
Transcriptomic Crosstalk between Fungal Invasive Pathogens and Their Host Cells: Opportunities and Challenges for Next-Generation Sequencing Methods

 ,
,  ,
, 

Abstract
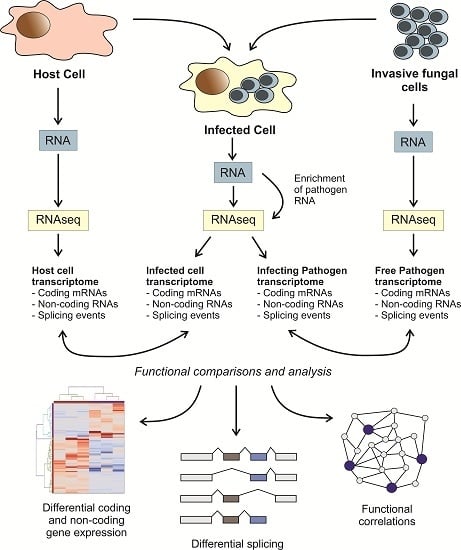
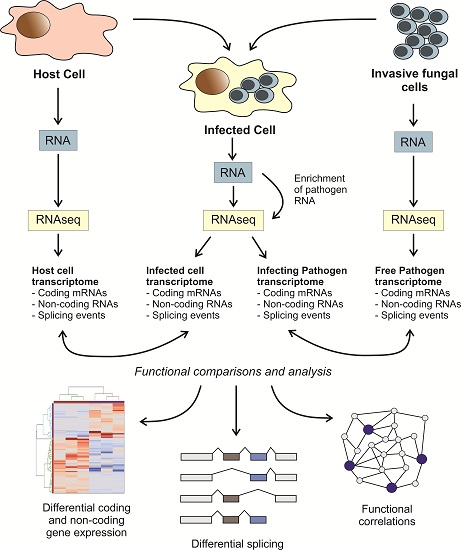
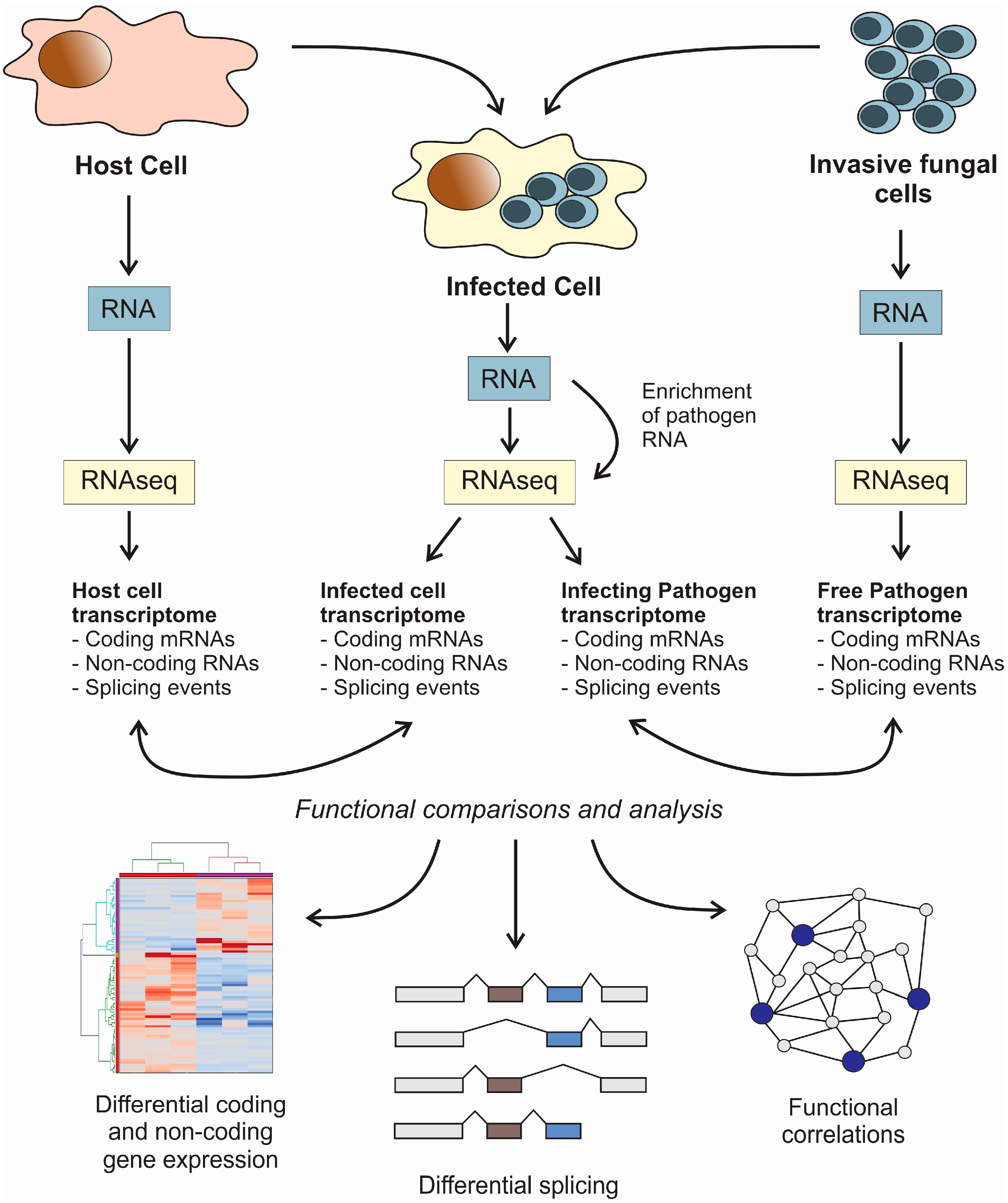
:
1. Introduction
2. Expression of Virulence Factors and Transcriptional Landscape of Infection in Fungal Pathogens
2.1. Invasive Fungi Causing Infections in Humans

{kind=link}
{kind=link}
| Database | Access Code | Subject | NGS Platform | Reference |
|---|---|---|---|---|
| GEO | GSE55663 | Transcriptomic analysis of antifungal activity by humidimycin over Aspergillus fumigatus | Illumina HiSeq 2000 | [20] |
| GEO | GSE32049 GSE32228 | Transcriptomic analysis of capsule regulation in Cryptococcus neoformans strains | Illumina Genome Analyzer IIx and Illumina HiSeq 2000 | [21] |
| GEO | GSE40425 | Transcriptomic analysis of the response of Tricophyton rubrun to acriflavine | AB SOLiD 4 System | [22] |
| GEO | GSE43189 | Cryptococcus neoformans gene expression associated with cell wall remodeling and evasion of the immune system | Illumina Genome Analyzer II | [23] |
| GEO | GSE43363 | RNAi-mediated genomic defense in Cryptococcus neoformans | Illumina Genome Analyzer II | [24] |
| GEO | GSE51573 | Cryptococcus neoformans transcriptome analysis at the site of human meningitis | Illumina HiSeq 2000 | [25] |
| GEO | GSE56091 | Characterization of transcriptome dynamics of Candida albicans in response to contact with host cells | Illumina HiSeq 2000 | [26] |
| GEO | GSE57217 | Cross talk between the cell wall integrity and cAMP/protein kinase A pathways in Cryptococcus neoformans | Illumina HiSeq 2000 | [9] |
| GEO | GSE60398 | Virulence regulation in Cryptococcus neoformans | Illumina HiSeq 2000 and Illumina Hiseq 2500 | [15] |
| GEO | GSE61550 | Epigenetic regulation of virulence and genomic specificity in Cryptococcus neoformans | Illumina HiSeq 2500 | [27] |
| GEO | GSE67688 | Transcriptomic analysis of vulvovaginal candidiasis in mouse | Illumina HiSeq 2000 | [18] |
| GEO | GSE70227 | Aspergillus fumigatus in blood infections | Illumina HiSeq 2000 | [28] |
| SRA | SRP055976 | Transcriptomic analysis of Pseudogymnoascus destructans infection in bats | Illumina HiSeq 2500 | [29] |
| SRA | SRP058281 | Candida albicans transcriptome during infection of mouse kidneys and Galleria mellonella | Illumina HiSeq 2500 | [30] |
| SRA | SRP028588 | Histoplasma capsulatum yeast and mycelia transcriptomes | Illumina Genome Analyzer II | [31] |
2.2. Plant Pathogens
| Database | Access Code | Subject | NGS Platform | Reference |
|---|---|---|---|---|
| GEO | GSE32010 | Genome-wide analysis of rice transcriptional change during the early stages of false smut formation caused by Ustilaginoidea virens | Illumina HiSeq 2000 | [49] |
| GEO | GSE40952 | Digital gene expression analysis of early root infection of Sporisorium reilianum f.sp. zeae in maize | Illumina Genome Analyzer | [50] |
| GEO | GSE57857 | Small RNA-seq identification and characterization the microRNA response from chickpea against Fusarium oxysporum f.sp. ciceris infection | Illumina Genome Analyzer IIx | [51] |
| GEO | GSE67191 | Comparative transcriptomics of Central Asian Vitis vinifera to characterize the distinct defense strategies against powdery mildew | Illumina HiSeq 2500 | [52] |
| GEO | GSE57587 GSE57586 | Transcriptional analysis of Botrytis cinerea targeting cells from grape and tomato | Illumina HiSeq 2000 | [53] |
| GEO | GSE58653 | Transcriptome analysis of fungal pathogen Neofusicoccum parvum in grapevine leaves | Illumina HiSeq 2500 | [54] |
| GEO | GSE58958 | Genomic structural variation in the grape mildew pathogen Erysiphe necator | Illumina HiSeq 2500 | [55] |
| GEO | GSE40581 | Transcriptome analysis of several pathogenic strains of Fusarium oxysporum | Illumina HiSeq 2000 | [46] |
| GEO | GSE51597 | Transcriptomic analysis of hypoxia-responsive genes in Magnaporthe oryzae | Illumina HiSeq 2000 | [48] |
| SRA | SRP035525 | Transcriptional analysis of Brassica napus infected by two Leptosphaeria species | Illumina HiSeq 2000 | [56] |
| SRA | SRP015912 | Transcriptome analysis of Lactuca sativa cv. Salinas after inoculation with Botrytis cinerea | Illumina HiSeq 2000 | [57] |
| SRA | SRP052276 | Response of Arabidopsis thaliana to Fusarium oxysporum | Illumina HiSeq 2000 | [58] |
3. Transcriptional Programs of the Host Cells in Response to a Fungal Infection
3.1. Human and Mammalian Host Cells
3.2. Plant Host Cells
3.3. The Emerging New Roles of Non-Coding RNAs in Fungal Infections
4. High-Throughput Transcriptomic Methods for the Analysis of the Molecular Mechanisms Involved in the Interaction between Fungi and Host Cells

5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Tortorano, A.M.; Dho, G.; Prigitano, A.; Breda, G.; Grancini, A.; Emmi, V.; Cavanna, C.; Marino, G.; Morero, S.; Ossi, C.; et al. Invasive fungal infections in the intensive care unit: A multicentre, prospective, observational study in Italy (2006–2008). Mycoses 2012, 55, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Tuite, N.L.; Lacey, K. Overview of invasive fungal infections. Methods Mol. Biol. 2013, 968, 1–23. [Google Scholar] [PubMed]
- Thornton, C.R.; Wills, O.E. Immunodetection of fungal and oomycete pathogens: Established and emerging threats to human health, animal welfare and global food security. Crit. Rev. Microbiol. 2015, 41, 27–51. [Google Scholar] [CrossRef] [PubMed]
- Mendes-Giannini, M.J.; Taylor, M.L.; Bouchara, J.B.; Burger, E.; Calich, V.L.; Escalante, E.D.; Hanna, S.A.; Lenzi, H.L.; Machado, M.P.; Miyaji, M.; et al. Pathogenesis II: Fungal responses to host responses: Interaction of host cells with fungi. Med. Mycol. 2000, 38, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Romani, L. Immunity to fungal infections. Nat. Rev. Immunol. 2011, 11, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Safdar, A. Antifungal immunity and adjuvant cytokine immune enhancement in cancer patients with invasive fungal infections. Clin. Microbiol. Infect. 2007, 13, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A. Antibody-mediated protection against intracellular pathogens. Trends Microbiol. 1998, 6, 102–107. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L.A. Host-pathogen interactions: Redefining the basic concepts of virulence and pathogenicity. Infect. Immun. 1999, 67, 3703–3713. [Google Scholar] [PubMed]
- Donlin, M.J.; Upadhya, R.; Gerik, K.J.; Lam, W.; VanArendonk, L.G.; Specht, C.A.; Sharma, N.K.; Lodge, J.K. Cross talk between the cell wall integrity and cyclic AMP/protein kinase A pathways in Cryptococcus neoformans. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Kriengkauykiat, J.; Ito, J.I.; Dadwal, S.S. Epidemiology and treatment approaches in management of invasive fungal infections. Clin. Epidemiol. 2011, 3, 175–191. [Google Scholar] [PubMed]
- Nucci, M.; Marr, K.A. Emerging fungal diseases. Clin. Infect. Dis. 2005, 41, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Ajesh, K.; Sreejith, K. Peptide antibiotics: An alternative and effective antimicrobial strategy to circumvent fungal infections. Peptides 2009, 30, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Gullo, F.P.; Rossi, S.A.; Sardi Jde, C.; Teodoro, V.L.; Mendes-Giannini, M.J.; Fusco-Almeida, A.M. Cryptococcosis: Epidemiology, fungal resistance, and new alternatives for treatment. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.L. Epidemiology and treatment of hematogenous candidiasis: A Brazilian perspective. Braz. J. Infect. Dis. 2000, 4, 113–118. [Google Scholar] [PubMed]
- Maier, E.J.; Haynes, B.C.; Gish, S.R.; Wang, Z.A.; Skowyra, M.L.; Marulli, A.L.; Doering, T.L.; Brent, M.R. Model-driven mapping of transcriptional networks reveals the circuitry and dynamics of virulence regulation. Genome Res. 2015, 25, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.; Minuzzi, F.; Bignell, E. The host-infecting fungal transcriptome. FEMS Microbiol. Lett. 2010, 307, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wartenberg, A.; Linde, J.; Martin, R.; Schreiner, M.; Horn, F.; Jacobsen, I.D.; Jenull, S.; Wolf, T.; Kuchler, K.; Guthke, R.; et al. Microevolution of Candida albicans in macrophages restores filamentation in a nonfilamentous mutant. PLoS Genet. 2014, 10, e1004824. [Google Scholar] [CrossRef] [PubMed]
- Bruno, V.M.; Shetty, A.C.; Yano, J.; Fidel, P.L., Jr.; Noverr, M.C.; Peters, B.M. Transcriptomic analysis of vulvovaginal candidiasis identifies a role for the NLRP3 inflammasome. MBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Clancy, C.J.; Xu, W.; Schneider, F.; Hao, B.; Mitchell, A.P.; Nguyen, M.H. Profiling of Candida albicans gene expression during intra-abdominal candidiasis identifies biologic processes involved in pathogenesis. J. Infect. Dis. 2013, 208, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Valiante, V.; Monteiro, M.C.; Martin, J.; Altwasser, R.; El Aouad, N.; Gonzalez, I.; Kniemeyer, O.; Mellado, E.; Palomo, S.; de Pedro, N.; et al. Hitting the caspofungin salvage pathway of human-pathogenic fungi with the novel lasso peptide humidimycin (MDN-0010). Antimicrob. Agents Chemother. 2015, 59, 5145–5153. [Google Scholar] [CrossRef] [PubMed]
- Haynes, B.C.; Skowyra, M.L.; Spencer, S.J.; Gish, S.R.; Williams, M.; Held, E.P.; Brent, M.R.; Doering, T.L. Toward an integrated model of capsule regulation in Cryptococcus neoformans. PLoS Pathog. 2011, 7, e1002411. [Google Scholar] [CrossRef] [PubMed]
- Persinoti, G.F.; de Aguiar Peres, N.T.; Jacob, T.R.; Rossi, A.; Vencio, R.Z.; Martinez-Rossi, N.M. RNA-sequencing analysis of Trichophyton rubrum transcriptome in response to sublethal doses of acriflavine. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [Green Version]
- O’Meara, T.R.; Holmer, S.M.; Selvig, K.; Dietrich, F.; Alspaugh, J.A. Cryptococcus neoformans Rim101 is associated with cell wall remodeling and evasion of the host immune responses. MBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, P.A.; Natarajan, P.; Chen, C.; Drinnenberg, I.A.; Schiller, B.J.; Thompson, J.; Moresco, J.J.; Yates, J.R., 3rd; Bartel, D.P.; Madhani, H.D. Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell 2013, 152, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Toffaletti, D.L.; Tenor, J.L.; Litvintseva, A.P.; Fang, C.; Mitchell, T.G.; McDonald, T.R.; Nielsen, K.; Boulware, D.R.; Bicanic, T.; et al. The Cryptococcus neoformans transcriptome at the site of human meningitis. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shetty, A.C.; Schwartz, J.A.; Bradford, L.L.; Xu, W.; Phan, Q.T.; Kumari, P.; Mahurkar, A.; Mitchell, A.P.; Ravel, J.; et al. New signaling pathways govern the host response to C. albicans infection in various niches. Genome Res. 2015, 25, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, P.A.; Homer, C.M.; Moresco, J.J.; Pack, L.R.; Shanle, E.K.; Coyle, S.M.; Strahl, B.D.; Fujimori, D.G.; Yates, J.R., 3rd; Madhani, H.D. Product binding enforces the genomic specificity of a yeast polycomb repressive complex. Cell 2015, 160, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Irmer, H.; Tarazona, S.; Sasse, C.; Olbermann, P.; Loeffler, J.; Krappmann, S.; Conesa, A.; Braus, G.H. RNAseq analysis of Aspergillus fumigatus in blood reveals a just wait and see resting stage behavior. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Field, K.A.; Johnson, J.S.; Lilley, T.M.; Reeder, S.M.; Rogers, E.J.; Behr, M.J.; Reeder, D.M. The white-nose syndrome transcriptome: Activation of anti-fungal host responses in wing tissue of hibernating little brown myotis. PLoS Pathog. 2015, 11, e1005168. [Google Scholar] [CrossRef] [PubMed]
- Amorim-Vaz, S.; Tran Vdu, T.; Pradervand, S.; Pagni, M.; Coste, A.T.; Sanglard, D. RNA enrichment method for quantitative transcriptional analysis of pathogens in vivo applied to the fungus Candida albicans. MBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.A.; Chen, C.; Kemski, M.M.; Hu, J.; Mitchell, T.K.; Rappleye, C.A. Histoplasma yeast and mycelial transcriptomes reveal pathogenic-phase and lineage-specific gene expression profiles. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Velagapudi, R.; Hsueh, Y.P.; Geunes-Boyer, S.; Wright, J.R.; Heitman, J. Spores as infectious propagules of Cryptococcus neoformans. Infect. Immun. 2009, 77, 4345–4355. [Google Scholar] [CrossRef] [PubMed]
- Brizendine, K.D.; Baddley, J.W.; Pappas, P.G. Predictors of mortality and differences in clinical features among patients with Cryptococcosis according to immune status. PLoS ONE 2013, 8, e60431. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A. Cryptococci at the brain gate: break and enter or use a Trojan horse? J. Clin. Investig. 2010, 120, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Vu, K.; Tham, R.; Uhrig, J.P.; Thompson, G.R., 3rd; Na Pombejra, S.; Jamklang, M.; Bautos, J.M.; Gelli, A. Invasion of the central nervous system by Cryptococcus neoformans requires a secreted fungal metalloprotease. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Deepe, G.S., Jr.; Gibbons, R.S. Interleukins 17 and 23 influence the host response to Histoplasma capsulatum. J. Infect. Dis. 2009, 200, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Kroetz, D.N.; Deepe, G.S. The role of cytokines and chemokines in Histoplasma capsulatum infection. Cytokine 2012, 58, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Adenis, A.; Nacher, M.; Hanf, M.; Vantilcke, V.; Boukhari, R.; Blachet, D.; Demar, M.; Aznar, C.; Carme, B.; Couppie, P. HIV-associated histoplasmosis early mortality and incidence trends: From neglect to priority. PLoS Negl. Trop. Dis. 2014, 8, e3100. [Google Scholar] [CrossRef] [PubMed]
- Bonifaz, A.; Vazquez-Gonzalez, D.; Perusquia-Ortiz, A.M. Endemic systemic mycoses: Coccidioidomycosis, histoplasmosis, paracoccidioidomycosis and blastomycosis. J. Dtsch. Dermatol. Ges. 2011, 9, 705–714. [Google Scholar] [CrossRef] [PubMed]
- DuBois, J.C.; Pasula, R.; Dade, J.E.; Smulian, A.G. Yeast transcriptome and in vivo hypoxia detection reveals Histoplasma capsulatum response to low oxygen tension. Med. Mycol. 2015, 54, 40–58. [Google Scholar] [PubMed]
- Gilmore, S.A.; Voorhies, M.; Gebhart, D.; Sil, A. Genome-wide reprogramming of transcript architecture by Temperature specifies the developmental states of the human pathogen Histoplasma. PLoS Genet. 2015, 11, e1005395. [Google Scholar] [CrossRef] [PubMed]
- Tavares, A.H.; Fernandes, L.; Bocca, A.L.; Silva-Pereira, I.; Felipe, M.S. Transcriptomic reprogramming of genus Paracoccidioides in dimorphism and host niches. Fungal Genet. Biol. 2015, 81, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Lima Pde, S.; Casaletti, L.; Bailao, A.M.; de Vasconcelos, A.T.; Fernandes Gda, R.; Soares, C.M. Transcriptional and proteomic responses to carbon starvation in Paracoccidioides. PLoS Negl. Trop. Dis. 2014, 8, e2855. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.C.; Studholme, D.J.; Talbot, N.J.; Haynes, K. New and improved techniques for the study of pathogenic fungi. Trends Microbiol. 2015, 24, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Kellner, N.; Heimel, K.; Obhof, T.; Finkernagel, F.; Kamper, J. The SPF27 homologue Num1 connects splicing and kinesin 1-dependent cytoplasmic trafficking in Ustilago maydis. PLoS Genet. 2014, 10, e1004046. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Han, L.; Yang, L.; Zeng, H.; Fan, D.; Zhu, Y.; Feng, Y.; Wang, G.; Peng, C.; Jiang, X.; et al. Genome and transcriptome analysis of the fungal pathogen Fusarium oxysporum f. sp. cubense causing banana vascular wilt disease. PLoS ONE 2014, 9, e95543. [Google Scholar] [PubMed]
- Kawahara, Y.; Oono, Y.; Kanamori, H.; Matsumoto, T.; Itoh, T.; Minami, E. Simultaneous RNA-seq analysis of a mixed transcriptome of rice and blast fungus interaction. PLoS ONE 2012, 7, e49423. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Chung, H.; Lee, G.W.; Koh, S.K.; Chae, S.K.; Lee, Y.H. Genome-wide analysis of hypoxia-responsive genes in the rice blast fungus, Magnaporthe oryzae. PLoS ONE 2015, 10, e0134939. [Google Scholar] [CrossRef] [PubMed]
- Chao, J.; Jin, J.; Wang, D.; Han, R.; Zhu, R.; Zhu, Y.; Li, S. Cytological and transcriptional dynamics analysis of host plant revealed stage-specific biological processes related to compatible rice-Ustilaginoidea virens interaction. PLoS ONE 2014, 9, e91391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Xiao, Y.; Zhao, J.; Wang, F.; Zheng, Y. Digital gene expression analysis of early root infection resistance to Sporisorium reilianum f. sp. zeae in maize. Mol. Genet. Genom. 2013, 288, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Kohli, D.; Joshi, G.; Deokar, A.A.; Bhardwaj, A.R.; Agarwal, M.; Katiyar-Agarwal, S.; Srinivasan, R.; Jain, P.K. Identification and characterization of Wilt and salt stress-responsive microRNAs in chickpea through high-throughput sequencing. PLoS ONE 2014, 9, e108851. [Google Scholar] [CrossRef] [PubMed]
- Amrine, K.C.; Blanco-Ulate, B.; Riaz, S.; Pap, D.; Jones, L.; Figueroa-Balderas, R.; Walker, M.A.; Cantu, D. Comparative transcriptomics of Central Asian Vitis vinifera accessions reveals distinct defense strategies against powdery mildew. Hortic. Res. 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Ulate, B.; Morales-Cruz, A.; Amrine, K.C.; Labavitch, J.M.; Powell, A.L.; Cantu, D. Genome-wide transcriptional profiling of Botrytis cinerea genes targeting plant cell walls during infections of different hosts. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Czemmel, S.; Galarneau, E.R.; Travadon, R.; McElrone, A.J.; Cramer, G.R.; Baumgartner, K. Genes expressed in grapevine leaves reveal latent wood infection by the fungal pathogen Neofusicoccum parvum. PLoS ONE 2015, 10, e0121828. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.; Riaz, S.; Morales-Cruz, A.; Amrine, K.C.; McGuire, B.; Gubler, W.D.; Walker, M.A.; Cantu, D. Adaptive genomic structural variation in the grape powdery mildew pathogen, Erysiphe necator. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.G.; Cassin, A.; Grandaubert, J.; Clark, B.L.; van de Wouw, A.P.; Rouxel, T.; Howlett, B.J. Genomes and transcriptomes of partners in plant-fungal-interactions between canola (Brassica napus) and two Leptosphaeria species. PLoS ONE 2014, 9, e103098. [Google Scholar] [PubMed]
- De Cremer, K.; Mathys, J.; Vos, C.; Froenicke, L.; Michelmore, R.W.; Cammue, B.P.; de Coninck, B. RNAseq-based transcriptome analysis of Lactuca sativa infected by the fungal necrotroph Botrytis cinerea. Plant Cell Environ. 2013, 36, 1992–2007. [Google Scholar] [PubMed]
- Lyons, R.; Rusu, A.; Stiller, J.; Powell, J.; Manners, J.M.; Kazan, K. Investigating the association between flowering time and defense in the Arabidopsis thaliana-Fusarium oxysporum interaction. PLoS ONE 2015, 10, e0127699. [Google Scholar] [CrossRef] [PubMed]
- Baxt, L.A.; Garza-Mayers, A.C.; Goldberg, M.B. Bacterial subversion of host innate immune pathways. Science 2013, 340, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Chaussabel, D.; Semnani, R.T.; McDowell, M.A.; Sacks, D.; Sher, A.; Nutman, T.B. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood 2003, 102, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Afonso-Grunz, F.; Hoffmeier, K.; Muller, S.; Westermann, A.J.; Rotter, B.; Vogel, J.; Winter, P.; Kahl, G. Dual 3’Seq using deepSuperSAGE uncovers transcriptomes of interacting Salmonella enterica Typhimurium and human host cells. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Rienksma, R.A.; Suarez-Diez, M.; Mollenkopf, H.J.; Dolganov, G.M.; Dorhoi, A.; Schoolnik, G.K.; Martins Dos Santos, V.A.; Kaufmann, S.H.; Schaap, P.J.; Gengenbacher, M. Comprehensive insights into transcriptional adaptation of intracellular mycobacteria by microbe-enriched dual RNA sequencing. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Oosthuizen, J.L.; Gomez, P.; Ruan, J.; Hackett, T.L.; Moore, M.M.; Knight, D.A.; Tebbutt, S.J. Dual organism transcriptomics of airway epithelial cells interacting with conidia of Aspergillus fumigatus. PLoS ONE 2011, 6, e20527. [Google Scholar] [CrossRef] [PubMed]
- Tierney, L.; Linde, J.; Muller, S.; Brunke, S.; Molina, J.C.; Hube, B.; Schock, U.; Guthke, R.; Kuchler, K. An interspecies regulatory network inferred from simultaneous RNA-seq of Candida albicans invading innate immune cells. Front. Microbiol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Agudelo-Romero, P.; Erban, A.; Rego, C.; Carbonell-Bejerano, P.; Nascimento, T.; Sousa, L.; Martinez-Zapater, J.M.; Kopka, J.; Fortes, A.M. Transcriptome and metabolome reprogramming in Vitis vinifera cv. Trincadeira berries upon infection with Botrytis cinerea. J. Exp. Bot. 2015, 66, 1769–1785. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Tu, Z.J.; Millett, B.P.; Bradeen, J.M. Insights into organ-specific pathogen defense responses in plants: RNA-seq analysis of potato tuber-Phytophthora infestans interactions. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Zemp, N.; Tavares, R.; Widmer, A. Fungal infection induces sex-specific transcriptional Changes and alters sexual dimorphism in the dioecious plant Silene latifolia. PLoS Genet. 2015, 11, e1005536. [Google Scholar] [CrossRef] [PubMed]
- Das Gupta, M.; Fliesser, M.; Springer, J.; Breitschopf, T.; Schlossnagel, H.; Schmitt, A.L.; Kurzai, O.; Hunniger, K.; Einsele, H.; Loffler, J. Aspergillus fumigatus induces microRNA-132 in human monocytes and dendritic cells. Int. J. Med. Microbiol. 2014, 304, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Halder, P.; Sahu, S.K.; Kumar, M.; Kumari, M.; Jana, K.; Ghosh, Z.; Sharma, P.; Kundu, M.; Basu, J. Identification of a novel role of ESAT-6-dependent miR-155 induction during infection of macrophages with Mycobacterium tuberculosis. Cell Microbiol. 2012, 14, 1620–1631. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol. 2010, 28, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, G.; Deng, X.; Yu, Q.; Hu, Y.; Sun, H.; Wang, Z.; Chen, H.; Jia, C.; Wang, D. Analysis of miRNA expression profiling in human macrophages responding to Mycobacterium infection: Induction of the immune regulator miR-146a. J. Infect. 2014, 68, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Pope, S.M.; Lasser, C. Toxoplasma gondii infection of fibroblasts causes the production of exosome-like vesicles containing a unique array of mRNA and miRNA transcripts compared to serum starvation. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef]
- Schnitger, A.K.; Machova, A.; Mueller, R.U.; Androulidaki, A.; Schermer, B.; Pasparakis, M.; Kronke, M.; Papadopoulou, N. Listeria monocytogenes infection in macrophages induces vacuolar-dependent host miRNA response. PLoS ONE 2011, 6, e27435. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Nunez, R.T.; Louafi, F.; Friedmann, P.S.; Sanchez-Elsner, T. MicroRNA-155 modulates the pathogen binding ability of dendritic cells (DCs) by down-regulation of DC-specific intercellular adhesion molecule-3 grabbing non-integrin (DC-SIGN). J. Biol. Chem. 2009, 284, 16334–16342. [Google Scholar] [CrossRef] [PubMed]
- Monk, C.E.; Hutvagner, G.; Arthur, J.S. Regulation of miRNA transcription in macrophages in response to Candida albicans. PLoS ONE 2010, 5, e13669. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Zheng, Y.; Liu, B.; Zhong, S.; Giovannoni, J.; Fei, Z. A cost-effective method for Illumina small RNA-Seq library preparation using T4 RNA ligase 1 adenylated adapters. Plant Methods 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Cui, J.; Zhai, J.; Li, J.; Han, L.; Meng, J. High-throughput sequencing reveals differential expression of miRNAs in tomato inoculated with Phytophthora infestans. Planta 2015, 241, 1405–1416. [Google Scholar] [CrossRef] [PubMed]
- Baldrich, P.; Kakar, K.; Sire, C.; Moreno, A.B.; Berger, A.; Garcia-Chapa, M.; Lopez-Moya, J.J.; Riechmann, J.L.; San Segundo, B. Small RNA profiling reveals regulation of Arabidopsis miR168 and heterochromatic siRNA415 in response to fungal elicitors. BMC Genom. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.M.; Clayton, C.L.; Tomita, T.; Wallace, D.M.; Robinson, P.A.; Crabtree, J.E. cDNA array analysis of cag pathogenicity island-associated Helicobacter pylori epithelial cell response genes. Infect. Immun. 2001, 69, 6970–6980. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Murthy, S.; Rangarajan, P.N. Identification and characterization of a virus-inducible non-coding RNA in mouse brain. J. Gen. Virol. 2006, 87, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, L.; Xue, Z.; Ye, Q.; Zhang, L.; Li, S.; Liu, Y. Transcription of the major Neurospora crassa microRNA-like small RNAs relies on RNA polymerase III. PLoS Genet. 2013, 9, e1003227. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Li, L.; Gu, W.; Xue, Z.; Crosthwaite, S.K.; Pertsemlidis, A.; Lewis, Z.A.; Freitag, M.; Selker, E.U.; Mello, C.C.; et al. Diverse pathways generate microRNA-like RNAs and Dicer-independent small interfering RNAs in fungi. Mol. Cell 2010, 38, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Chow, W.N.; Wong, A.Y.; Yeung, J.M.; Bao, J.; Zhang, N.; Lok, S.; Woo, P.C.; Yuen, K.Y. Identification of microRNA-like RNAs in mycelial and yeast phases of the thermal dimorphic fungus Penicillium marneffei. PLoS Negl. Trop. Dis. 2013, 7, e2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Lan, F.; Yang, W.; Zhang, F.; Yang, K.; Li, Z.; Gao, P.; Wang, S. sRNA profiling in Aspergillus flavus reveals differentially expressed miRNA-like RNAs response to water activity and temperature. Fungal Genet. Biol. 2015, 81, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Yang, Y.P.; Janbon, G.; Pan, J.; Zhu, X.D. Identification and functional demonstration of miRNAs in the fungus Cryptococcus neoformans. PLoS ONE 2012, 7, e52734. [Google Scholar] [CrossRef] [PubMed]
- Peres da Silva, R.; Puccia, R.; Rodrigues, M.L.; Oliveira, D.L.; Joffe, L.S.; Cesar, G.V.; Nimrichter, L.; Goldenberg, S.; Alves, L.R. Extracellular vesicle-mediated export of fungal RNA. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Chacko, N.; Zhao, Y.; Yang, E.; Wang, L.; Cai, J.J.; Lin, X. The lncRNA RZE1 controls cryptococcal morphological transition. PLoS Genet. 2015, 11, e1005692. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Chai, J.C.; Kim, S.H.; Park, K.S.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. Dual RNA sequencing reveals the expression of unique transcriptomic signatures in lipopolysaccharide-induced BV-2 microglial cells. PLoS ONE 2015, 10, e0121117. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Next-generation sequencing platforms. Annu. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Strong, M.J.; Lacey, M.R.; Baribault, C.; Flemington, E.K.; Taylor, C.M. RNA CoMPASS: A dual approach for pathogen and host transcriptome analysis of RNA-seq datasets. PLoS ONE 2014, 9, e89445. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Henkel, S.G.; Vlaic, S.; Guthke, R.; van Zoelen, E.J.; Driesch, D. Inference of dynamical gene-regulatory networks based on time-resolved multi-stimuli multi-experiment data applying NetGenerator V2.0. BMC Syst. Biol. 2013, 7. [Google Scholar] [CrossRef] [PubMed]
- Schulze, S.; Henkel, S.G.; Driesch, D.; Guthke, R.; Linde, J. Computational prediction of molecular pathogen-host interactions based on dual transcriptome data. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Vilain, C.; Libert, F.; Venet, D.; Costagliola, S.; Vassart, G. Small amplified RNA-SAGE: An alternative approach to study transcriptome from limiting amount of mRNA. Nucleic Acids Res. 2003, 31. [Google Scholar] [CrossRef]
- Bhavsar, A.P.; Guttman, J.A.; Finlay, B.B. Manipulation of host-cell pathways by bacterial pathogens. Nature 2007, 449, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Diaz, E.; Jorda, M.; Peinado, M.A.; Rivero, A. Epigenetics of host-pathogen interactions: The road ahead and the road behind. PLoS Pathog. 2012, 8, e1003007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silmon de Monerri, N.C.; Kim, K. Pathogens hijack the epigenome: A new twist on host-pathogen interactions. Am. J. Pathol. 2014, 184, 897–911. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enguita, F.J.; Costa, M.C.; Fusco-Almeida, A.M.; Mendes-Giannini, M.J.; Leitão, A.L. Transcriptomic Crosstalk between Fungal Invasive Pathogens and Their Host Cells: Opportunities and Challenges for Next-Generation Sequencing Methods. J. Fungi 2016, 2, 7. https://doi.org/10.3390/jof2010007
Enguita FJ, Costa MC, Fusco-Almeida AM, Mendes-Giannini MJ, Leitão AL. Transcriptomic Crosstalk between Fungal Invasive Pathogens and Their Host Cells: Opportunities and Challenges for Next-Generation Sequencing Methods. Journal of Fungi. 2016; 2(1):7. https://doi.org/10.3390/jof2010007
Chicago/Turabian StyleEnguita, Francisco J., Marina C. Costa, Ana Marisa Fusco-Almeida, Maria José Mendes-Giannini, and Ana Lúcia Leitão. 2016. "Transcriptomic Crosstalk between Fungal Invasive Pathogens and Their Host Cells: Opportunities and Challenges for Next-Generation Sequencing Methods" Journal of Fungi 2, no. 1: 7. https://doi.org/10.3390/jof2010007
APA StyleEnguita, F. J., Costa, M. C., Fusco-Almeida, A. M., Mendes-Giannini, M. J., & Leitão, A. L. (2016). Transcriptomic Crosstalk between Fungal Invasive Pathogens and Their Host Cells: Opportunities and Challenges for Next-Generation Sequencing Methods. Journal of Fungi, 2(1), 7. https://doi.org/10.3390/jof2010007