Stem Endophytic Mycobiota in Wild and Domesticated Wheat: Structural Differences and Hidden Resources for Wheat Improvement


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Extraction of Genomic DNA
2.3. Amplicon Library Preparation and Sequencing
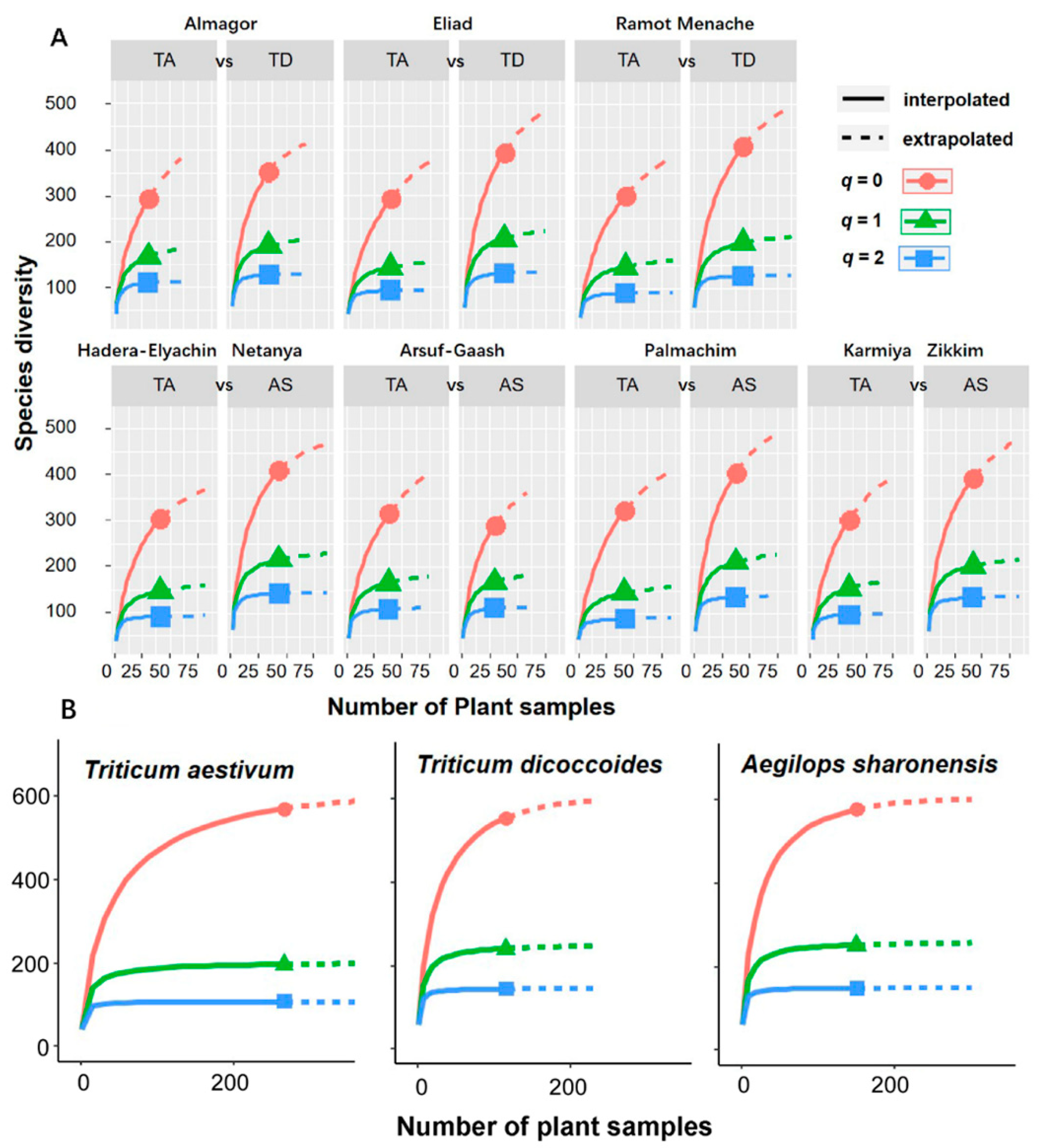
2.4. Data Analyses
2.4.1. Quality Control and Data Preparation
2.4.2. Variation within FECs
2.4.3. Variation among FECs of Separate Plants within a Population
2.4.4. Variation among FECs of Host Species and Locations
2.4.5. Co-Occurrence Analysis
3. Results
3.1. Composition and Taxonomy of the Fungal Endophytes
3.2. Effect of Plant Species on Variation within FECs
3.3. Variation among FECs of Individual Plants within a Population
3.4. Effect of Species on FEC Structural Variation among the Populations of Plants
3.5. Co-Occurrence Network Analysis and Identification of Putative Hub Fungal Taxa
3.6. Differentially Abundant Endophytes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gdanetz, K.; Noel, Z.; Trail, F. The phytobiomes of a three-crop rotation: Influence of land management, host, and plant organ on microbial diversity. BioRxiv 2020. [Google Scholar] [CrossRef]
- Sun, X.; Kosman, E.; Sharon, O.; Ezrati, S.; Sharon, A. Significant host- and environment-dependent differentiation among highly sporadic fungal endophyte communities in cereal crops-related wild grasses. Environ. Microbiol. 2020, 22, 3357–3374. [Google Scholar] [CrossRef]
- Gdanetz, K.; Benucci, G.M.N.; Pol, N.V.; Bonito, G. CONSTAX: A tool for improved taxonomic resolution of environmental fungal ITS sequences. BMC Bioinform. 2017, 18, 538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.J.; Bonth, A.C.M.D.; Briggs, L.R.; Caradus, J.R.; Finch, S.C.; Fleetwood, D.J.; Fletcher, L.R.; Hume, D.E.; Johnson, R.D.; Popay, A.J.; et al. The exploitation of epichloae endophytes for agricultural benefit. Fungal Divers. 2013, 60, 171–188. [Google Scholar] [CrossRef]
- Johnson, J.M.; Alex, T.; Oelmüller, R. Piriformospora indica: The versatile and multifunctional root endophytic fungus for enhanced yield and tolerance to biotic and abiotic stress in crop plants. J. Trop. Agric. 2014, 52, 103–122. [Google Scholar]
- Zapalski, M.K. Is absence of proof a proof of absence? Comments on commensalism. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2011, 302, 484–488. [Google Scholar] [CrossRef]
- Hardoim, P.R.; Overbeek, L.S.V.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Döring, M.; Sessitsch, A. The hidden world within plants: Ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shade, A.; Handelsman, J. Beyond the Venn diagram: The hunt for a core microbiome. Environ. Microbiol. 2011, 14, 4–12. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Van, A.L.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Toju, H.; Peay, K.G.; Yamamichi, M.; Narisawa, K.; Hiruma, K.; Naito, K.; Fukuda, S.; Ushio, M.; Nakaoka, S.; Onoda, Y.; et al. Core microbiomes for sustainable agroecosystems. Nat. Plants 2018, 4, 247–257. [Google Scholar] [CrossRef]
- Agler, M.T.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.-T.; Weigel, D.; Kemen, E.M. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016, 14, e1002352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toju, H.; Tanabe, A.S.; Sato, H. Network hubs in root-associated fungal metacommunities. Microbiome 2018, 6, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, R.J.; White, J.F., Jr.; Arnold, A.E.; Redman, R.S. Fungal endophytes: Diversity and functional roles. New Phytol. 2009, 182, 314–330. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Walder, F.; Büchi, L.; Meyer, M.; Held, A.Y.; Gattinger, A.; Keller, T.; Charles, R.; Heijden, M.G.A.V.D. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 2019, 13, 1722–1736. [Google Scholar] [CrossRef] [Green Version]
- Hassani, M.A.; Özkurt, E.; Franzenburg, S.; Stukenbrock, E.H. Ecological assembly processes of the bacterial and fungal microbiota of wild and domesticated wheat species. Phytobiomes J. 2020. [Google Scholar] [CrossRef] [Green Version]
- Gardes, M.; Bruns, T.D. Its primers with enhanced specificity for basidiomycetes—Application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef]
- Smith, D.P.; Peay, K.G. Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PLoS ONE 2014, 9, e90234. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Kõljalg, U.; Nilsson, R.H.; Abarenkov, K.; Tedersoo, L.; Taylor, A.F.S.; Bahram, M.; Bates, S.T.; Bruns, T.D.; Bengtsson-Palme, J.; Callaghan, T.M.; et al. Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 2013, 22, 5271–5277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, K.S.; Kirkegaard, R.H.; Karst, S.M.; Albertsen, M. ampvis2: An R package to analyse and visualise 16S rRNA amplicon data. BioRxiv 2018, 299537. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package. Available online: https://CRAN.R-project.org/package=vegan (accessed on 1 September 2020).
- Reese, G.C.; Wilson, K.R.; Flather, C.H. Performance of species richness estimators across assemblage types and survey parameters. Global Ecol. Biogeogr. 2014, 23, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.H.; Chao, A. Estimating and comparing microbial diversity in the presence of sequencing errors. PeerJ 2016, 4, e1634. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.C.; Ma, K.H.; Chao, A. iNEXT: An R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol. 2016, 7, 1451–1456. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Dice, L.R. Measures of the amount of ecologic association between species. Ecology 1945, 26, 297–302. [Google Scholar] [CrossRef]
- Kosman, E. Measuring diversity: From individuals to populations. Eur. J. Plant Pathol. 2014, 138, 467–486. [Google Scholar] [CrossRef]
- Kosman, E. Difference and diversity of plant pathogen populations: A new approach for measuring. Phytopathology 1996, 86, 1152–1155. [Google Scholar]
- Kosman, E.; Leonard, K.J. Conceptual analysis of methods applied to assessment of diversity within and distance between populations with asexual or mixed mode of reproduction. New Phytol. 2007, 174, 683–696. [Google Scholar] [CrossRef]
- Anderson, M.J.; Walsh, D.C.I. PERMANOVA, ANOSIM, and the Mantel test in the face of heterogeneous dispersions: What null hypothesis are you testing? Ecol. Monogr. 2013, 83, 557–574. [Google Scholar] [CrossRef]
- Kosman, E.; Ben-Yehuda, P.; Manisterski, J. Diversity of virulence phenotypes among annual populations of wheat leaf rust in Israel from 1993 to 2008. Plant. Pathol. 2014, 63, 563–571. [Google Scholar] [CrossRef]
- Scheiner, S.M.; Kosman, E.; Presley, S.J.; Willig, M.R. Decomposing functional diversity. Methods Ecol. Evol. 2017, 8, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Kosman, E.; Chen, X.; Dreiseitl, A.; McCallum, B.; Lebeda, A.; Ben-Yehuda, P.; Gultyaeva, E.; Manisterski, J. Functional variation of plant-pathogen interactions: New concept and methods for virulence data analyses. Phytopathology 2019, 109, 1324–1330. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R_Core_Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 1 September 2020).
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef] [Green Version]
- Arnold, A.E.; Mejía, L.C.; Kyllo, D.; Rojas, E.I.; Maynard, Z.; Robbins, N.; Herre, E.A. Fungal endophytes limit pathogen damage in a tropical tree. Proc. Natl. Acad. Sci. USA 2003, 100, 15649–15654. [Google Scholar] [CrossRef] [Green Version]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Ofek-Lalzar, M.; Gur, Y.; Ben-Moshe, S.; Sharon, O.; Kosman, E.; Mochli, E.; Sharon, A. Diversity of fungal endophytes in recent and ancient wheat ancestors Triticum dicoccoides and Aegilops sharonensis. FEMS Microbiol. Ecol. 2016, 92, fiw152. [Google Scholar] [CrossRef] [Green Version]
- Kosiak, B.; Torp, M.; Skjerve, E.; Andersen, B. Alternaria and Fusarium in Norwegian grains of reduced quality—A matched pair sample study. Int. J. Food Microbiol. 2004, 93, 51–62. [Google Scholar] [CrossRef]
- Perello, A.; Moreno, M.; Sisterna, M. Alternaria infectoria species-group associated with black point of wheat in Argentina. Plant Pathol. 2008, 57, 379. [Google Scholar] [CrossRef] [Green Version]
- Serdani, M.; Crous, P.W.; Holz, G.; Petrini, O. Endophytic fungi associated with core rot of apples in South Africa, with specific reference to Alternaria species. Sydowia 1998, 50, 257–271. [Google Scholar]
- Scholtysik, A.; Unterseher, M.; Otto, P.; Wirth, C. Spatio-temporal dynamics of endophyte diversity in the canopy of European ash (Fraxinus excelsior). Mycol. Prog. 2013, 12, 291–304. [Google Scholar] [CrossRef]
- Sapkota, R.; Knorr, K.; Jørgensen, L.N.; O’Hanlon, K.A.; Nicolaisen, M. Host genotype is an important determinant of the cereal phyllosphere mycobiome. New Phytol. 2015, 207, 1134–1144. [Google Scholar] [CrossRef]
- Gdanetz, K.; Trail, F. The wheat microbiome under four management strategies, and potential for endophytes in disease protection. Phytobiomes 2017, 1, 158–168. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title | Latitude | Longitude | Wild Plant Species | Wheat |
|---|---|---|---|---|
| Arsuf Ga’ash | 32.22142 | 34.82036 | Aegilops sharonensis (29/44) | Triticum aestivum (37/42) |
| Zikkim | 31.61472 | 34.52537 | Aegilops sharonensis (41/42) | ─ ─ ─ |
| Palmachim | 31.93258 | 34.72893 | Aegilops sharonensis (37/40) | Triticum aestivum (42/43) |
| Netanya | 32.41755 | 34.88858 | Aegilops sharonensis (43/44) | ─ ─ ─ |
| Almagor | 32.90316 | 35.60003 | Triticum dicoccoides (34/42) | Triticum aestivum (30/43) |
| Eliad | 32.79048 | 35.74926 | Triticum dicoccoides (37/42) | Triticum aestivum (39/44) |
| Ramot Menache | 32.6035 | 35.06862 | Triticum dicoccoides (44/44) | Triticum aestivum (42/42) |
| Karmiya | 31.60292 | 34.54686 | ─ ─ ─ | Triticum aestivum (34/42) |
| Hadera Elyachin | 32.41429 | 34.91244 | ─ ─ ─ | Triticum aestivum (41/42) |
| Site | Diversity | Sample Size | Observed Diversity | Asymptotic Estimator |
|---|---|---|---|---|
| Almagor TA | q = 0 | 30 | 292 | 488.569 |
| q = 1 | 30 | 166.624 | 200.933 | |
| q = 2 | 30 | 109.883 | 116.258 | |
| Eliad TA | q = 0 | 39 | 292 | 461.16 |
| q = 1 | 39 | 144.162 | 166.665 | |
| q = 2 | 39 | 94.358 | 97.718 | |
| Ramot Menache TA | q = 0 | 42 | 301 | 454.842 |
| q = 1 | 42 | 146.45 | 170.794 | |
| q = 2 | 42 | 88.982 | 92.284 | |
| Hadera Elyachin TA | q = 0 | 41 | 304 | 397.478 |
| q = 1 | 41 | 147.23 | 166.459 | |
| q = 2 | 41 | 91.8 | 94.781 | |
| Arsuf Gaash TA | q = 0 | 37 | 316 | 483.901 |
| q = 1 | 37 | 165.651 | 191.492 | |
| q = 2 | 37 | 108.939 | 113.407 | |
| Palmachim TA | q = 0 | 42 | 325 | 455.817 |
| q = 1 | 42 | 145.019 | 165.682 | |
| q = 2 | 42 | 88.323 | 90.728 | |
| Karmiya TA | q = 0 | 34 | 303 | 457.091 |
| q = 1 | 34 | 153.679 | 180.549 | |
| q = 2 | 34 | 95.708 | 99.516 | |
| Almagor TD | q = 0 | 34 | 351 | 439.704 |
| q = 1 | 34 | 190.752 | 212.173 | |
| q = 2 | 34 | 128.701 | 133.344 | |
| Eliad TD | q = 0 | 37 | 393 | 548.44 |
| q = 1 | 37 | 206.382 | 236.2 | |
| q = 2 | 37 | 130.888 | 136.023 | |
| Ramot Menache TD | q = 0 | 44 | 408 | 542.891 |
| q = 1 | 44 | 198.574 | 220.889 | |
| q = 2 | 44 | 127.402 | 130.991 | |
| Netanya AS | q = 0 | 43 | 411 | 489.032 |
| q = 1 | 43 | 217.492 | 237.221 | |
| q = 2 | 43 | 144.463 | 148.872 | |
| Arsuf Gaash AS | q = 0 | 29 | 290 | 404.916 |
| q = 1 | 29 | 166.35 | 194.229 | |
| q = 2 | 29 | 110.598 | 116.609 | |
| Palmachim AS | q = 0 | 37 | 406 | 542.282 |
| q = 1 | 37 | 211.862 | 238.726 | |
| q = 2 | 37 | 136.279 | 141.049 | |
| Zikkim AS | q = 0 | 41 | 395 | 535.214 |
| q = 1 | 41 | 204.404 | 227.225 | |
| q = 2 | 41 | 134.336 | 138.495 |
| Al a | El | Ra | HE- Ne | AG | Pa | Ka- Zi | All Locations e | |
|---|---|---|---|---|---|---|---|---|
| AS b | 0.653 | 0.708 | 0.640 | 0.652 | 0.702 d | |||
| TA | 0.736 c | 0.673 | 0.698 | 0.656 | 0.699 | 0.608 | 0.655 | 0.734 |
| TD | 0.638 | 0.678 | 0.627 | 0.700 |
| AS d | TA | TD | |
|---|---|---|---|
| Number of FECs | 4 | 7 | 3 |
| Da | 0.125 | 0.190 | 0.151 |
| b | 2.637 | 4.251 | 2.093 |
| c | 0.546 | 0.542 | 0.547 |
| AS d | TA | TD | |
|---|---|---|---|
| Number of FECs | 4 | 7 | 3 |
| Da | 0.314 | 0.367 | 0.288 |
| b | 2.138 | 3.200 | 1.749 |
| c | 0.379 | 0.367 | 0.375 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Kosman, E.; Sharon, A. Stem Endophytic Mycobiota in Wild and Domesticated Wheat: Structural Differences and Hidden Resources for Wheat Improvement. J. Fungi 2020, 6, 180. https://doi.org/10.3390/jof6030180
Sun X, Kosman E, Sharon A. Stem Endophytic Mycobiota in Wild and Domesticated Wheat: Structural Differences and Hidden Resources for Wheat Improvement. Journal of Fungi. 2020; 6(3):180. https://doi.org/10.3390/jof6030180
Chicago/Turabian StyleSun, Xiang, Evsey Kosman, and Amir Sharon. 2020. "Stem Endophytic Mycobiota in Wild and Domesticated Wheat: Structural Differences and Hidden Resources for Wheat Improvement" Journal of Fungi 6, no. 3: 180. https://doi.org/10.3390/jof6030180
APA StyleSun, X., Kosman, E., & Sharon, A. (2020). Stem Endophytic Mycobiota in Wild and Domesticated Wheat: Structural Differences and Hidden Resources for Wheat Improvement. Journal of Fungi, 6(3), 180. https://doi.org/10.3390/jof6030180