.P.D.; Zismann, V.; Sivaprakasam, K.; Legendre, C.; Poorman, K.; Tembe, W.; Perdigones, N.; Kiefer, J.; Liang, W.; DeLuca, V.; et al. Somatic inactivating PTPRJ mutations and dysregulated pathways identified in canine malignant melanoma by integrated comparative genomic analysis. PLoS Genet. 2018, 14, e1007589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porcellato, I.; Brachelente, C.; De Paolis, L.; Menchetti, L.; Silvestri, S.; Sforna, M.; Vichi, G.; Iussich, S.; Mechelli, L. FoxP3 and IDO in Canine Melanocytic Tumors. Vet. Pathol. 2019, 56, 189–199. [Google Scholar] [CrossRef]
- Wong, K.; van der Weyden, L.; Schott, C.R.; Foote, A.; Constantino-Casas, F.; Smith, S.; Dobson, J.M.; Murchison, E.P.; Wu, H.; Yeh, I.; et al. Cross-species genomic landscape comparison of human mucosal melanoma with canine oral and equine melanoma. Nat. Commun. 2019, 10, 353. [Google Scholar] [CrossRef]
- Flisikowski, K.; Flisikowska, T.; Sikorska, A.; Perkowska, A.; Kind, A.; Schnieke, A.; Switonski, M. Germline gene polymorphisms predisposing domestic mammals to carcinogenesis. Vet. Comp. Oncol. 2017, 15, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Dobson, J.M. Breed-predispositions to cancer in pedigree dogs. ISRN Vet. Sci. 2013, 2013, 941275. [Google Scholar] [CrossRef] [PubMed]
- Bergman, P.J. Canine oral melanoma. Clin. Tech. Small Anim. Pract. 2007, 22, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Esplin, D.G. Survival of dogs following surgical excision of histologically well-differentiated melanocytic neoplasms of the mucous membranes of the lips and oral cavity. Vet. Pathol. 2008, 45, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Spangler, W.L.; Kass, P.H. The histologic and epidemiologic bases for prognostic considerations in canine melanocytic neoplasia. Vet. Pathol. 2006, 43, 136–149. [Google Scholar] [CrossRef] [Green Version]
- Prouteau, A.; André, C. Canine Melanomas as Models for Human Melanomas: Clinical, Histological, and Genetic Comparison. Genes 2019, 10, 501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iussich, S.; Maniscalco, L.; Di Sciuva, A.; Iotti, B.; Morello, E.; Martano, M.; Gattino, F.; Buracco, P.; De Maria, R. PDGFRs expression in dogs affected by malignant oral melanomas: Correlation with prognosis. Vet. Comp. Oncol. 2017, 15, 462–469. [Google Scholar] [CrossRef]
- Taylor, K.H.; Smith, A.N.; Higginbotham, M.; Schwartz, D.D.; Carpenter, D.M.; Whitley, E.M. Expression of vascular endothelial growth factor in canine oral malignant melanoma. Vet. Comp. Oncol. 2007, 5, 208–218. [Google Scholar] [CrossRef]
- Kawabe, M.; Mori, T.; Ito, Y.; Murakami, M.; Sakai, H.; Yanai, T.; Maruo, K. Outcomes of dogs undergoing radiotherapy for treatment of oral malignant melanoma: 111 cases (2006–2012). J. Am. Vet. Med. Assoc. 2015, 247, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, N.; Konnai, S.; Takagi, S.; Kagawa, Y.; Okagawa, T.; Nishimori, A.; Ikebuchi, R.; Izumi, Y.; Deguchi, T.; Nakajima, C.; et al. A canine chimeric monoclonal antibody targeting PD-L1 and its clinical efficacy in canine oral malignant melanoma or undifferentiated sarcoma. Sci. Rep. 2017, 7, 8951. [Google Scholar] [CrossRef]
- Almela, R.M.; Ansón, A. A Review of Immunotherapeutic Strategies in Canine Malignant Melanoma. Vet. Sci. 2019, 6, 15. [Google Scholar] [CrossRef] [Green Version]
- Adams, V.J.; Evans, K.M.; Sampson, J.; Wood, J.L. Methods and mortality results of a health survey of purebred dogs in the UK. J. Small Anim. Pract. 2010, 51, 512–524. [Google Scholar] [CrossRef]
- Beltrán Hernández, I.; Kromhout, J.Z.; Teske, E.; Hennink, W.E.; van Nimwegen, S.A.; Oliveira, S. Molecular targets for anticancer therapies in companion animals and humans: What can we learn from each other? Theranostics 2021, 11, 3882–3897. [Google Scholar] [CrossRef] [PubMed]
- Khanna, C.; Lindblad-Toh, K.; Vail, D.; London, C.; Bergman, P.; Barber, L.; Breen, M.; Kitchell, B.; McNeil, E.; Modiano, J.F.; et al. The dog as a cancer model. Nat. Biotechnol. 2006, 24, 1065–1066. [Google Scholar] [CrossRef]
- Harrison, B.M.; Loukopoulos, P. Genomics and transcriptomics in veterinary oncology. Oncol. Lett. 2021, 21, 336. [Google Scholar] [CrossRef]
- Gray, M.; Meehan, J.; Turnbull, A.K.; Martínez-Pérez, C.; Kay, C.; Pang, L.Y.; Argyle, D.J. The Importance of the Tumor Microenvironment and Hypoxia in Delivering a Precision Medicine Approach to Veterinary Oncology. Front. Vet. Sci. 2020, 7, 598338. [Google Scholar] [CrossRef] [PubMed]
- Miedema, J.; Andea, A.A. Through the looking glass and what you find there: Making sense of comparative genomic hybridization and fluorescence in situ hybridization for melanoma diagnosis. Mod. Pathol. 2020, 33, 1318–1330. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Xiao, H. Perspectives of DNA microarray and next-generation DNA sequencing technologies. Sci. China Ser. C Life Sci. 2009, 52, 7–16. [Google Scholar] [CrossRef]
- Costa, C.; Giménez-Capitán, A.; Karachaliou, N.; Rosell, R. Comprehensive molecular screening: From the RT-PCR to the RNA-seq. Transl. Lung Cancer Res. 2013, 2, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Porcellato, I.; Brachelente, C.; Cappelli, K.; Menchetti, L.; Silvestri, S.; Sforna, M.; Mecocci, S.; Iussich, S.; Leonardi, L.; Mechelli, L. FoxP3, CTLA-4, and IDO in Canine Melanocytic Tumors. Vet. Pathol. 2021, 58, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Starkey, M.P.; Compston-Garnett, L.; Malho, P.; Dunn, K.; Dubielzig, R. Metastasis-associated microRNA expression in canine uveal melanoma. Vet. Comp. Oncol. 2018, 16, 81–89. [Google Scholar] [CrossRef]
- Ramos-Vara, J.A.; Beissenherz, M.E.; Miller, M.A.; Johnson, G.C.; Pace, L.W.; Fard, A.; Kottler, S.J. Retrospective study of 338 canine oral melanomas with clinical, histologic, and immunohistochemical review of 129 cases. Vet. Pathol. 2000, 37, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Smedley, R.C.; Lamoureux, J.; Sledge, D.G.; Kiupel, M. Immunohistochemical diagnosis of canine oral amelanotic melanocytic neoplasms. Vet. Pathol. 2011, 48, 32–40. [Google Scholar] [CrossRef]
- Bergin, I.L.; Smedley, R.C.; Esplin, D.G.; Spangler, W.L.; Kiupel, M. Prognostic evaluation of Ki67 threshold value in canine oral melanoma. Vet. Pathol. 2011, 48, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Fedchenko, N.; Reifenrath, J. Different approaches for interpretation and reporting of immunohistochemistry analysis results in the bone tissue—A review. Diagn. Pathol. 2014, 9, 221. [Google Scholar] [CrossRef] [Green Version]
- Segaoula, Z.; Primot, A.; Lepretre, F.; Hedan, B.; Bouchaert, E.; Minier, K.; Marescaux, L.; Serres, F.; Galiègue-Zouitina, S.; André, C.; et al. Isolation and characterization of two canine melanoma cell lines: New models for comparative oncology. BMC Cancer 2018, 18, 1219. [Google Scholar] [CrossRef]
- Inoue, K.; Ohashi, E.; Kadosawa, T.; Hong, S.H.; Matsunaga, S.; Mochizuki, M.; Nishimura, R.; Sasaki, N. Establishment and characterization of four canine melanoma cell lines. J. Vet. Med. Sci. 2004, 66, 1437–1440. [Google Scholar] [CrossRef] [Green Version]
- Aina, O.H.; Maeda, Y.; Harrison, M.; Zwingenberger, A.L.; Walker, N.J.; Lam, K.S.; Kent, M.S. Canine malignant melanoma alpha-3 integrin binding peptides. Vet. Immunol. Immunopathol. 2011, 143, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Ohashi, E.; Hong, S.H.; Takahashi, T.; Nakagawa, T.; Mochizuki, M.; Nishimura, R.; Sasak, N. Effect of retinoids on growth inhibition of two canine melanoma cell lines. J. Vet. Med. Sci. 2001, 63, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Touil, Y.; Segaoula, Z.; Thuru, X.; Galiègue-Zouitina, S.; Tierny, D.; Quesnel, B. Aggressiveness Potential of Spontaneous Canine Mucosal Melanoma Can Dictate Distinct Cancer Stem Cell Compartment Behaviors in Regard to Their Initial Size and Expansion Abilities. Stem Cells Dev. 2020, 29, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.A.; Lu, Z.; Jia, S.; Assumpção, A.; Van Hesteren, M.A.; Huelsmeyer, M.K.; Vail, D.M.; Pan, X. Pevonedistat targeted therapy inhibits canine melanoma cell growth through induction of DNA re-replication and senescence. Vet. Comp. Oncol. 2020, 18, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.N.; Huelsmeyer, M.K.; Mitzey, A.; Dubielzig, R.R.; Kurzman, I.D.; Macewen, E.G.; Vail, D.M. Development of an allogeneic whole-cell tumor vaccine expressing xenogeneic gp100 and its implementation in a phase II clinical trial in canine patients with malignant melanoma. Cancer Immunol. Immunother. CII 2006, 55, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Ritt, M.G.; Wojcieszyn, J.; Modiano, J.F. Functional loss of p21/Waf-1 in a case of benign canine multicentric melanoma. Vet. Pathol. 1998, 35, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.R.; Michael, H.T.; Halsey, C.H.; Peer, C.J.; Adhikari, A.; Dwyer, J.E.; Hoover, S.B.; El Meskini, R.; Kozlov, S.; Weaver Ohler, Z.; et al. Synergistic targeted inhibition of MEK and dual PI3K/mTOR diminishes viability and inhibits tumor growth of canine melanoma underscoring its utility as a preclinical model for human mucosal melanoma. Pigment Cell Melanoma Res. 2016, 29, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Koenig, A.; Wojcieszyn, J.; Weeks, B.R.; Modiano, J.F. Expression of S100a, vimentin, NSE, and melan A/MART-1 in seven canine melanoma cells lines and twenty-nine retrospective cases of canine melanoma. Vet. Pathol. 2001, 38, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Enjoji, S.; Yabe, R.; Fujiwara, N.; Tsuji, S.; Vitek, M.P.; Mizuno, T.; Nakagawa, T.; Usui, T.; Ohama, T.; Sato, K. The therapeutic effects of SET/I2PP2A inhibitors on canine melanoma. J. Vet. Med. Sci. 2015, 77, 1451–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.M.; Lai, Y.C.; Husna, A.A.; Chen, H.W.; Tanaka, Y.; Kawaguchi, H.; Hatai, H.; Miyoshi, N.; Nakagawa, T.; Fukushima, R.; et al. Transcriptome analysis of dog oral melanoma and its oncogenic analogy with human melanoma. Oncol. Rep. 2020, 43, 16–30. [Google Scholar] [CrossRef] [Green Version]
- Brachelente, C.; Cappelli, K.; Capomaccio, S.; Porcellato, I.; Silvestri, S.; Bongiovanni, L.; De Maria, R.; Verini Supplizi, A.; Mechelli, L.; Sforna, M. Transcriptome Analysis of Canine Cutaneous Melanoma and Melanocytoma Reveals a Modulation of Genes Regulating Extracellular Matrix Metabolism and Cell Cycle. Sci. Rep. 2017, 7, 6386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowles, J.S.; Denton, C.L.; Gustafson, D.L. Comparative analysis of MAPK and PI3K/AKT pathway activation and inhibition in human and canine melanoma. Vet. Comp. Oncol. 2015, 13, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Poorman, K.; Borst, L.; Moroff, S.; Roy, S.; Labelle, P.; Motsinger-Reif, A.; Breen, M. Comparative cytogenetic characterization of primary canine melanocytic lesions using array CGH and fluorescence in situ hybridization. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2015, 23, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Hörsting, N.; Wohlsein, P.; Reimann, N.; Bartnitzke, S.; Bullerdiek, J.; Nolte, I. Cytogenetic analysis of three oropharyngeal malignant melanomas in dogs. Res. Vet. Sci. 1999, 67, 149–151. [Google Scholar] [CrossRef]
- Brocca, G.; Ferraresso, S.; Zamboni, C.; Martinez-Merlo, E.M.; Ferro, S.; Goldschmidt, M.H.; Castagnaro, M. Array Comparative Genomic Hybridization Analysis Reveals Significantly Enriched Pathways in Canine Oral Melanoma. Front. Oncol. 2019, 9, 1397. [Google Scholar] [CrossRef] [Green Version]
- Giannuzzi, D.; Marconato, L.; Elgendy, R.; Ferraresso, S.; Scarselli, E.; Fariselli, P.; Nicosia, A.; Pegolo, S.; Leoni, G.; Laganga, P.; et al. Longitudinal transcriptomic and genetic landscape of radiotherapy response in canine melanoma. Vet. Comp. Oncol. 2019, 17, 308–316. [Google Scholar] [CrossRef]
- Prouteau, A.; Chocteau, F.; de Brito, C.; Cadieu, E.; Primot, A.; Botherel, N.; Degorce, F.; Cornevin, L.; Lagadic, M.A.; Cabillic, F.; et al. Prognostic value of somatic focal amplifications on chromosome 30 in canine oral melanoma. Vet. Comp. Oncol. 2020, 18, 214–223. [Google Scholar] [CrossRef]
- Prouteau, A.; Denis, J.A.; De Fornel, P.; Cadieu, E.; Derrien, T.; Kergal, C.; Botherel, N.; Ulvé, R.; Rault, M.; Bouzidi, A.; et al. Circulating tumor DNA is detectable in canine histiocytic sarcoma, oral malignant melanoma, and multicentric lymphoma. Sci. Rep. 2021, 11, 877. [Google Scholar] [CrossRef]
- Hitte, C.; Le Béguec, C.; Cadieu, E.; Wucher, V.; Primot, A.; Prouteau, A.; Botherel, N.; Hédan, B.; Lindblad-Toh, K.; André, C.; et al. Genome-Wide Analysis of Long Non-Coding RNA Profiles in Canine Oral Melanomas. Genes 2019, 10, 477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, A.E.; Torrisi, E.; Bevelacqua, Y.; Perrotta, R.; Libra, M.; McCubrey, J.A.; Spandidos, D.A.; Stivala, F.; Malaponte, G. Melanoma: Molecular pathogenesis and emerging target therapies (Review). Int. J. Oncol. 2009, 34, 1481–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenig, A.; Bianco, S.R.; Fosmire, S.; Wojcieszyn, J.; Modiano, J.F. Expression and significance of p53, rb, p21/waf-1, p16/ink-4a, and PTEN tumor suppressors in canine melanoma. Vet. Pathol. 2002, 39, 458–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelly, S.; Chien, M.B.; Yip, B.; Kent, M.S.; Theon, A.P.; McCallan, J.L.; London, C.A. Exon 15 BRAF mutations are uncommon in canine oral malignant melanomas. Mamm. Genome 2005, 16, 211–217. [Google Scholar] [CrossRef]
- Mochizuki, H.; Breen, M. Comparative Aspects of BRAF Mutations in Canine Cancers. Vet. Sci. 2015, 2, 231–245. [Google Scholar] [CrossRef]
- Zamboni, C.; Brocca, G.; Ferraresso, S.; Ferro, S.; Sammarco, A.; Dal Corso, C.; Iussich, S.; de Andres, P.J.; Martìnez de Merlo, E.M.; Cavicchioli, L.; et al. Cyclin D1 immunohistochemical expression and somatic mutations in canine oral melanoma. Vet. Comp. Oncol. 2020, 18, 231–238. [Google Scholar] [CrossRef]
- London, C.A. Tyrosine kinase inhibitors in veterinary medicine. Top. Companion Anim. Med. 2009, 24, 106–112. [Google Scholar] [CrossRef] [Green Version]
- Simpson, R.M.; Bastian, B.C.; Michael, H.T.; Webster, J.D.; Prasad, M.L.; Conway, C.M.; Prieto, V.M.; Gary, J.M.; Goldschmidt, M.H.; Esplin, D.G.; et al. Sporadic naturally occurring melanoma in dogs as a preclinical model for human melanoma. Pigment Cell Melanoma Res. 2014, 27, 37–47. [Google Scholar] [CrossRef]
- Chu, P.Y.; Pan, S.L.; Liu, C.H.; Lee, J.; Yeh, L.S.; Liao, A.T. KIT gene exon 11 mutations in canine malignant melanoma. Vet. J. 2013, 196, 226–230. [Google Scholar] [CrossRef]
- Brocca, G.; Poncina, B.; Sammarco, A.; Cavicchioli, L.; Castagnaro, M. KIT Somatic Mutations and Immunohistochemical Expression in Canine Oral Melanoma. Animals 2020, 10, 2370. [Google Scholar] [CrossRef]
- Murakami, A.; Mori, T.; Sakai, H.; Murakami, M.; Yanai, T.; Hoshino, Y.; Maruo, K. Analysis of KIT expression and KIT exon 11 mutations in canine oral malignant melanomas. Vet. Comp. Oncol. 2011, 9, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Smedley, R.C.; Thaiwong, T.; Deeth, L.E.; Kiupel, M. Correlation Between KIT Expression and c-Kit Mutations in 2 Subtypes of Canine Oral Melanocytic Neoplasms. Vet. Pathol. 2021, 58, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, A.; Dobson, J. Prospective clinical trial of masitinib mesylate treatment for advanced stage III and IV canine malignant melanoma. J. Small Anim. Pract. 2020, 61, 190–194. [Google Scholar] [CrossRef]
- Tani, H.; Miyamoto, R.; Noguchi, S.; Kurita, S.; Nagashima, T.; Michishita, M.; Yayoshi, N.; Tamura, K.; Bonkobara, M. A canine case of malignant melanoma carrying a KIT c.1725_1733del mutation treated with toceranib: A case report and in vitro analysis. BMC Vet. Res. 2021, 17, 147. [Google Scholar] [CrossRef] [PubMed]
- Xavier, P.L.P.; Müller, S.; Fukumasu, H. Epigenetic Mechanisms in Canine Cancer. Front. Oncol. 2020, 10, 591843. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Bird, A.P. CpG islands—A rough guide. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [Green Version]
- Scattone, N.V.; Epiphanio, T.M.F.; Caddrobi, K.G.; Ferrão, J.S.P.; Hernandez-Blazquez, F.J.; Loureiro, A.P.M.; Massoco, C.O.; Dagli, M.L.Z. Quantification of Global DNA Methylation in Canine Melanotic and Amelanotic Oral Mucosal Melanomas and Peripheral Blood Leukocytes From the Same Patients with OMM: First Study. Front. Vet. Sci. 2021, 8, 680181. [Google Scholar] [CrossRef]
- Ishizaki, T.; Yamazaki, J.; Jelinek, J.; Aoshima, K.; Kimura, T. Genome-wide DNA methylation analysis identifies promoter hypermethylation in canine malignant melanoma. Res. Vet. Sci. 2020, 132, 521–526. [Google Scholar] [CrossRef]
- Noguchi, S.; Mori, T.; Igase, M.; Mizuno, T. A novel apoptosis-inducing mechanism of 5-aza-2'-deoxycitidine in melanoma cells: Demethylation of TNF-α and activation of FOXO1. Cancer Lett. 2015, 369, 344–353. [Google Scholar] [CrossRef]
- Ishizaki, T.; Yamazaki, J.; Meagawa, S.; Yokoyama, N.; Aoshima, K.; Takiguchi, M.; Kimura, T. Long interspersed nucleotide element-1 hypomethylation in canine malignant mucosal melanoma. Vet. Comp. Oncol. 2020, 18, 854–860. [Google Scholar] [CrossRef]
- Tobin, S.J.; Chang, H.; Kent, M.S.; Davies, A.E. JARID1-targeted histone H3 demethylase inhibitors exhibit anti-proliferative activity and overcome cisplatin resistance in canine oral melanoma cell lines. Vet. Comp. Oncol. 2021, 19, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Lai, Y.C.; Husna, A.A.; Chen, H.W.; Tanaka, Y.; Kawaguchi, H.; Miyoshi, N.; Nakagawa, T.; Fukushima, R.; Miura, N. Micro RNA Transcriptome Profile in Canine Oral Melanoma. Int. J. Mol. Sci. 2019, 20, 4832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio, N.; Rahman, M.M.; Maemura, T.; Lai, Y.C.; Iwanaga, T.; Kawaguchi, H.; Miyoshi, N.; Momoi, Y.; Miura, N. Identification of dysregulated microRNAs in canine malignant melanoma. Oncol. Lett. 2019, 17, 1080–1088. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Gao, G.; Yan, D.; Chen, X.; Yao, X.; Guo, S.; Li, G.; Zhao, Y. Effects of miR-145-5p through NRAS on the cell proliferation, apoptosis, migration, and invasion in melanoma by inhibiting MAPK and PI3K/AKT pathways. Cancer Med. 2017, 6, 819–833. [Google Scholar] [CrossRef]
- Yoshikawa, R.; Mori, T.; Noguchi, S.; Akao, Y.; Maruo, K.; Kitade, Y. Synthetic microRNA-205 exhibited tumour suppression in spontaneous canine malignant melanoma by intratumoral injection. Vet. Comp. Oncol. 2019, 17, 407–412. [Google Scholar] [CrossRef]
- Noguchi, S.; Mori, T.; Hoshino, Y.; Yamada, N.; Maruo, K.; Akao, Y. MicroRNAs as tumour suppressors in canine and human melanoma cells and as a prognostic factor in canine melanomas. Vet. Comp. Oncol. 2013, 11, 113–123. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Noguchi, S.; Mori, T.; Hoshino, Y.; Yamada, N.; Nakagawa, T.; Sasaki, N.; Akao, Y.; Maruo, K. Comparative study of anti-oncogenic microRNA-145 in canine and human malignant melanoma. J. Vet. Med. Sci. 2012, 74, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zamarian, V.; Catozzi, C.; Ressel, L.; Finotello, R.; Ceciliani, F.; Vilafranca, M.; Altimira, J.; Lecchi, C. MicroRNA Expression in Formalin-Fixed, Paraffin-Embedded Samples of Canine Cutaneous and Oral Melanoma by RT-qPCR. Vet. Pathol. 2019, 56, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Zhang, Z.W. Expression of miR-203 is decreased and associated with the prognosis of melanoma patients. Int. J. Clin. Exp. Pathol. 2015, 8, 13249–13254. [Google Scholar] [PubMed]
- Noguchi, S.; Mori, T.; Nakagawa, T.; Itamoto, K.; Haraguchi, T.; Mizuno, T. DNA methylation contributes toward silencing of antioncogenic microRNA-203 in human and canine melanoma cells. Melanoma Res. 2015, 25, 390–398. [Google Scholar] [CrossRef]
- Noguchi, S.; Mori, T.; Otsuka, Y.; Yamada, N.; Yasui, Y.; Iwasaki, J.; Kumazaki, M.; Maruo, K.; Akao, Y. Anti-oncogenic microRNA-203 induces senescence by targeting E2F3 protein in human melanoma cells. J. Biol. Chem. 2012, 287, 11769–11777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, S.; Kumazaki, M.; Mori, T.; Baba, K.; Okuda, M.; Mizuno, T.; Akao, Y. Analysis of microRNA-203 function in CREB/MITF/RAB27a pathway: Comparison between canine and human melanoma cells. Vet. Comp. Oncol. 2016, 14, 384–394. [Google Scholar] [CrossRef]
- Liu, S.; Tetzlaff, M.T.; Liu, A.; Liegl-Atzwanger, B.; Guo, J.; Xu, X. Loss of microRNA-205 expression is associated with melanoma progression. Lab. Investig. 2012, 92, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Iwasaki, J.; Kumazaki, M.; Mori, T.; Maruo, K.; Sakai, H.; Yamada, N.; Shimada, K.; Naoe, T.; Kitade, Y.; et al. Chemically modified synthetic microRNA-205 inhibits the growth of melanoma cells in vitro and in vivo. Mol. Ther. 2013, 21, 1204–1211. [Google Scholar] [CrossRef] [Green Version]
- Heishima, K.; Ichikawa, Y.; Yoshida, K.; Iwasaki, R.; Sakai, H.; Nakagawa, T.; Tanaka, Y.; Hoshino, Y.; Okamura, Y.; Murakami, M.; et al. Circulating microRNA-214 and -126 as potential biomarkers for canine neoplastic disease. Sci. Rep. 2017, 7, 2301. [Google Scholar] [CrossRef]
- Husna, A.A.; Rahman, M.M.; Lai, Y.C.; Chen, H.W.; Hasan, M.N.; Nakagawa, T.; Miura, N. Identification of melanoma-specific exosomal miRNAs as the potential biomarker for canine oral melanoma. Pigment Cell Melanoma Res. 2021, 34, 1062–1073. [Google Scholar] [CrossRef]
- Hino, Y.; Rahman, M.M.; Lai, Y.C.; Husna, A.A.; Chen, H.W.; Hasan, M.N.; Nakagawa, T.; Miura, N. Hypoxic miRNAs expression are different between primary and metastatic melanoma cells. Gene 2021, 782, 145552. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Ahern, T.E.; Bird, R.C.; Bird, A.E.; Wolfe, L.G. Overexpression of c-erbB-2 and c-myc but not c-ras, in canine melanoma cell lines, is associated with metastatic potential in nude mice. Anticancer Res. 1993, 13, 1365–1371. [Google Scholar] [PubMed]
- Veloso, E.S.; Gonçalves, I.N.N.; Silveira, T.L.; Oliveira, F.S.; Vieira, D.S.; Cassali, G.D.; Del Puerto, H.L.; Ferreira, E. Diverse roles of epidermal growth factors receptors in oral and cutaneous canine melanomas. BMC Vet. Res. 2020, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Thamm, D.H.; Huelsmeyer, M.K.; Mitzey, A.M.; Qurollo, B.; Rose, B.J.; Kurzman, I.D. RT-PCR-based tyrosine kinase display profiling of canine melanoma: IGF-1 receptor as a potential therapeutic target. Melanoma Res. 2010, 20, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Kent, M.S.; Collins, C.J.; Ye, F. Activation of the AKT and mammalian target of rapamycin pathways and the inhibitory effects of rapamycin on those pathways in canine malignant melanoma cell lines. Am. J. Vet. Res. 2009, 70, 263–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, S.; Ogusu, R.; Wada, Y.; Matsuyama, S.; Mori, T. PTEN, A Target of Microrna-374b, Contributes to the Radiosensitivity of Canine Oral Melanoma Cells. Int. J. Mol. Sci. 2019, 20, 4631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Goel, V.; Haluska, F.G. PTEN signaling pathways in melanoma. Oncogene 2003, 22, 3113–3122. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, X.; Kent, M.S.; Rodriguez, C.O.; Chen, X. Establishment of a dog model for the p53 family pathway and identification of a novel isoform of p21 cyclin-dependent kinase inhibitor. Mol. Cancer Res. MCR 2009, 7, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Schmid, F.; Brodesser, D.; Reifinger, M.; Forte, S.; Semp, P.; Eberspächer-Schweda, M.C.; Wolschek, M.; Brandt, S.; Kleiter, M.; Pratscher, B. Canine oral primary melanoma cells exhibit shift to mesenchymal phenotype and phagocytic behaviour. Vet. Comp. Oncol. 2019, 17, 211–220. [Google Scholar] [CrossRef]
- Veloso, E.S.; Gonçalves, I.N.N.; Silveira, T.L.; Espirito Santo, J.T.; Figueiredo, L.V.; Varaschin, M.S.; Cassali, G.D.; Del Puerto, H.L.; Ferreira, E. ZEB and Snail expression indicates epithelial-mesenchymal transition in canine melanoma. Res. Vet. Sci. 2020, 131, 7–14. [Google Scholar] [CrossRef]
- Pisamai, S.; Rungsipipat, A.; Kalpravidh, C.; Suriyaphol, G. Gene expression profiles of cell adhesion molecules, matrix metalloproteinases and their tissue inhibitors in canine oral tumors. Res. Vet. Sci. 2017, 113, 94–100. [Google Scholar] [CrossRef]
- Han, J.I.; Kim, Y.; Kim, D.Y.; Na, K.J. Alteration in E-cadherin/β-catenin expression in canine melanotic tumors. Vet. Pathol. 2013, 50, 274–280. [Google Scholar] [CrossRef] [Green Version]
- Chon, E.; Thompson, V.; Schmid, S.; Stein, T.J. Activation of the canonical Wnt/β-catenin signalling pathway is rare in canine malignant melanoma tissue and cell lines. J. Comp. Pathol. 2013, 148, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Yamada, N.; Mori, T.; Murakami, M.; Noguchi, S.; Sakai, H.; Akao, Y.; Maruo, K. Fascin-1 expression in canine cutaneous and oral melanocytic tumours. Vet. Comp. Oncol. 2012, 10, 303–311. [Google Scholar] [CrossRef]
- Shinada, M.; Kato, D.; Kamoto, S.; Yoshimoto, S.; Tsuboi, M.; Yoshitake, R.; Eto, S.; Ikeda, N.; Saeki, K.; Hashimoto, Y.; et al. PDPN Is Expressed in Various Types of Canine Tumors and Its Silencing Induces Apoptosis and Cell Cycle Arrest in Canine Malignant Melanoma. Cells 2020, 9, 1136. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Rabanal, R.M.; Miquel, L.; Domenzain, C.; Bassols, A. Differential expression of CD44 in canine melanocytic tumours. J. Comp. Pathol. 2004, 130, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Flickinger, I.; Rütgen, B.C.; Gerner, W.; Calice, I.; Tichy, A.; Saalmüller, A.; Kleiter, M. Radiation up-regulates the expression of VEGF in a canine oral melanoma cell line. J. Vet. Sci. 2013, 14, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peggs, K.S.; Quezada, S.A.; Allison, J.P. Cancer immunotherapy: Co-stimulatory agonists and co-inhibitory antagonists. Clin. Exp. Immunol. 2009, 157, 9–19. [Google Scholar] [CrossRef]
- Porcellato, I.; Silvestri, S.; Menchetti, L.; Recupero, F.; Mechelli, L.; Sforna, M.; Iussich, S.; Bongiovanni, L.; Lepri, E.; Brachelente, C. Tumour-infiltrating lymphocytes in canine melanocytic tumours: An investigation on the prognostic role of CD3(+) and CD20(+) lymphocytic populations. Vet. Comp. Oncol. 2020, 18, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Pires, I.; Garcia, A.; Prada, J.; Queiroga, F.L. COX-1 and COX-2 expression in canine cutaneous, oral and ocular melanocytic tumours. J. Comp. Pathol. 2010, 143, 142–149. [Google Scholar] [CrossRef]
- Mohammed, S.I.; Coffman, K.; Glickman, N.W.; Hayek, M.G.; Waters, D.J.; Schlittler, D.; DeNicola, D.B.; Knapp, D.W. Prostaglandin E2 concentrations in naturally occurring canine cancer. Prostaglandins Leukot. Essent. Fat. Acids 2001, 64, 1–4. [Google Scholar] [CrossRef]
- Chwirot, B.W.; Kuźbicki, Ł. Cyclooxygenase-2 (COX-2): First immunohistochemical marker distinguishing early cutaneous melanomas from benign melanocytic skin tumours. Melanoma Res. 2007, 17, 139–145. [Google Scholar] [CrossRef]
- Martínez, C.M.; Peñafiel-Verdú, C.; Vilafranca, M.; Ramírez, G.; Méndez-Gallego, M.; Buendía, A.J.; Sánchez, J. Cyclooxygenase-2 expression is related with localization, proliferation, and overall survival in canine melanocytic neoplasms. Vet. Pathol. 2011, 48, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; An, J.H.; Park, S.M.; Lee, J.H.; Chae, H.K.; Lee, K.M.; Song, W.J.; Youn, H.Y. Enhanced expression of cyclooxygenase-2 related multi-drug resistance gene in melanoma and osteosarcoma cell lines by TSG-6 secreted from canine adipose-derived mesenchymal stem/stromal cells. Vet. Med. Sci. 2021, 7, 968–978. [Google Scholar] [CrossRef]
- Kitanaka, N.; Nakano, R.; Kitanaka, T.; Namba, S.; Konno, T.; Nakayama, T.; Sugiya, H. NF-κB p65 and p105 implicate in interleukin 1β-mediated COX-2 expression in melanoma cells. PLoS ONE 2018, 13, e0208955. [Google Scholar] [CrossRef]
- Gregório, H.; Raposo, T.P.; Queiroga, F.L.; Prada, J.; Pires, I. Investigating associations of cyclooxygenase-2 expression with angiogenesis, proliferation, macrophage and T-lymphocyte infiltration in canine melanocytic tumours. Melanoma Res. 2016, 26, 338–347. [Google Scholar] [CrossRef]
- Silveira, T.L.; Veloso, E.S.; Gonçalves, I.N.N.; Costa, R.F.; Rodrigues, M.A.; Cassali, G.D.; Del Puerto, H.L.; Pang, L.Y.; Argyle, D.J.; Ferreira, E. Cyclooxygenase-2 expression is associated with infiltration of inflammatory cells in oral and skin canine melanomas. Vet. Comp. Oncol. 2020, 18, 727–738. [Google Scholar] [CrossRef]
- Seo, K.W.; Coh, Y.R.; Rebhun, R.B.; Ahn, J.O.; Han, S.M.; Lee, H.W.; Youn, H.Y. Antitumor effects of celecoxib in COX-2 expressing and non-expressing canine melanoma cell lines. Res. Vet. Sci. 2014, 96, 482–486. [Google Scholar] [CrossRef] [Green Version]
- Yoshitake, R.; Saeki, K.; Watanabe, M.; Nakaoka, N.; Ong, S.M.; Hanafusa, M.; Choisunirachon, N.; Fujita, N.; Nishimura, R.; Nakagawa, T. Molecular investigation of the direct anti-tumour effects of nonsteroidal anti-inflammatory drugs in a panel of canine cancer cell lines. Vet. J. 2017, 221, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Yasumaru, C.C.; Xavier, J.G.; Strefezzi, R.F.; Salles-Gomes, C.O.M. Intratumoral T-Lymphocyte Subsets in Canine Oral Melanoma and Their Association With Clinical and Histopathological Parameters. Vet. Pathol. 2021, 58, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, V.B.; Perry, S.N.; Todd, M.; Huckle, W.R.; LeRoith, T. PD-1, PD-L1, and PD-L2 Gene Expression and Tumor Infiltrating Lymphocytes in Canine Melanoma. Vet. Pathol. 2021, 3009858211011939. [Google Scholar] [CrossRef]
- Horiuchi, Y.; Tominaga, M.; Ichikawa, M.; Yamashita, M.; Okano, K.; Jikumaru, Y.; Nariai, Y.; Nakajima, Y.; Kuwabara, M.; Yukawa, M. Relationship between regulatory and type 1 T cells in dogs with oral malignant melanoma. Microbiol. Immunol. 2010, 54, 152–159. [Google Scholar] [CrossRef]
- Takeuchi, H.; Konnai, S.; Maekawa, N.; Takagi, S.; Ohta, H.; Sasaki, N.; Kim, S.; Okagawa, T.; Suzuki, Y.; Murata, S.; et al. Canine Transforming Growth Factor-β Receptor 2-Ig: A Potential Candidate Biologic for Melanoma Treatment That Reverses Transforming Growth Factor-β1 Immunosuppression. Front. Vet. Sci. 2021, 8, 656715. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Horiuchi, Y.; Ichikawa, M.; Yamashita, M.; Okano, K.; Jikumaru, Y.; Nariai, Y.; Kadosawa, T. Flow cytometric analysis of peripheral blood and tumor-infiltrating regulatory T cells in dogs with oral malignant melanoma. J. Vet. Diagn. Investig. 2010, 22, 438–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, I.L.; Chung, T.F.; Huang, W.H.; Hsu, C.H.; Liu, C.C.; Chiu, Y.H.; Huang, K.C.; Liao, A.T.; Lin, C.S. Kynurenine 3-monooxygenase (KMO), and signal transducer and activator of transcription 3 (STAT3) expression is involved in tumour proliferation and predicts poor survival in canine melanoma. Vet. Comp. Oncol. 2021, 19, 79–91. [Google Scholar] [CrossRef]
- Camacho, L.H. CTLA-4 blockade with ipilimumab: Biology, safety, efficacy, and future considerations. Cancer Med. 2015, 4, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, N.; Konnai, S.; Ikebuchi, R.; Okagawa, T.; Adachi, M.; Takagi, S.; Kagawa, Y.; Nakajima, C.; Suzuki, Y.; Murata, S.; et al. Expression of PD-L1 on canine tumor cells and enhancement of IFN-γ production from tumor-infiltrating cells by PD-L1 blockade. PLoS ONE 2014, 9, e98415. [Google Scholar] [CrossRef]
- Maekawa, N.; Konnai, S.; Okagawa, T.; Nishimori, A.; Ikebuchi, R.; Izumi, Y.; Takagi, S.; Kagawa, Y.; Nakajima, C.; Suzuki, Y.; et al. Immunohistochemical Analysis of PD-L1 Expression in Canine Malignant Cancers and PD-1 Expression on Lymphocytes in Canine Oral Melanoma. PLoS ONE 2016, 11, e0157176. [Google Scholar] [CrossRef]
- Maekawa, N.; Konnai, S.; Nishimura, M.; Kagawa, Y.; Takagi, S.; Hosoya, K.; Ohta, H.; Kim, S.; Okagawa, T.; Izumi, Y.; et al. PD-L1 immunohistochemistry for canine cancers and clinical benefit of anti-PD-L1 antibody in dogs with pulmonary metastatic oral malignant melanoma. NPJ Precis. Oncol. 2021, 5, 10. [Google Scholar] [CrossRef]
- Takeuchi, H.; Konnai, S.; Maekawa, N.; Minato, E.; Ichikawa, Y.; Kobayashi, A.; Okagawa, T.; Murata, S.; Ohashi, K. Expression Analysis of Canine CMTM6 and CMTM4 as Potential Regulators of the PD-L1 Protein in Canine Cancers. Front. Vet. Sci. 2020, 7, 330. [Google Scholar] [CrossRef]
- Ariyarathna, H.; Thomson, N.A.; Aberdein, D.; Perrott, M.R.; Munday, J.S. Increased programmed death ligand (PD-L1) and cytotoxic T-lymphocyte antigen-4 (CTLA-4) expression is associated with metastasis and poor prognosis in malignant canine mammary gland tumours. Vet. Immunol. Immunopathol. 2020, 230, 110142. [Google Scholar] [CrossRef]
- Lee, B.H.; Neela, P.H.; Kent, M.S.; Zehnder, A.M. IQGAP1 is an oncogenic target in canine melanoma. PLoS ONE 2017, 12, e0176370. [Google Scholar] [CrossRef] [Green Version]
- Modiano, J.F.; Ritt, M.G.; Wojcieszyn, J. The molecular basis of canine melanoma: Pathogenesis and trends in diagnosis and therapy. J. Vet. Intern. Med. 1999, 13, 163–174. [Google Scholar] [CrossRef]
- Watanabe, Y.; Kano, R.; Maruyama, H.; Hasegawa, A.; Kamata, H. Small interfering RNA (siRNA) against the Bcl-2 gene increases apoptosis in a canine melanoma cell line. J. Vet. Med. Sci. 2010, 72, 383–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uwano, M.; Kano, R.; Maruyama, H.; Hasegawa, A.; Kamata, H. Therapeutic efficacy of ABT-737, a Bcl-2 inhibitor, in a canine melanoma cell line. J. Vet. Med. Sci. 2012, 74, 783–785. [Google Scholar] [CrossRef] [Green Version]
- Atherton, M.J.; Morris, J.S.; McDermott, M.R.; Lichty, B.D. Cancer immunology and canine malignant melanoma: A comparative review. Vet. Immunol. Immunopathol. 2016, 169, 15–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igase, M.; Nemoto, Y.; Itamoto, K.; Tani, K.; Nakaichi, M.; Sakurai, M.; Sakai, Y.; Noguchi, S.; Kato, M.; Tsukui, T.; et al. A pilot clinical study of the therapeutic antibody against canine PD-1 for advanced spontaneous cancers in dogs. Sci. Rep. 2020, 10, 18311. [Google Scholar] [CrossRef]
- Bergman, P.J.; McKnight, J.; Novosad, A.; Charney, S.; Farrelly, J.; Craft, D.; Wulderk, M.; Jeffers, Y.; Sadelain, M.; Hohenhaus, A.E.; et al. Long-term survival of dogs with advanced malignant melanoma after DNA vaccination with xenogeneic human tyrosinase: A phase I trial. Clin. Cancer Res. 2003, 9, 1284–1290. [Google Scholar] [PubMed]
- Verganti, S.; Berlato, D.; Blackwood, L.; Amores-Fuster, I.; Polton, G.A.; Elders, R.; Doyle, R.; Taylor, A.; Murphy, S. Use of Oncept melanoma vaccine in 69 canine oral malignant melanomas in the UK. J. Small Anim. Pract. 2017, 58, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Riccardo, F.; Iussich, S.; Maniscalco, L.; Lorda Mayayo, S.; La Rosa, G.; Arigoni, M.; De Maria, R.; Gattino, F.; Lanzardo, S.; Lardone, E.; et al. CSPG4-specific immunity and survival prolongation in dogs with oral malignant melanoma immunized with human CSPG4 DNA. Clin. Cancer Res. 2014, 20, 3753–3762. [Google Scholar] [CrossRef] [Green Version]
- Piras, L.A.; Riccardo, F.; Iussich, S.; Maniscalco, L.; Gattino, F.; Martano, M.; Morello, E.; Lorda Mayayo, S.; Rolih, V.; Garavaglia, F.; et al. Prolongation of survival of dogs with oral malignant melanoma treated by en bloc surgical resection and adjuvant CSPG4-antigen electrovaccination. Vet. Comp. Oncol. 2017, 15, 996–1013. [Google Scholar] [CrossRef] [Green Version]
- Kamoto, S.; Shinada, M.; Kato, D.; Yoshimoto, S.; Ikeda, N.; Tsuboi, M.; Yoshitake, R.; Eto, S.; Hashimoto, Y.; Takahashi, Y.; et al. Phase I/II Clinical Trial of the Anti-Podoplanin Monoclonal Antibody Therapy in Dogs with Malignant Melanoma. Cells 2020, 9, 2529. [Google Scholar] [CrossRef]
- Hoffmann, R.M.; Crescioli, S.; Mele, S.; Sachouli, E.; Cheung, A.; Chui, C.K.; Andriollo, P.; Jackson, P.J.M.; Lacy, K.E.; Spicer, J.F.; et al. A Novel Antibody-Drug Conjugate (ADC) Delivering a DNA Mono-Alkylating Payload to Chondroitin Sulfate Proteoglycan (CSPG4)-Expressing Melanoma. Cancers 2020, 12, 1029. [Google Scholar] [CrossRef]
- Giudice, C.; Ceciliani, F.; Rondena, M.; Stefanello, D.; Grieco, V. Immunohistochemical investigation of PNL2 reactivity of canine melanocytic neoplasms and comparison with Melan A. J. Vet. Diagn. Investig. 2010, 22, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou Asa, S. Immunohistochemical Expression of MCAM/CD146 in Canine Melanoma. J. Comp. Pathol. 2017, 157, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Mayayo, S.L.; Prestigio, S.; Maniscalco, L.; Rosa, G.; Aricò, A.; Maria, R.; Cavallo, F.; Ferrone, S.; Buracco, P.; Iussich, S. Chondroitin sulfate proteoglycan-4: A biomarker and a potential immunotherapeutic target for canine malignant melanoma. Vet. J. 2011, 190, e26–e30. [Google Scholar] [CrossRef]
- Bowlt Blacklock, K.L.; Birand, Z.; Selmic, L.E.; Nelissen, P.; Murphy, S.; Blackwood, L.; Bass, J.; McKay, J.; Fox, R.; Beaver, S.; et al. Genome-wide analysis of canine oral malignant melanoma metastasis-associated gene expression. Sci. Rep. 2019, 9, 6511. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cell Line | Derivation | Reference |
|---|---|---|
| KMeC | Primary oral melanoma | [32] |
| LMe-C | Lymph node metastasis of oral melanoma | [32] |
| UCDK9M1 | Skin metastasis of oral melanoma | [33] |
| UCDK9M2 | Lymph node metastasis of oral melanoma | [33] |
| UCDK9M3 | Primary oral melanoma | [33] |
| UCDK9M4 | Primary oral melanoma | [33] |
| UCDK9M5 | Lymph node metastasis of oral melanoma | [33] |
| CMM1 | Primary oral melanoma from patient with lymph node metastasis | [34] |
| CMM2 | Primary oral melanoma from patient with no metastasis | [34] |
| Ocr_OCMM1X | Xenograft derived from primary stage IV oral melanoma | [31,35] |
| Ocr_OCMM2X | Xenograft derived from primary stage III oral melanoma | [31,35] |
| CML-1 | Primary oral melanoma | [36] |
| CML-10C2 | Primary oral melanoma | [36] |
| 17CM98 | Lymph node metastasis of primary oral melanoma | [37] |
| TLM-1 | Primary oral melanoma | [38] |
| JONES | Primary oral melanoma | [39] |
| JENNY | Primary oral melanoma | [40] |
| SCOOTER | Primary oral melanoma | [40] |
| SHADOW | Pulmonary metastasis of primary oral melanoma | [40] |
| CMGD-2 | Primary oral melanoma | [41] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardwick, L. A Comparative View on Molecular Alterations and Potential Therapeutic Strategies for Canine Oral Melanoma. Vet. Sci. 2021, 8, 286. https://doi.org/10.3390/vetsci8110286
Hardwick L. A Comparative View on Molecular Alterations and Potential Therapeutic Strategies for Canine Oral Melanoma. Veterinary Sciences. 2021; 8(11):286. https://doi.org/10.3390/vetsci8110286
Chicago/Turabian StyleHardwick, Laura. 2021. "A Comparative View on Molecular Alterations and Potential Therapeutic Strategies for Canine Oral Melanoma" Veterinary Sciences 8, no. 11: 286. https://doi.org/10.3390/vetsci8110286
APA StyleHardwick, L. (2021). A Comparative View on Molecular Alterations and Potential Therapeutic Strategies for Canine Oral Melanoma. Veterinary Sciences, 8(11), 286. https://doi.org/10.3390/vetsci8110286