

The Roles of Histone Lysine Methyltransferases in Heart Development and Disease


Abstract
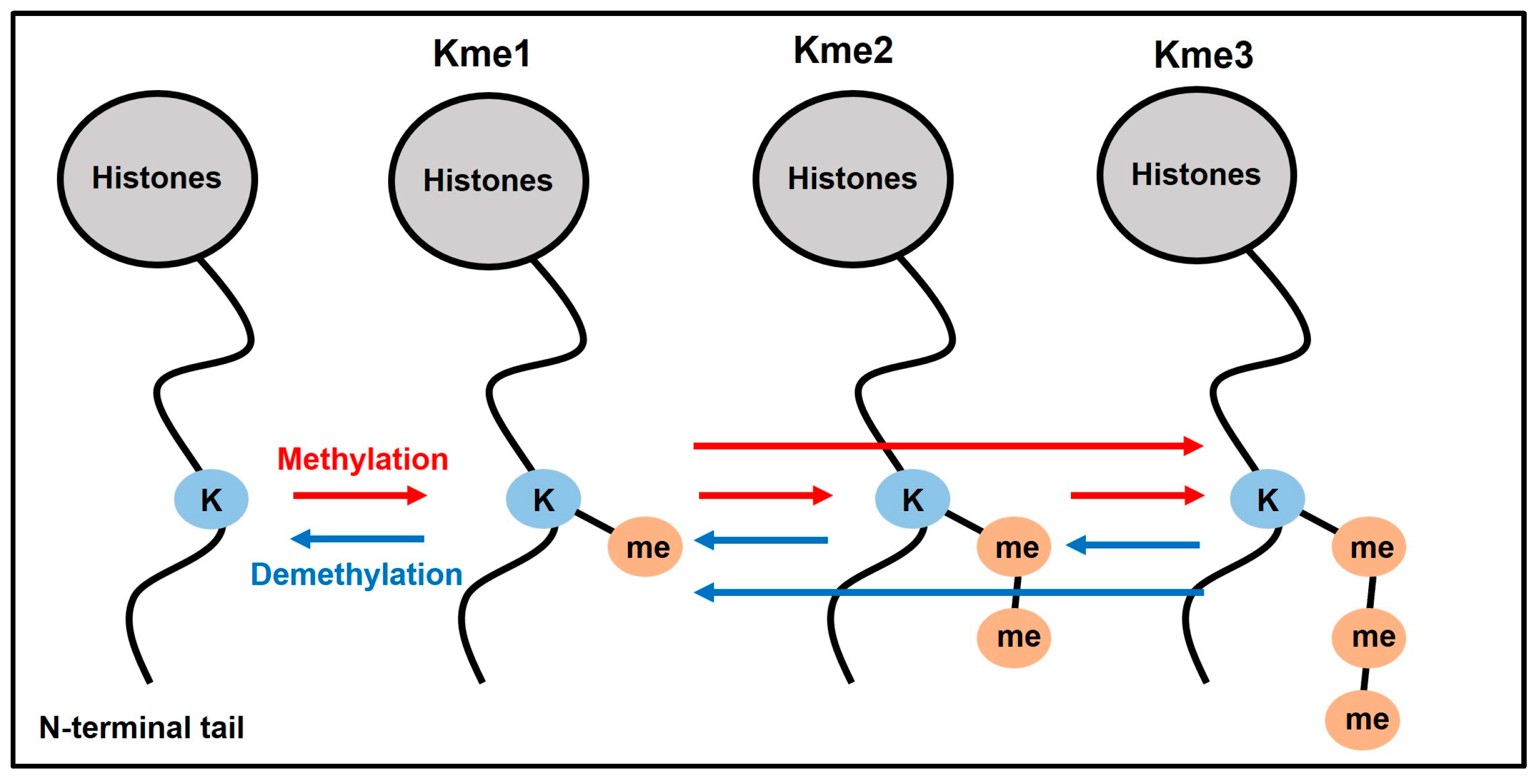
:1. Introduction
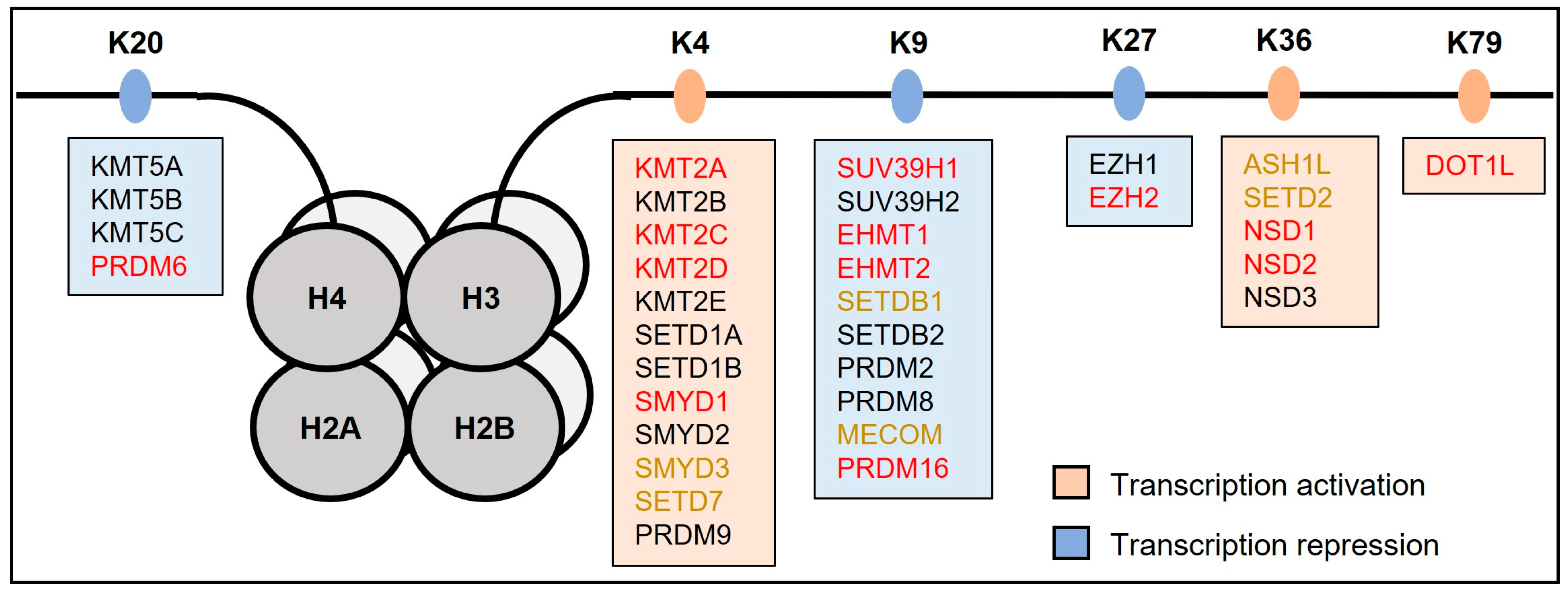
2. Lysine-Specific Methylation in Heart Development and Disease
2.1. H3K4 Methylation
2.2. H3K9 Methylation
2.3. H3K27 Methylation
2.4. H3K36 Methylation
2.5. Additional Methylation Marks

{kind=link}
{kind=link}
| KMT | Variant | Cardiac Phenotype | Reference(s) (Footnote 1) |
|---|---|---|---|
| KMT2A | N/A | Congenital heart disease; Structural abnormalities | Sheppard et al., 2021 [31] |
| KMT2D | 12 variants | Congenital heart defects; Kabuki syndrome | Van Laarhoven et al., 2015 [33] |
| 19 variants | Congenital heart defects; Kabuki syndrome | Digilio et al., 2017 [35] | |
| 1 variant | Congenital cardiovascular malformation; Left-sided lesions | Li et al., 2017 [36] | |
| L3542P; G3553V | Congenital heart disease | Cuvertino et al., 2020 [37] | |
| R2860H; T1710M; V1561G | Congenital heart disease; Heterotaxy | Liang et al., 2020 [38] | |
| 7 variants | Cardiac left-sided lesions; Hypoplastic left heart syndrome | Sun et al., 2020 [39] | |
| G3465Dfs * 37 | Congenital heart defect; Left heart hypoplasia | Luo et al., 2021 [40] | |
| KMT2C | N/A | Dilated cardiomyopathy | Jiang et al., 2017 [44] |
| Q4753L (footnote 2) | Congenital heart disease; Ventricular septal defect | Szot et al., 2018 [45] | |
| SMYD1 | S91S; S321S (footnote 3) | Hypertrophic cardiomyopathy | Abaci et al., 2010 [52] |
| EHMT1 | 10 variants | Heart defects, unspecified; Kleefstra syndrome | Willemsen et al., 2012 [68] |
| PRDM16 | R525Pfs * 79; K702 *; N816S | Left ventricular noncompaction; Deletion 1p36 syndrome | Arndt et al., 2013 [75] |
| E271K; P291L; L887P; V1101M | Dilated cardiomyopathy; Deletion 1p36 syndrome | Arndt et al., 2013 [75] | |
| S350fs * 48 | Dilated cardiomyopathy, pediatric | Long et al., 2017 [77] | |
| Q353 * | Cardiomyopathy; Left ventricular noncompaction | Delplancq et al., 2020 [76] | |
| ASH1L | L1346 * | Anomalous coronary branching; Single left coronary | Homsy et al., 2015 [99]; Jin et al., 2017 [25]; Ji et al., 2020 [100] |
| N/A | Congenital heart disease | Ji et al., 2020 [100] | |
| NSD1 | 7 variants | Congenital heart defect, unspecified/Heart conduction defect | Cecconi et al., 2005 [108] |
| A933P; R361S | Congenital heart disease; Atrioventricular septal defects | Priest et al., 2016 [110] | |
| NSD2 | N/A | Congenital heart disease | Ji et al., 2020 [100] |
| 10+ variants | Congenital heart defects; Wolf–Hirschhorn syndrome | Zanoni et al., 2021 [112] |
3. Future Perspective
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mansisidor, A.R.; Risca, V.I. Chromatin Accessibility: Methods, Mechanisms, and Biological Insights. Nucleus 2022, 13, 236–276. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, V.; Sarkar, S.; Tan, D. Histone Variants and Chromatin Structure, Update of Advances. Comput. Struct. Biotechnol. J. 2023, 21, 299–311. [Google Scholar] [CrossRef]
- Shirvaliloo, M. The Landscape of Histone Modifications in Epigenomics since 2020. Epigenomics 2022, 14, 1465–1477. [Google Scholar] [CrossRef]
- Del Rizzo, P.A.; Trievel, R.C. Substrate and Product Specificities of SET Domain Methyltransferases. Epigenetics 2011, 6, 1059–1067. [Google Scholar] [CrossRef]
- Husmann, D.; Gozani, O. Histone Lysine Methyltransferases in Biology and Disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baral, I.; Varghese, P.C.; Dutta, D. Epigenetics as “Conductor” in “Orchestra” of Pluripotent States. Cell Tissue Res. 2022, 390, 141–172. [Google Scholar] [CrossRef] [PubMed]
- Petrossian, T.C.; Clarke, S.G. Uncovering the Human Methyltransferasome. Mol. Cell. Proteom. 2011, 10, M110.000976. [Google Scholar] [CrossRef] [Green Version]
- Kooistra, S.M.; Helin, K. Molecular Mechanisms and Potential Functions of Histone Demethylases. Nat. Rev. Mol. Cell Biol. 2012, 13, 297–311. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [Green Version]
- Faundes, V.; Newman, W.G.; Bernardini, L.; Canham, N.; Clayton-Smith, J.; Dallapiccola, B.; Davies, S.J.; Demos, M.K.; Goldman, A.; Gill, H.; et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018, 102, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The Expanding Landscape of “oncohistone” Mutations in Human Cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Torres-Arciga, K.; Flores-León, M.; Ruiz-Pérez, S.; Trujillo-Pineda, M.; González-Barrios, R.; Herrera, L.A. Histones and Their Chaperones: Adaptive Remodelers of an Ever-Changing Chromatinic Landscape. Front. Genet. 2022, 13, 1057846. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-J.; Liu, Z.-P. Histone Methylations in Heart Development, Congenital and Adult Heart Diseases. Epigenomics 2015, 7, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szulik, M.W.; Davis, K.; Bakhtina, A.; Azarcon, P.; Bia, R.; Horiuchi, E.; Franklin, S. Transcriptional Regulation by Methyltransferases and Their Role in the Heart: Highlighting Novel Emerging Functionality. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H847–H865. [Google Scholar] [CrossRef]
- Davis, K.; Azarcon, P.; Hickenlooper, S.; Bia, R.; Horiuchi, E.; Szulik, M.W.; Franklin, S. The Role of Demethylases in Cardiac Development and Disease. J. Mol. Cell. Cardiol. 2021, 158, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.; Bi, Y.; Bu, P.; Zhang, M. The Histone Demethylase PHF8 Represses Cardiac Hypertrophy upon Pressure Overload. Exp. Cell Res. 2015, 335, 123–134. [Google Scholar] [CrossRef]
- Akerberg, A.A.; Henner, A.; Stewart, S.; Stankunas, K. Histone Demethylases Kdm6ba and Kdm6bb Redundantly Promote Cardiomyocyte Proliferation during Zebrafish Heart Ventricle Maturation. Dev. Biol. 2017, 426, 84–96. [Google Scholar] [CrossRef]
- El-Nachef, D.; Oyama, K.; Wu, Y.-Y.; Freeman, M.; Zhang, Y.; MacLellan, W.R. Repressive Histone Methylation Regulates Cardiac Myocyte Cell Cycle Exit. J. Mol. Cell. Cardiol. 2018, 121, 1–12. [Google Scholar] [CrossRef]
- Huo, J.-L.; Jiao, L.; An, Q.; Chen, X.; Qi, Y.; Wei, B.; Zheng, Y.; Shi, X.; Gao, E.; Liu, H.-M.; et al. Myofibroblast Deficiency of LSD1 Alleviates TAC-Induced Heart Failure. Circ. Res. 2021, 129, 400–413. [Google Scholar] [CrossRef]
- Yang, K.-C.; Yamada, K.A.; Patel, A.Y.; Topkara, V.K.; George, I.; Cheema, F.H.; Ewald, G.A.; Mann, D.L.; Nerbonne, J.M. Deep RNA Sequencing Reveals Dynamic Regulation of Myocardial Noncoding RNAs in Failing Human Heart and Remodeling with Mechanical Circulatory Support. Circulation 2014, 129, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Morley, M.; Brandimarto, J.; Hannenhalli, S.; Hu, Y.; Ashley, E.A.; Tang, W.H.W.; Moravec, C.S.; Margulies, K.B.; Cappola, T.P.; et al. RNA-Seq Identifies Novel Myocardial Gene Expression Signatures of Heart Failure. Genomics 2015, 105, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Sweet, M.E.; Cocciolo, A.; Slavov, D.; Jones, K.L.; Sweet, J.R.; Graw, S.L.; Reece, T.B.; Ambardekar, A.V.; Bristow, M.R.; Mestroni, L.; et al. Transcriptome Analysis of Human Heart Failure Reveals Dysregulated Cell Adhesion in Dilated Cardiomyopathy and Activated Immune Pathways in Ischemic Heart Failure. BMC Genom. 2018, 19, 812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, S.; Choi, M.; Wakimoto, H.; Ma, L.; Jiang, J.; Overton, J.D.; Romano-Adesman, A.; Bjornson, R.D.; Breitbart, R.E.; Brown, K.K.; et al. De Novo Mutations in Histone-Modifying Genes in Congenital Heart Disease. Nature 2013, 498, 220–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.C.; Homsy, J.; Zaidi, S.; Lu, Q.; Morton, S.; DePalma, S.R.; Zeng, X.; Qi, H.; Chang, W.; Sierant, M.C.; et al. Contribution of Rare Inherited and de Novo Variants in 2871 Congenital Heart Disease Probands. Nat. Genet. 2017, 49, 1593–1601. [Google Scholar] [CrossRef] [Green Version]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The SET-Domain Protein Superfamily: Protein Lysine Methyltransferases. Genome Biol. 2005, 6, 227. [Google Scholar] [CrossRef] [Green Version]
- Baumbusch, L.O.; Thorstensen, T.; Krauss, V.; Fischer, A.; Naumann, K.; Assalkhou, R.; Schulz, I.; Reuter, G.; Aalen, R.B. The Arabidopsis Thaliana Genome Contains at Least 29 Active Genes Encoding SET Domain Proteins That Can Be Assigned to Four Evolutionarily Conserved Classes. Nucleic Acids Res. 2001, 29, 4319–4333. [Google Scholar] [CrossRef]
- Alvarez-Venegas, R. Bacterial SET Domain Proteins and Their Role in Eukaryotic Chromatin Modification. Front. Genet. 2014, 5, 65. [Google Scholar] [CrossRef] [Green Version]
- Jones, W.D.; Dafou, D.; McEntagart, M.; Woollard, W.J.; Elmslie, F.V.; Holder-Espinasse, M.; Irving, M.; Saggar, A.K.; Smithson, S.; Trembath, R.C.; et al. De Novo Mutations in MLL Cause Wiedemann-Steiner Syndrome. Am. J. Hum. Genet. 2012, 91, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Miyake, N.; Tsurusaki, Y.; Koshimizu, E.; Okamoto, N.; Kosho, T.; Brown, N.J.; Tan, T.Y.; Yap, P.J.J.; Suzumura, H.; Tanaka, T.; et al. Delineation of Clinical Features in Wiedemann-Steiner Syndrome Caused by KMT2A Mutations. Clin. Genet. 2016, 89, 115–119. [Google Scholar] [CrossRef]
- Sheppard, S.E.; Campbell, I.M.; Harr, M.H.; Gold, N.; Li, D.; Bjornsson, H.T.; Cohen, J.S.; Fahrner, J.A.; Fatemi, A.; Harris, J.R.; et al. Expanding the Genotypic and Phenotypic Spectrum in a Diverse Cohort of 104 Individuals with Wiedemann-Steiner Syndrome. Am. J. Med. Genet. A 2021, 185, 1649–1665. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-Y.; Fu, Y.; Nettleton, M.; Richman, A.; Han, Z. High Throughput in Vivo Functional Validation of Candidate Congenital Heart Disease Genes in Drosophila. Elife 2017, 6, e22617. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaikh, T.H. Kabuki Syndrome Genes KMT2D and KDM6A: Functional Analyses Demonstrate Critical Roles in Craniofacial, Heart and Brain Development. Hum. Mol. Genet. 2015, 24, 4443–4453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, S.-Y.; Uebersohn, A.; Spencer, C.I.; Huang, Y.; Lee, J.-E.; Ge, K.; Bruneau, B.G. KMT2D Regulates Specific Programs in Heart Development via Histone H3 Lysine 4 Di-Methylation. Development 2016, 143, 810–821. [Google Scholar] [CrossRef] [Green Version]
- Digilio, M.C.; Gnazzo, M.; Lepri, F.; Dentici, M.L.; Pisaneschi, E.; Baban, A.; Passarelli, C.; Capolino, R.; Angioni, A.; Novelli, A.; et al. Congenital Heart Defects in Molecularly Proven Kabuki Syndrome Patients. Am. J. Med. Genet. A 2017, 173, 2912–2922. [Google Scholar] [CrossRef]
- Li, A.H.; Hanchard, N.A.; Furthner, D.; Fernbach, S.; Azamian, M.; Nicosia, A.; Rosenfeld, J.; Muzny, D.; D’Alessandro, L.C.A.; Morris, S.; et al. Whole Exome Sequencing in 342 Congenital Cardiac Left Sided Lesion Cases Reveals Extensive Genetic Heterogeneity and Complex Inheritance Patterns. Genome Med. 2017, 9, 95. [Google Scholar] [CrossRef]
- Cuvertino, S.; Hartill, V.; Colyer, A.; Garner, T.; Nair, N.; Al-Gazali, L.; Canham, N.; Faundes, V.; Flinter, F.; Hertecant, J.; et al. A Restricted Spectrum of Missense KMT2D Variants Cause a Multiple Malformations Disorder Distinct from Kabuki Syndrome. Genet. Med. 2020, 22, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Shi, X.; Yu, C.; Shao, X.; Zhou, H.; Li, X.; Chang, C.; Lai, K.S.; Ma, J.; Zhang, R. Identification of Novel Candidate Genes in Heterotaxy Syndrome Patients with Congenital Heart Diseases by Whole Exome Sequencing. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165906. [Google Scholar] [CrossRef]
- Sun, H.; Yi, T.; Hao, X.; Yan, H.; Wang, J.; Li, Q.; Gu, X.; Zhou, X.; Wang, S.; Wang, X.; et al. Contribution of Single-Gene Defects to Congenital Cardiac Left-Sided Lesions in the Prenatal Setting. Ultrasound Obstet. Gynecol. 2020, 56, 225–232. [Google Scholar] [CrossRef]
- Luo, S.; Chen, L.; Wei, W.; Tan, L.; Zhang, M.; Duan, Z.; Cao, J.; Zhou, Y.; Zhou, A.; He, X. Prenatal Genetic Diagnosis in Three Fetuses with Left Heart Hypoplasia (LHH) From Three Unrelated Families. Front. Cardiovasc. Med. 2021, 8, 631374. [Google Scholar] [CrossRef]
- Schwenty-Lara, J.; Nürnberger, A.; Borchers, A. Loss of Function of Kmt2d, a Gene Mutated in Kabuki Syndrome, Affects Heart Development in Xenopus Laevis. Dev. Dyn. 2019, 248, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.D.L.A.; Demarest, B.L.; Tone-Pah-Hote, T.; Tristani-Firouzi, M.; Yost, H.J. Inhibition of Notch Signaling Rescues Cardiovascular Development in Kabuki Syndrome. PLoS Biol. 2019, 17, e3000087. [Google Scholar] [CrossRef]
- Huang, W.; Zhu, J.-Y.; Fu, Y.; van de Leemput, J.; Han, Z. Lpt, Trr, and Hcf Regulate Histone Mono- and Dimethylation That Are Essential for Drosophila Heart Development. Dev. Biol. 2022, 490, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.-S.; Yi, X.; Li, R.; Su, Y.-S.; Wang, J.; Chen, M.-L.; Liu, L.-G.; Hu, M.; Cheng, C.; Zheng, P.; et al. The Histone Methyltransferase Mixed Lineage Leukemia (MLL) 3 May Play a Potential Role on Clinical Dilated Cardiomyopathy. Mol. Med. 2017, 23, 196–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szot, J.O.; Cuny, H.; Blue, G.M.; Humphreys, D.T.; Ip, E.; Harrison, K.; Sholler, G.F.; Giannoulatou, E.; Leo, P.; Duncan, E.L.; et al. A Screening Approach to Identify Clinically Actionable Variants Causing Congenital Heart Disease in Exome Data. Circ. Genom. Precis. Med. 2018, 11, e001978. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Borchers, C.; Tempst, P.; Zhang, Y. Purification and Functional Characterization of a Histone H3-Lysine 4-Specific Methyltransferase. Mol. Cell 2001, 8, 1207–1217. [Google Scholar] [CrossRef]
- Kim, J.-D.; Kim, E.; Koun, S.; Ham, H.-J.; Rhee, M.; Kim, M.-J.; Huh, T.-L. Proper Activity of Histone H3 Lysine 4 (H3K4) Methyltransferase Is Required for Morphogenesis during Zebrafish Cardiogenesis. Mol. Cells 2015, 38, 580–586. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Shao, N.-Y.; Paik, D.T.; Wu, H.; Guo, H.; Termglinchan, V.; Churko, J.M.; Kim, Y.; Kitani, T.; Zhao, M.-T.; et al. SETD7 Drives Cardiac Lineage Commitment through Stage-Specific Transcriptional Activation. Cell Stem Cell 2018, 22, 428–444.e5. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.-J.; Xu, P.-F.; Zhou, T.; Hu, M.; Fu, C.-T.; Zhang, Y.; Jin, Y.; Chen, Y.; Chen, S.-J.; Huang, Q.-H.; et al. Genome-Wide Survey and Developmental Expression Mapping of Zebrafish SET Domain-Containing Genes. PLoS ONE 2008, 3, e1499. [Google Scholar] [CrossRef]
- Fujii, T.; Tsunesumi, S.-I.; Yamaguchi, K.; Watanabe, S.; Furukawa, Y. Smyd3 Is Required for the Development of Cardiac and Skeletal Muscle in Zebrafish. PLoS ONE 2011, 6, e23491. [Google Scholar] [CrossRef] [Green Version]
- Borlak, J.; Thum, T. Hallmarks of Ion Channel Gene Expression in End-Stage Heart Failure. FASEB J. 2003, 17, 1592–1608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abaci, N.; Güleç, C.; Bayrak, F.; Kömürcü Bayrak, E.; Kahveci, G.; Erginel Unaltuna, N. The Variations of BOP Gene in Hypertrophic Cardiomyopathy. Anadolu Kardiyol. Derg. 2010, 10, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, P.D.; Pierce, S.A.; Sims, R.J.; Yamagishi, H.; Weihe, E.K.; Harriss, J.V.; Maika, S.D.; Kuziel, W.A.; King, H.L.; Olson, E.N.; et al. Bop Encodes a Muscle-Restricted Protein Containing MYND and SET Domains and Is Essential for Cardiac Differentiation and Morphogenesis. Nat. Genet. 2002, 31, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Rotllant, J.; Li, H.; De Deyne, P.; Du, S.J. SmyD1, a Histone Methyltransferase, Is Required for Myofibril Organization and Muscle Contraction in Zebrafish Embryos. Proc. Natl. Acad. Sci. USA 2006, 103, 2713–2718. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Han, L.; Liu, L.; He, F.; Chu, W.; Zhang, J.; Tian, Z.; Du, S. Defective Sarcomere Assembly in Smyd1a and Smyd1b Zebrafish Mutants. FASEB J. 2019, 33, 6209–6225. [Google Scholar] [CrossRef]
- Jiao, S.; Xu, R.; Du, S. Smyd1 Is Essential for Myosin Expression and Sarcomere Organization in Craniofacial, Extraocular, and Cardiac Muscles. J. Genet. Genom. 2021, 48, 208–218. [Google Scholar] [CrossRef]
- Rasmussen, T.L.; Ma, Y.; Park, C.Y.; Harriss, J.; Pierce, S.A.; Dekker, J.D.; Valenzuela, N.; Srivastava, D.; Schwartz, R.J.; Stewart, M.D.; et al. Smyd1 Facilitates Heart Development by Antagonizing Oxidative and ER Stress Responses. PLoS ONE 2015, 10, e0121765. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Schwartz, R.J.; Liu, J.; Sun, F.; Li, Q.; Ma, Y. Smyd1 Orchestrates Early Heart Development through Positive and Negative Gene Regulation. Front. Cell Dev. Biol. 2021, 9, 654682. [Google Scholar] [CrossRef]
- Franklin, S.; Kimball, T.; Rasmussen, T.L.; Rosa-Garrido, M.; Chen, H.; Tran, T.; Miller, M.R.; Gray, R.; Jiang, S.; Ren, S.; et al. The Chromatin-Binding Protein Smyd1 Restricts Adult Mammalian Heart Growth. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1234–H1247. [Google Scholar] [CrossRef] [Green Version]
- Warren, J.S.; Tracy, C.M.; Miller, M.R.; Makaju, A.; Szulik, M.W.; Oka, S.-I.; Yuzyuk, T.N.; Cox, J.E.; Kumar, A.; Lozier, B.K.; et al. Histone Methyltransferase Smyd1 Regulates Mitochondrial Energetics in the Heart. Proc. Natl. Acad. Sci. USA 2018, 115, E7871–E7880. [Google Scholar] [CrossRef] [Green Version]
- Paigen, K.; Petkov, P.M. PRDM9 and Its Role in Genetic Recombination. Trends Genet. 2018, 34, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Brown, M.A.; van Amerongen, M.J.; Novoyatleva, T.; Wietelmann, A.; Harriss, J.; Ferrazzi, F.; Böttger, T.; Harvey, R.P.; Tucker, P.W.; et al. Cardiac Deletion of Smyd2 Is Dispensable for Mouse Heart Development. PLoS ONE 2010, 5, e9748. [Google Scholar] [CrossRef] [Green Version]
- Ito, E.; Miyagawa, S.; Fukushima, S.; Yoshikawa, Y.; Saito, S.; Saito, T.; Harada, A.; Takeda, M.; Kashiyama, N.; Nakamura, Y.; et al. Histone Modification Is Correlated with Reverse Left Ventricular Remodeling in Nonischemic Dilated Cardiomyopathy. Ann. Thorac. Surg. 2017, 104, 1531–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, S.; Yang, S.; Du, P.; Gao, K.; Cao, Y.; Yao, B.; Guo, R.; Zhao, M. Regulatory Factor X1 Downregulation Contributes to Monocyte Chemoattractant Protein-1 Overexpression in CD14+ Monocytes via Epigenetic Mechanisms in Coronary Heart Disease. Front. Genet. 2019, 10, 1098. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Chi, X.; Zhang, X.; Feng, X.; Chu, W.; Zhang, S.; Wu, J.; Song, Y.; Zhang, Y.; Kong, W.; et al. Kindlin-2 Suppresses Transcription Factor GATA4 through Interaction with SUV39H1 to Attenuate Hypertrophy. Cell Death Dis. 2019, 10, 890. [Google Scholar] [CrossRef] [Green Version]
- Tao, Y.; Neppl, R.L.; Huang, Z.-P.; Chen, J.; Tang, R.-H.; Cao, R.; Zhang, Y.; Jin, S.-W.; Wang, D.-Z. The Histone Methyltransferase Set7/9 Promotes Myoblast Differentiation and Myofibril Assembly. J. Cell Biol. 2011, 194, 551–565. [Google Scholar] [CrossRef]
- Koemans, T.S.; Kleefstra, T.; Chubak, M.C.; Stone, M.H.; Reijnders, M.R.F.; de Munnik, S.; Willemsen, M.H.; Fenckova, M.; Stumpel, C.T.R.M.; Bok, L.A.; et al. Functional Convergence of Histone Methyltransferases EHMT1 and KMT2C Involved in Intellectual Disability and Autism Spectrum Disorder. PLoS Genet. 2017, 13, e1006864. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, M.H.; Vulto-van Silfhout, A.T.; Nillesen, W.M.; Wissink-Lindhout, W.M.; van Bokhoven, H.; Philip, N.; Berry-Kravis, E.M.; Kini, U.; van Ravenswaaij-Arts, C.M.A.; Delle Chiaie, B.; et al. Update on Kleefstra Syndrome. Mol. Syndromol. 2012, 2, 202–212. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Gu, X.; Li, H.; Xu, P.; Li, M.; Zhu, Y.; Zuo, Q.; Li, B. H3K9me2 Regulates Early Transcription Factors to Promote Mesenchymal Stem-cell Differentiation into Cardiomyocytes. Mol. Med. Rep. 2021, 24, 616. [Google Scholar] [CrossRef]
- Papait, R.; Serio, S.; Pagiatakis, C.; Rusconi, F.; Carullo, P.; Mazzola, M.; Salvarani, N.; Miragoli, M.; Condorelli, G. Histone Methyltransferase G9a Is Required for Cardiomyocyte Homeostasis and Hypertrophy. Circulation 2017, 136, 1233–1246. [Google Scholar] [CrossRef]
- Kaur, K.; Yang, J.; Edwards, J.G.; Eisenberg, C.A.; Eisenberg, L.M. G9a Histone Methyltransferase Inhibitor BIX01294 Promotes Expansion of Adult Cardiac Progenitor Cells without Changing Their Phenotype or Differentiation Potential. Cell Prolif. 2016, 49, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Mysliwiec, M.R.; Carlson, C.D.; Tietjen, J.; Hung, H.; Ansari, A.Z.; Lee, Y. Jarid2 (Jumonji, AT Rich Interactive Domain 2) Regulates NOTCH1 Expression via Histone Modification in the Developing Heart. J. Biol. Chem. 2012, 287, 1235–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyt, P.R.; Bartholomew, C.; Davis, A.J.; Yutzey, K.; Gamer, L.W.; Potter, S.S.; Ihle, J.N.; Mucenski, M.L. The Evi1 Proto-Oncogene Is Required at Midgestation for Neural, Heart, and Paraxial Mesenchyme Development. Mech. Dev. 1997, 65, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Bard-Chapeau, E.A.; Szumska, D.; Jacob, B.; Chua, B.Q.L.; Chatterjee, G.C.; Zhang, Y.; Ward, J.M.; Urun, F.; Kinameri, E.; Vincent, S.D.; et al. Mice Carrying a Hypomorphic Evi1 Allele Are Embryonic Viable but Exhibit Severe Congenital Heart Defects. PLoS ONE 2014, 9, e89397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, A.-K.; Schafer, S.; Drenckhahn, J.-D.; Sabeh, M.K.; Plovie, E.R.; Caliebe, A.; Klopocki, E.; Musso, G.; Werdich, A.A.; Kalwa, H.; et al. Fine Mapping of the 1p36 Deletion Syndrome Identifies Mutation of PRDM16 as a Cause of Cardiomyopathy. Am. J. Hum. Genet. 2013, 93, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Delplancq, G.; Tarris, G.; Vitobello, A.; Nambot, S.; Sorlin, A.; Philippe, C.; Carmignac, V.; Duffourd, Y.; Denis, C.; Eicher, J.C.; et al. Cardiomyopathy Due to PRDM16 Mutation: First Description of a Fetal Presentation, with Possible Modifier Genes. In American Journal of Medical Genetics Part C: Seminars in Medical Genetics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2020; Volume 184, pp. 129–135. [Google Scholar]
- Long, P.A.; Evans, J.M.; Olson, T.M. Diagnostic Yield of Whole Exome Sequencing in Pediatric Dilated Cardiomyopathy. J. Cardiovasc. Dev. Dis. 2017, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Cibi, D.M.; Bi-Lin, K.W.; Shekeran, S.G.; Sandireddy, R.; Tee, N.; Singh, A.; Wu, Y.; Srinivasan, D.K.; Kovalik, J.-P.; Ghosh, S.; et al. Prdm16 Deficiency Leads to Age-Dependent Cardiac Hypertrophy, Adverse Remodeling, Mitochondrial Dysfunction, and Heart Failure. Cell Rep. 2020, 33, 108288. [Google Scholar] [CrossRef]
- Nam, J.M.; Lim, J.E.; Ha, T.W.; Oh, B.; Kang, J.-O. Cardiac-Specific Inactivation of Prdm16 Effects Cardiac Conduction Abnormalities and Cardiomyopathy-Associated Phenotypes. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H764–H777. [Google Scholar] [CrossRef]
- Wu, T.; Liang, Z.; Zhang, Z.; Liu, C.; Zhang, L.; Gu, Y.; Peterson, K.L.; Evans, S.M.; Fu, X.-D.; Chen, J. PRDM16 Is a Compact Myocardium-Enriched Transcription Factor Required to Maintain Compact Myocardial Cardiomyocyte Identity in Left Ventricle. Circulation 2022, 145, 586–602. [Google Scholar] [CrossRef]
- Wells, Q.S.; Veatch, O.J.; Fessel, J.P.; Joon, A.Y.; Levinson, R.T.; Mosley, J.D.; Held, E.P.; Lindsay, C.S.; Shaffer, C.M.; Weeke, P.E.; et al. Genome-Wide Association and Pathway Analysis of Left Ventricular Function after Anthracycline Exposure in Adults. Pharmacogenetics Genom. 2017, 27, 247–254. [Google Scholar] [CrossRef]
- Yuan, J.-L.; Yin, C.-Y.; Li, Y.-Z.; Song, S.; Fang, G.-J.; Wang, Q.-S. EZH2 as an Epigenetic Regulator of Cardiovascular Development and Diseases. J. Cardiovasc. Pharmacol. 2021, 78, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Tschirner, A.; Palus, S.; Hetzer, R.; Meyer, R.; Anker, S.D.; Springer, J. Six1 Is Down-Regulated in End-Stage Human Dilated Cardiomyopathy Independently of Ezh2. ESC Heart Fail. 2014, 1, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Zhang, R.; Mo, B.; Chen, L.; Liu, L.; Yu, Y.; Cao, W.; Fang, G.; Wan, Y.; Gu, Y.; et al. EZH2 as a Novel Therapeutic Target for Atrial Fibrosis and Atrial Fibrillation. J. Mol. Cell. Cardiol. 2019, 135, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dai, C.; Lei, Y.; Wu, W.; Liu, W. Inhibition of EZH2 Attenuates Coronary Heart Disease by Interacting with MicroRNA-22 to Regulate the TXNIP/Nuclear Factor-ΚB Pathway. Exp. Physiol. 2020, 105, 2038–2050. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; You, T.; Lu, Y.; Lin, S.; Li, F.; Xu, H. Elevated EZH2 in Ischemic Heart Disease Epigenetically Mediates Suppression of NaV1.5 Expression. J. Mol. Cell. Cardiol. 2021, 153, 95–103. [Google Scholar] [CrossRef]
- Ai, S.; Yu, X.; Li, Y.; Peng, Y.; Li, C.; Yue, Y.; Tao, G.; Li, C.; Pu, W.T.; He, A. Divergent Requirements for EZH1 in Heart Development Versus Regeneration. Circ. Res. 2017, 121, 106–112. [Google Scholar] [CrossRef]
- He, A.; Ma, Q.; Cao, J.; von Gise, A.; Zhou, P.; Xie, H.; Zhang, B.; Hsing, M.; Christodoulou, D.C.; Cahan, P.; et al. Polycomb Repressive Complex 2 Regulates Normal Development of the Mouse Heart. Circ. Res. 2012, 110, 406–415. [Google Scholar] [CrossRef]
- Chen, L.; Ma, Y.; Kim, E.Y.; Yu, W.; Schwartz, R.J.; Qian, L.; Wang, J. Conditional Ablation of Ezh2 in Murine Hearts Reveals Its Essential Roles in Endocardial Cushion Formation, Cardiomyocyte Proliferation and Survival. PLoS ONE 2012, 7, e31005. [Google Scholar] [CrossRef] [Green Version]
- Leimeister, C.; Externbrink, A.; Klamt, B.; Gessler, M. Hey Genes: A Novel Subfamily of Hairy- and Enhancer of Split Related Genes Specifically Expressed during Mouse Embryogenesis. Mech. Dev. 1999, 85, 173–177. [Google Scholar] [CrossRef]
- Gessler, M.; Knobeloch, K.-P.; Helisch, A.; Amann, K.; Schumacher, N.; Rohde, E.; Fischer, A.; Leimeister, C. Mouse Gridlock: No Aortic Coarctation or Deficiency, but Fatal Cardiac Defects in Hey2 -/- Mice. Curr. Biol. 2002, 12, 1601–1604. [Google Scholar] [CrossRef] [Green Version]
- van Walree, E.S.; Dombrowsky, G.; Jansen, I.E.; Mirkov, M.U.; Zwart, R.; Ilgun, A.; Guo, D.; Clur, S.-A.B.; Amin, A.S.; Savage, J.E.; et al. Germline Variants in HEY2 Functional Domains Lead to Congenital Heart Defects and Thoracic Aortic Aneurysms. Genet. Med. 2021, 23, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Chen, J.; Lian, H.; Pei, J.; Li, Y.; Chen, X.; Song, S.; Xia, J.; Zhou, B.; Feng, J.; et al. PDGFR-β Signaling Regulates Cardiomyocyte Proliferation and Myocardial Regeneration. Cell Rep. 2019, 28, 966–978.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Z.; Yin, C.; Li, Y.; Tian, D.; Xiang, Y.; Li, Q.; Tang, Y.; Zhang, Y. Long Noncoding RNA NEAT1 Promotes Cardiac Fibrosis in Heart Failure through Increased Recruitment of EZH2 to the Smad7 Promoter Region. J. Transl. Med. 2022, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Olguín, P.; Huang, Y.; Li, X.; Christodoulou, D.; Seidman, C.E.; Seidman, J.G.; Tarakhovsky, A.; Bruneau, B.G. Epigenetic Repression of Cardiac Progenitor Gene Expression by Ezh2 Is Required for Postnatal Cardiac Homeostasis. Nat. Genet. 2012, 44, 343–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Katagiri, Z.-I.; Kawahashi, K.; Kioussis, D.; Kitajima, S. Trithorax-Group Protein ASH1 Methylates Histone H3 Lysine 36. Gene 2007, 397, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, Q.; Wong, S.H.K.; Huang, M.; Klein, B.J.; Shen, J.; Ikenouye, L.; Onishi, M.; Schneidawind, D.; Buechele, C.; et al. ASH1L Links Histone H3 Lysine 36 Dimethylation to MLL Leukemia. Cancer Discov. 2016, 6, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Yang, F.; Zhang, Z.; Zhang, J.; Cai, G.; Li, L.; Zheng, Y.; Chen, S.; Xi, R.; Zhu, B. Mrg15 Stimulates Ash1 H3K36 Methyltransferase Activity and Facilitates Ash1 Trithorax Group Protein Function in Drosophila. Nat. Commun. 2017, 8, 1649. [Google Scholar] [CrossRef] [Green Version]
- Homsy, J.; Zaidi, S.; Shen, Y.; Ware, J.S.; Samocha, K.E.; Karczewski, K.J.; DePalma, S.R.; McKean, D.; Wakimoto, H.; Gorham, J.; et al. De Novo Mutations in Congenital Heart Disease with Neurodevelopmental and Other Congenital Anomalies. Science 2015, 350, 1262–1266. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.; Ferdman, D.; Copel, J.; Scheinost, D.; Shabanova, V.; Brueckner, M.; Khokha, M.K.; Ment, L.R. De Novo Damaging Variants Associated with Congenital Heart Diseases Contribute to the Connectome. Sci. Rep. 2020, 10, 7046. [Google Scholar] [CrossRef]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic Histone H3 Methylation during Gene Induction: HYPB/Setd2 Mediates All H3K36 Trimethylation. EMBO J. 2008, 27, 406–420. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Chen, J.; Wang, H.; Tang, H.; Huang, L.; Wang, S.; Wang, X.; Fang, X.; Liu, J.; Li, L.; et al. Histone Lysine Methyltransferase SETD2 Regulates Coronary Vascular Development in Embryonic Mouse Hearts. Front. Cell Dev. Biol. 2021, 9, 651655. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Trojer, P.; Xu, C.-F.; Cheung, P.; Kuo, A.; Drury, W.J., 3rd; Qiao, Q.; Neubert, T.A.; Xu, R.-M.; Gozani, O.; et al. The Target of the NSD Family of Histone Lysine Methyltransferases Depends on the Nature of the Substrate. J. Biol. Chem. 2009, 284, 34283–34295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucio-Eterovic, A.K.; Singh, M.M.; Gardner, J.E.; Veerappan, C.S.; Rice, J.C.; Carpenter, P.B. Role for the Nuclear Receptor-Binding SET Domain Protein 1 (NSD1) Methyltransferase in Coordinating Lysine 36 Methylation at Histone 3 with RNA Polymerase II Function. Proc. Natl. Acad. Sci. USA. 2010, 107, 16952–16957. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.J.; Cheung, P.; Chen, K.; Zee, B.M.; Kioi, M.; Lauring, J.; Xi, Y.; Park, B.H.; Shi, X.; Garcia, B.A.; et al. NSD2 Links Dimethylation of Histone H3 at Lysine 36 to Oncogenic Programming. Mol. Cell 2011, 44, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, R.; Zhang, Z.; Xu, Z.; Ouyang, H.; Wang, L.; Ouyang, H.; Greenblatt, M.; Chen, X.; Zou, W. H3K36 Methyltransferase NSD1 Regulates Chondrocyte Differentiation for Skeletal Development and Fracture Repair. Bone Res. 2021, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, M.; Murakami, K.; Iino, H.; Tateishi, H.; Fujita, K.; Uchida, M. Congenital Heart Defects in Sotos Syndrome. Am. J. Med. Genet. 1999, 84, 172. [Google Scholar] [CrossRef]
- Cecconi, M.; Forzano, F.; Milani, D.; Cavani, S.; Baldo, C.; Selicorni, A.; Pantaleoni, C.; Silengo, M.; Ferrero, G.B.; Scarano, G.; et al. Mutation Analysis of the NSD1 Gene in a Group of 59 Patients with Congenital Overgrowth. Am. J. Med. Genet. A 2005, 134, 247–253. [Google Scholar] [CrossRef]
- Baujat, G.; Cormier-Daire, V. Sotos Syndrome. Orphanet J. Rare Dis. 2007, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Priest, J.R.; Osoegawa, K.; Mohammed, N.; Nanda, V.; Kundu, R.; Schultz, K.; Lammer, E.J.; Girirajan, S.; Scheetz, T.; Waggott, D.; et al. De Novo and Rare Variants at Multiple Loci Support the Oligogenic Origins of Atrioventricular Septal Heart Defects. PLoS Genet. 2016, 12, e1005963. [Google Scholar] [CrossRef] [Green Version]
- Bergemann, A.D.; Cole, F.; Hirschhorn, K. The Etiology of Wolf-Hirschhorn Syndrome. Trends Genet. 2005, 21, 188–195. [Google Scholar] [CrossRef]
- Zanoni, P.; Steindl, K.; Sengupta, D.; Joset, P.; Bahr, A.; Sticht, H.; Lang-Muritano, M.; van Ravenswaaij-Arts, C.M.A.; Shinawi, M.; Andrews, M.; et al. Loss-of-Function and Missense Variants in NSD2 Cause Decreased Methylation Activity and Are Associated with a Distinct Developmental Phenotype. Genet. Med. 2021, 23, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Nimura, K.; Ura, K.; Shiratori, H.; Ikawa, M.; Okabe, M.; Schwartz, R.J.; Kaneda, Y. A Histone H3 Lysine 36 Trimethyltransferase Links Nkx2-5 to Wolf-Hirschhorn Syndrome. Nature 2009, 460, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-L.; Zhu, R.-R.; Wu, X.; Xu, H.; Li, Y.-Y.; Xu, Q.-R.; Liu, S.; Huang, H.; Xu, X.; Wan, L.; et al. NSD2 Promotes Ventricular Remodelling Mediated by the Regulation of H3K36me2. J. Cell. Mol. Med. 2019, 23, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.T.; Xiao, B.; Neppl, R.L.; Kallin, E.M.; Li, J.; Chen, T.; Wang, D.-Z.; Xiao, X.; Zhang, Y. DOT1L Regulates Dystrophin Expression and Is Critical for Cardiac Function. Genes Dev. 2011, 25, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, P.; Kunderfranco, P.; Greco, C.; Guffanti, A.; Stirparo, G.G.; Rusconi, F.; Rizzi, R.; Di Pasquale, E.; Locatelli, S.L.; Latronico, M.V.G.; et al. DOT1L-Mediated H3K79me2 Modification Critically Regulates Gene Expression during Cardiomyocyte Differentiation. Cell Death Differ. 2016, 23, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Hong, K.-W.; Lim, J.E.; Kim, J.W.; Tabara, Y.; Ueshima, H.; Miki, T.; Matsuda, F.; Cho, Y.S.; Kim, Y.; Oh, B. Identification of Three Novel Genetic Variations Associated with Electrocardiographic Traits (QRS Duration and PR Interval) in East Asians. Hum. Mol. Genet. 2014, 23, 6659–6667. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Ferguson, J.E., 3rd; Wang, H.; Kelley, R.; Ren, R.; McDonough, H.; Meeker, J.; Charles, P.C.; Wang, H.; Patterson, C. PRDM6 Is Enriched in Vascular Precursors during Development and Inhibits Endothelial Cell Proliferation, Survival, and Differentiation. J. Mol. Cell. Cardiol. 2008, 44, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Li, N.; Gasque, V.; Mehta, S.; Ye, L.; Wu, Y.; Li, J.; Gewies, A.; Ruland, J.; Hirschi, K.K.; et al. Prdm6 Controls Heart Development by Regulating Neural Crest Cell Differentiation and Migration. JCI Insight 2022, 7, e156046. [Google Scholar] [CrossRef]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.E.; Ding, H.; Wylie, J.N.; Pico, A.R.; Capra, J.A.; et al. Dynamic and Coordinated Epigenetic Regulation of Developmental Transitions in the Cardiac Lineage. Cell 2012, 151, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Paige, S.L.; Thomas, S.; Stoick-Cooper, C.L.; Wang, H.; Maves, L.; Sandstrom, R.; Pabon, L.; Reinecke, H.; Pratt, G.; Keller, G.; et al. A Temporal Chromatin Signature in Human Embryonic Stem Cells Identifies Regulators of Cardiac Development. Cell 2012, 151, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L Inhibitor Pinometostat Reduces H3K79 Methylation and Has Modest Clinical Activity in Adult Acute Leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The Timeline of Epigenetic Drug Discovery: From Reality to Dreams. Clin. Epigenetics 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulati, N.; Béguelin, W.; Giulino-Roth, L. Enhancer of Zeste Homolog 2 (EZH2) Inhibitors. Leuk. Lymphoma 2018, 59, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.-Y.; Fu, Y.; Richman, A.; Zhao, Z.; Ray, P.E.; Han, Z. A Personalized Model of COQ2 Nephropathy Rescued by the Wild-Type COQ2 Allele or Dietary Coenzyme Q10 Supplementation. J. Am. Soc. Nephrol. 2017, 28, 2607–2617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.-Y.; Huang, X.; Fu, Y.; Wang, Y.; Zheng, P.; Liu, Y.; Han, Z. Pharmacological or Genetic Inhibition of Hypoxia Signaling Attenuates Oncogenic RAS-Induced Cancer Phenotypes. Dis. Model. Mech. 2021, 15, dmm048953. [Google Scholar] [CrossRef]
- Zhu, J.-Y.; Wang, G.; Huang, X.; Lee, H.; Lee, J.-G.; Yang, P.; van de Leemput, J.; Huang, W.; Kane, M.A.; Yang, P.; et al. SARS-CoV-2 Nsp6 Damages Drosophila Heart and Mouse Cardiomyocytes through MGA/MAX Complex-Mediated Increased Glycolysis. Commun. Biol. 2022, 5, 1039. [Google Scholar] [CrossRef]
- Zhu, J.-Y.; Lee, J.-G.; van de Leemput, J.; Lee, H.; Han, Z. Functional Analysis of SARS-CoV-2 Proteins in Drosophila Identifies Orf6-Induced Pathogenic Effects with Selinexor as an Effective Treatment. Cell Biosci. 2021, 11, 59. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, J.-y.; van de Leemput, J.; Han, Z. The Roles of Histone Lysine Methyltransferases in Heart Development and Disease. J. Cardiovasc. Dev. Dis. 2023, 10, 305. https://doi.org/10.3390/jcdd10070305
Zhu J-y, van de Leemput J, Han Z. The Roles of Histone Lysine Methyltransferases in Heart Development and Disease. Journal of Cardiovascular Development and Disease. 2023; 10(7):305. https://doi.org/10.3390/jcdd10070305
Chicago/Turabian StyleZhu, Jun-yi, Joyce van de Leemput, and Zhe Han. 2023. "The Roles of Histone Lysine Methyltransferases in Heart Development and Disease" Journal of Cardiovascular Development and Disease 10, no. 7: 305. https://doi.org/10.3390/jcdd10070305
APA StyleZhu, J. -y., van de Leemput, J., & Han, Z. (2023). The Roles of Histone Lysine Methyltransferases in Heart Development and Disease. Journal of Cardiovascular Development and Disease, 10(7), 305. https://doi.org/10.3390/jcdd10070305