Differential Regulation of Echinocandin Targets Fks1 and Fks2 in Candida glabrata by the Post-Transcriptional Regulator Ssd1

 , , and
, , and 
Abstract
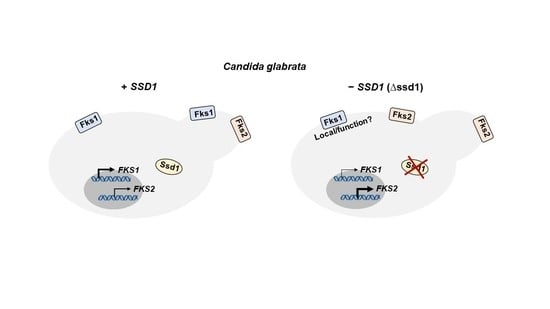
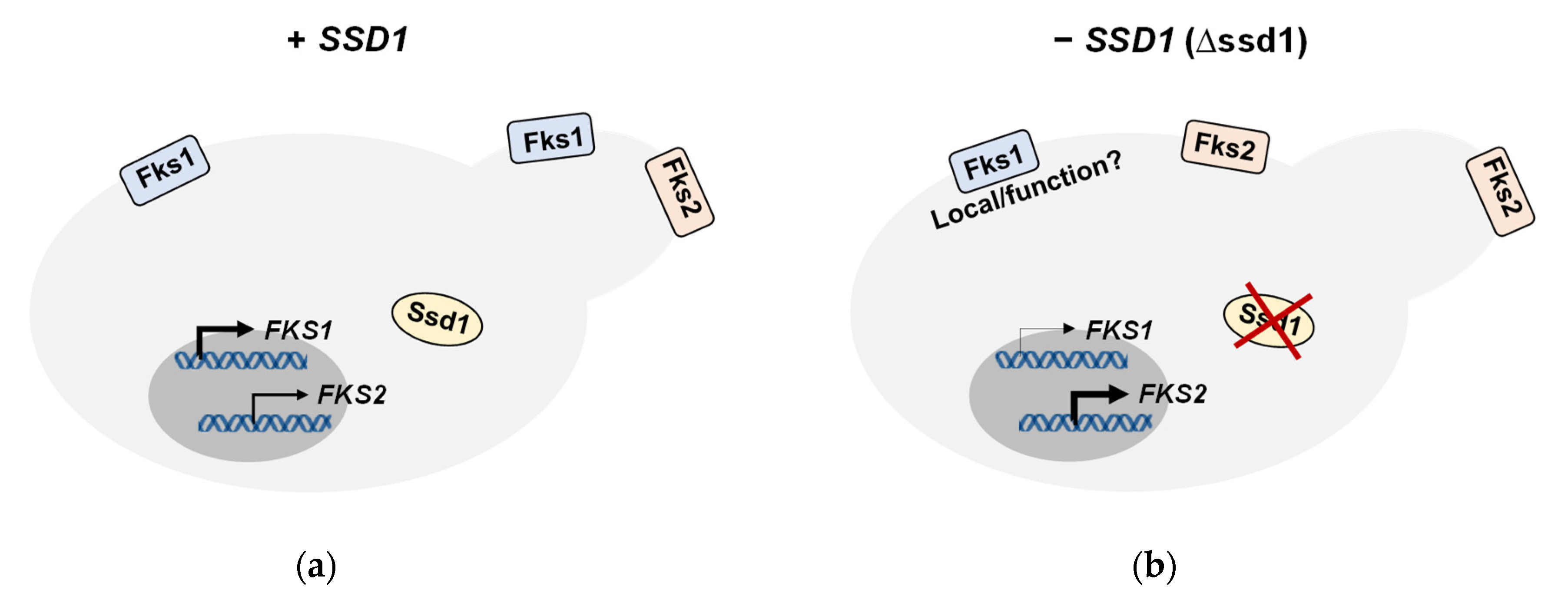
:
1. Introduction
2. Materials and Methods
2.1. Strain Construction and Media
2.2. Drug Susceptibility Assays
2.3. RNA Isolation and Quantitative RT-PCR
2.4. Glucan Synthase Preparation and Western Blotting
3. Results
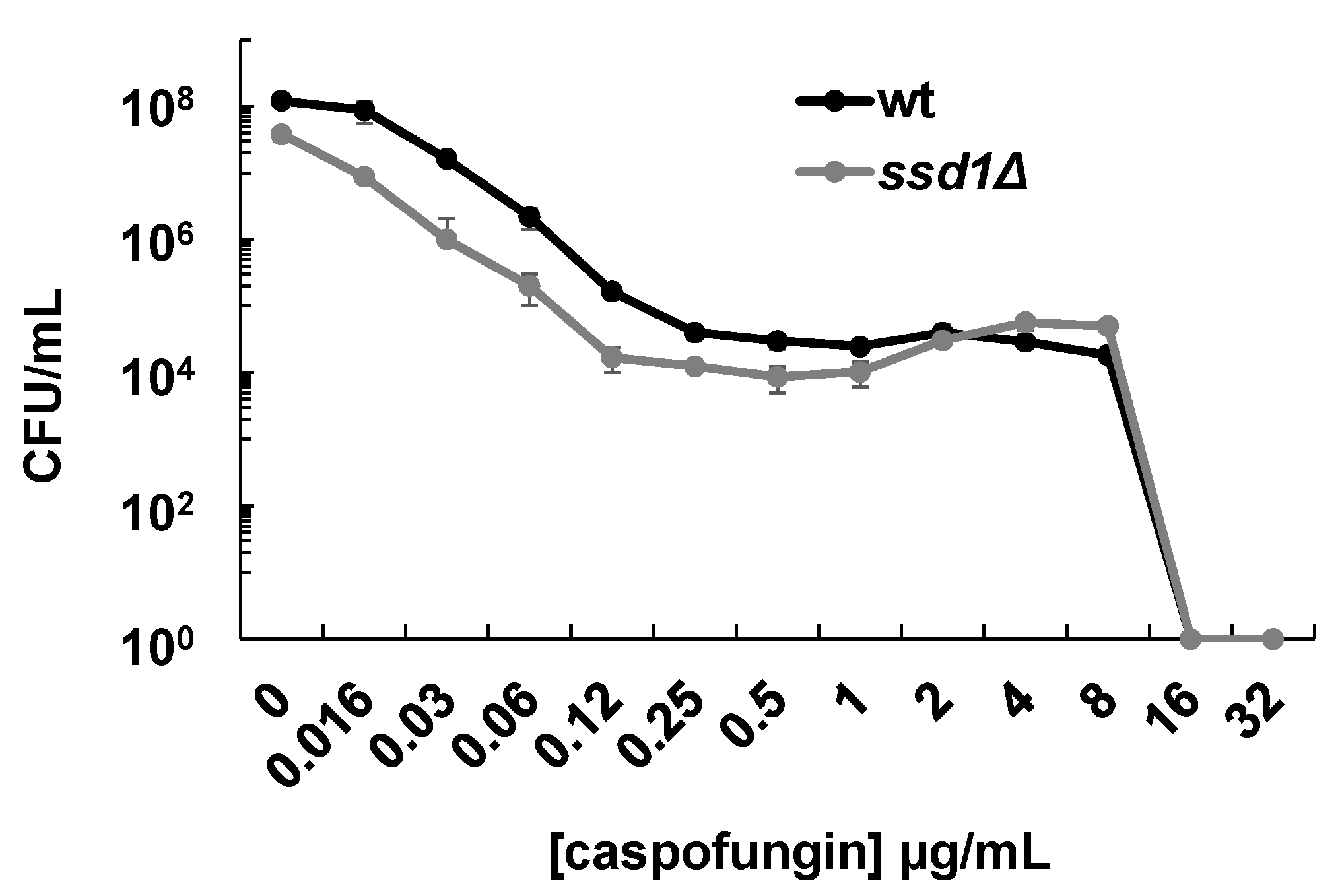
3.1. SSD1 Disruption Reverses FKS1-Mediated Resistance in C. glabrata
3.2. SSD1 Disruption Causes Increased Sensitivity to FK506
3.3. Loss of SSD1 Leads to Decreases in FKS1:FKS2 Gene and Fks1:Fks2 Protein Expression Ratios
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Castanheira, M.; Messer, S.A.; Jones, R.N.; Farrell, D.J.; Pfaller, M.A. Activity of echinocandins and triazoles against a contemporary (2012) worldwide collection of yeast and moulds collected from invasive infections. Int. J. Antimicrob. Agents 2014, 44, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.D.; Johnson, M.D.; Pfeiffer, C.D.; Jimenez-Ortigosa, C.; Catania, J.; Booker, R.; Castanheira, M.; Messer, S.A.; Perlin, D.S.; Pfaller, M.A. Increasing echinocandin resistance in Candida glabrata: Clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin. Infect. Dis. 2013, 56, 1724–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamoth, F.; Lockhart, S.R.; Berkow, E.L.; Calandra, T. Changes in the epidemiological landscape of invasive candidiasis. J. Antimicrob. Chemother. 2018, 73, i4–i13. [Google Scholar] [CrossRef] [Green Version]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.R.; Clancy, C.J.; Marr, K.A.; Ostrosky-Zeichner, L.; Reboli, A.C.; Schuster, M.G.; Vazquez, J.A.; Walsh, T.J.; et al. Clinical Practice Guideline for the Management of Candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2016, 62, e1–e50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlin, D.S. Mechanisms of echinocandin antifungal drug resistance. Ann. N. Y. Acad. Sci. 2015, 1354, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.; Morin, N.; Baginsky, W.; el-Sherbeini, M.; Clemas, J.A.; Nielsen, J.B.; Foor, F. Differential expression and function of two homologous subunits of yeast 1,3-beta-D-glucan synthase. Mol. Cell. Biol. 1995, 15, 5671–5681. [Google Scholar] [CrossRef] [Green Version]
- Katiyar, S.K.; Alastruey-Izquierdo, A.; Healey, K.R.; Johnson, M.E.; Perlin, D.S.; Edlind, T.D. Fks1 and Fks2 are functionally redundant but differentially regulated in Candida glabrata: Implications for echinocandin resistance. Antimicrob. Agents Chemother. 2012, 56, 6304–6309. [Google Scholar] [CrossRef] [Green Version]
- Gomar-Alba, M.; Morcillo-Parra, M.A.; Olmo, M.L. Response of yeast cells to high glucose involves molecular and physiological differences when compared to other osmostress conditions. FEMS Yeast Res. 2015, 15, fov039. [Google Scholar] [CrossRef]
- Zhao, C.; Jung, U.S.; Garrett-Engele, P.; Roe, T.; Cyert, M.S.; Levin, D.E. Temperature-induced expression of yeast FKS2 is under the dual control of protein kinase C and calcineurin. Mol. Cell. Biol 1998, 18, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Brisse, S.; Pannier, C.; Angoulvant, A.; de Meeus, T.; Diancourt, L.; Faure, O.; Muller, H.; Peman, J.; Viviani, M.A.; Grillot, R.; et al. Uneven distribution of mating types among genotypes of Candida glabrata isolates from clinical samples. Eukaryot. Cell 2009, 8, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Muller, H.; Hennequin, C.; Gallaud, J.; Dujon, B.; Fairhead, C. The asexual yeast Candida glabrata maintains distinct a and alpha haploid mating types. Eukaryot. Cell 2008, 7, 848–858. [Google Scholar] [CrossRef] [Green Version]
- Gabaldon, T.; Fairhead, C. Genomes shed light on the secret life of Candida glabrata: Not so asexual, not so commensal. Curr. Genet. 2019, 65, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Effron, G.; Katiyar, S.K.; Park, S.; Edlind, T.D.; Perlin, D.S. A naturally occurring proline-to-alanine amino acid change in Fks1p in Candida parapsilosis, Candida orthopsilosis, and Candida metapsilosis accounts for reduced echinocandin susceptibility. Antimicrob. Agents Chemother. 2008, 52, 2305–2312. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Effron, G.; Lee, S.; Park, S.; Cleary, J.D.; Perlin, D.S. Effect of Candida glabrata FKS1 and FKS2 mutations on echinocandin sensitivity and kinetics of 1,3-beta-D-glucan synthase: Implication for the existing susceptibility breakpoint. Antimicrob. Agents Chemother. 2009, 53, 3690–3699. [Google Scholar] [CrossRef] [Green Version]
- Niimi, K.; Woods, M.A.; Maki, K.; Nakayama, H.; Hatakenaka, K.; Chibana, H.; Ikeda, F.; Ueno, K.; Niimi, M.; Cannon, R.D.; et al. Reconstitution of high-level micafungin resistance detected in a clinical isolate of Candida glabrata identifies functional homozygosity in glucan synthase gene expression. J. Antimicrob. Chemother. 2012, 67, 1666–1676. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Chen, X.; Cai, L.; Tang, L.; Liu, L. Transcription factors Asg1p and Hal9p regulate pH homeostasis in Candida glabrata. Front. Microbiol. 2015, 6, 843. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, R.T.; Cunha, D.V.; Wang, C.; Pereira, L.; Silva, S.; Salazar, S.B.; Schroder, M.S.; Okamoto, M.; Takahashi-Nakaguchi, A.; Chibana, H.; et al. The CgHaa1-Regulon Mediates Response and Tolerance to Acetic Acid Stress in the Human Pathogen Candida glabrata. G3 (Bethesda) 2017, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Hogan, D.J.; Riordan, D.P.; Gerber, A.P.; Herschlag, D.; Brown, P.O. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008, 6, e255. [Google Scholar] [CrossRef] [Green Version]
- Jansen, J.M.; Wanless, A.G.; Seidel, C.W.; Weiss, E.L. Cbk1 regulation of the RNA-binding protein Ssd1 integrates cell fate with translational control. Curr. Biol. 2009, 19, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kurischko, C.; Kuravi, V.K.; Herbert, C.J.; Luca, F.C. Nucleocytoplasmic shuttling of Ssd1 defines the destiny of its bound mRNAs. Mol. Microbiol. 2011, 81, 831–849. [Google Scholar] [CrossRef] [Green Version]
- Kurischko, C.; Kim, H.K.; Kuravi, V.K.; Pratzka, J.; Luca, F.C. The yeast Cbk1 kinase regulates mRNA localization via the mRNA-binding protein Ssd1. J. Cell Biol 2011, 192, 583–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zordan, R.E.; Ren, Y.; Pan, S.J.; Rotondo, G.; De Las Penas, A.; Iluore, J.; Cormack, B.P. Expression plasmids for use in Candida glabrata. G3 (Bethesda) 2013, 3, 1675–1686. [Google Scholar] [CrossRef] [Green Version]
- Healey, K.R.; Katiyar, S.K.; Raj, S.; Edlind, T.D. CRS-MIS in Candida glabrata: Sphingolipids modulate echinocandin-Fks interaction. Mol. Microbiol. 2012, 86, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.E.; Edlind, T.D. Topological and mutational analysis of Saccharomyces cerevisiae Fks1. Eukaryot. Cell 2012, 11, 952–960. [Google Scholar] [CrossRef] [Green Version]
- National Committee for Clinical Laboratory Standards. Reference Method for Broth Dilution ANTIFUNGAL Susceptibility Testing of Yeasts. Clinical and Laboratory Standards Institute Document M27, 4th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- Healey, K.R.; Nagasaki, Y.; Zimmerman, M.; Kordalewska, M.; Park, S.; Zhao, Y.; Perlin, D.S. The Gastrointestinal Tract Is a Major Source of Echinocandin Drug Resistance in a Murine Model of Candida glabrata Colonization and Systemic Dissemination. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Li, Q.Q.; Skinner, J.; Bennett, J.E. Evaluation of reference genes for real-time quantitative PCR studies in Candida glabrata following azole treatment. BMC Mol. Biol. 2012, 13, 22. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Kelly, R.; Kahn, J.N.; Robles, J.; Hsu, M.J.; Register, E.; Li, W.; Vyas, V.; Fan, H.; Abruzzo, G.; et al. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob. Agents Chemother. 2005, 49, 3264–3273. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Healey, K.R.; Shor, E.; Kordalewska, M.; Ortigosa, C.J.; Paderu, P.; Xiao, M.; Wang, H.; Zhao, Y.; Lin, L.Y.; et al. Novel FKS1 and FKS2 modifications in a high-level echinocandin resistant clinical isolate of Candida glabrata. Emerg. Microbes Infect. 2019, 8, 1619–1625. [Google Scholar] [CrossRef] [Green Version]
- Schwarzmuller, T.; Ma, B.; Hiller, E.; Istel, F.; Tscherner, M.; Brunke, S.; Ames, L.; Firon, A.; Green, B.; Cabral, V.; et al. Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog. 2014, 10, e1004211. [Google Scholar] [CrossRef] [Green Version]
- Healey, K.R.; Perlin, D.S. Fungal Resistance to Echinocandins and the MDR Phenomenon in Candida glabrata. J. Fungi 2018, 4. [Google Scholar] [CrossRef]
- Shields, R.K.; Nguyen, M.H.; Press, E.G.; Kwa, A.L.; Cheng, S.; Du, C.; Clancy, C.J. The presence of an FKS mutation rather than MIC is an independent risk factor for failure of echinocandin therapy among patients with invasive candidiasis due to Candida glabrata. Antimicrob. Agents Chemother. 2012, 56, 4862–4869. [Google Scholar] [CrossRef] [Green Version]
- Kaeberlein, M.; Guarente, L. Saccharomyces cerevisiae MPT5 and SSD1 function in parallel pathways to promote cell wall integrity. Genetics 2002, 160, 83–95. [Google Scholar]
- Arias, P.; Diez-Muniz, S.; Garcia, R.; Nombela, C.; Rodriguez-Pena, J.M.; Arroyo, J. Genome-wide survey of yeast mutations leading to activation of the yeast cell integrity MAPK pathway: Novel insights into diverse MAPK outcomes. BMC Genom. 2011, 12, 390. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 24 h MICs (µg/mL) | ||||
|---|---|---|---|---|
| Strain | CSF | MCF | FK506 | |
| wild type | 0.03 | 0.03 | 32 | |
| ∆ssd1 | 0.016 | 0.008 | ≤0.5 | |
| ∆fks1 | 0.008 | 0.016 | ≤0.5 | |
| ∆fks2 | 0.016 | 0.03 | 32 | |
| Fks1- | ||||
| 625delF | 2 | 1 | 16 | |
| 625delF ∆ssd1 | 0.03 | 0.008 | ≤0.5 | |
| S629P | 2 | 1 | 32 | |
| S629P ∆ssd1 | 0.25 | 0.03 | 1 | |
| Fks2- | ||||
| 659delF | 2 | 1 | 32 | |
| 659delF ∆ssd1 | 2 | 0.5 | ≤0.5 | |
| S663P | 2 | 1 | 64 | |
| S663P ∆ssd1 | 2 | 1 | 2 | |
| 24 h MICs (µg/mL) | ||
|---|---|---|
| Strain | CSF | MCF |
| WT + empty | 0.016 | 0.016 |
| WT + pSSD1 | 0.016 | 0.016 |
| ∆ssd1 + empty | 0.016 | 0.008 |
| 625delF + empty | 4 | 1 |
| 625delF + pSSD1 | 4 | 1 |
| 625delF ∆ssd1 + empty | 0.12 | 0.016 |
| 625delF ∆ssd1 + pSSD1 | 4 | 0.5 |
| S629P + empty | 4 | 2 |
| S629P + pSSD1 | 2 | 2 |
| S629P ∆ssd1 + empty | 0.06 | 0.016 |
| S629P ∆ssd1 + pSSD1 | 2 | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Healey, K.R.; Paderu, P.; Hou, X.; Jimenez Ortigosa, C.; Bagley, N.; Patel, B.; Zhao, Y.; Perlin, D.S. Differential Regulation of Echinocandin Targets Fks1 and Fks2 in Candida glabrata by the Post-Transcriptional Regulator Ssd1. J. Fungi 2020, 6, 143. https://doi.org/10.3390/jof6030143
Healey KR, Paderu P, Hou X, Jimenez Ortigosa C, Bagley N, Patel B, Zhao Y, Perlin DS. Differential Regulation of Echinocandin Targets Fks1 and Fks2 in Candida glabrata by the Post-Transcriptional Regulator Ssd1. Journal of Fungi. 2020; 6(3):143. https://doi.org/10.3390/jof6030143
Chicago/Turabian StyleHealey, Kelley R., Padmaja Paderu, Xin Hou, Cristina Jimenez Ortigosa, Nicole Bagley, Biren Patel, Yanan Zhao, and David S. Perlin. 2020. "Differential Regulation of Echinocandin Targets Fks1 and Fks2 in Candida glabrata by the Post-Transcriptional Regulator Ssd1" Journal of Fungi 6, no. 3: 143. https://doi.org/10.3390/jof6030143
APA StyleHealey, K. R., Paderu, P., Hou, X., Jimenez Ortigosa, C., Bagley, N., Patel, B., Zhao, Y., & Perlin, D. S. (2020). Differential Regulation of Echinocandin Targets Fks1 and Fks2 in Candida glabrata by the Post-Transcriptional Regulator Ssd1. Journal of Fungi, 6(3), 143. https://doi.org/10.3390/jof6030143