ATAC-Seq Identifies Chromatin Landscapes Linked to the Regulation of Oxidative Stress in the Human Fungal Pathogen Candida albicans


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strains, Media, and Growth Conditions
2.2. Nuclei and Genomic DNA (gDNA) Isolation, Tagmentation, ATAC-Seq Libraries, and Sequencing
2.2.1. gDNA Isolation
2.2.2. Tagmentation
2.2.3. ATAC-seq Library Amplification and Size Selection
2.2.4. Next-Generation Sequencing
2.3. ATAC-Seq Data Analysis Workflow
2.3.1. Pre-Processing and Read Alignment
2.3.2. Prediction of Nucleosomal Positions and Genomic Coverage of ATAC-seq Signals
2.3.3. Peak Calling and Genomic Annotation of ATAC-seq Peaks
2.3.4. Analysis of Differential ATAC-seq Peaks
2.3.5. Motif Search
2.3.6. Data Plotting
2.4. External Datasets
2.5. Code Availability
2.6. Data Availability
3. Results and Discussion
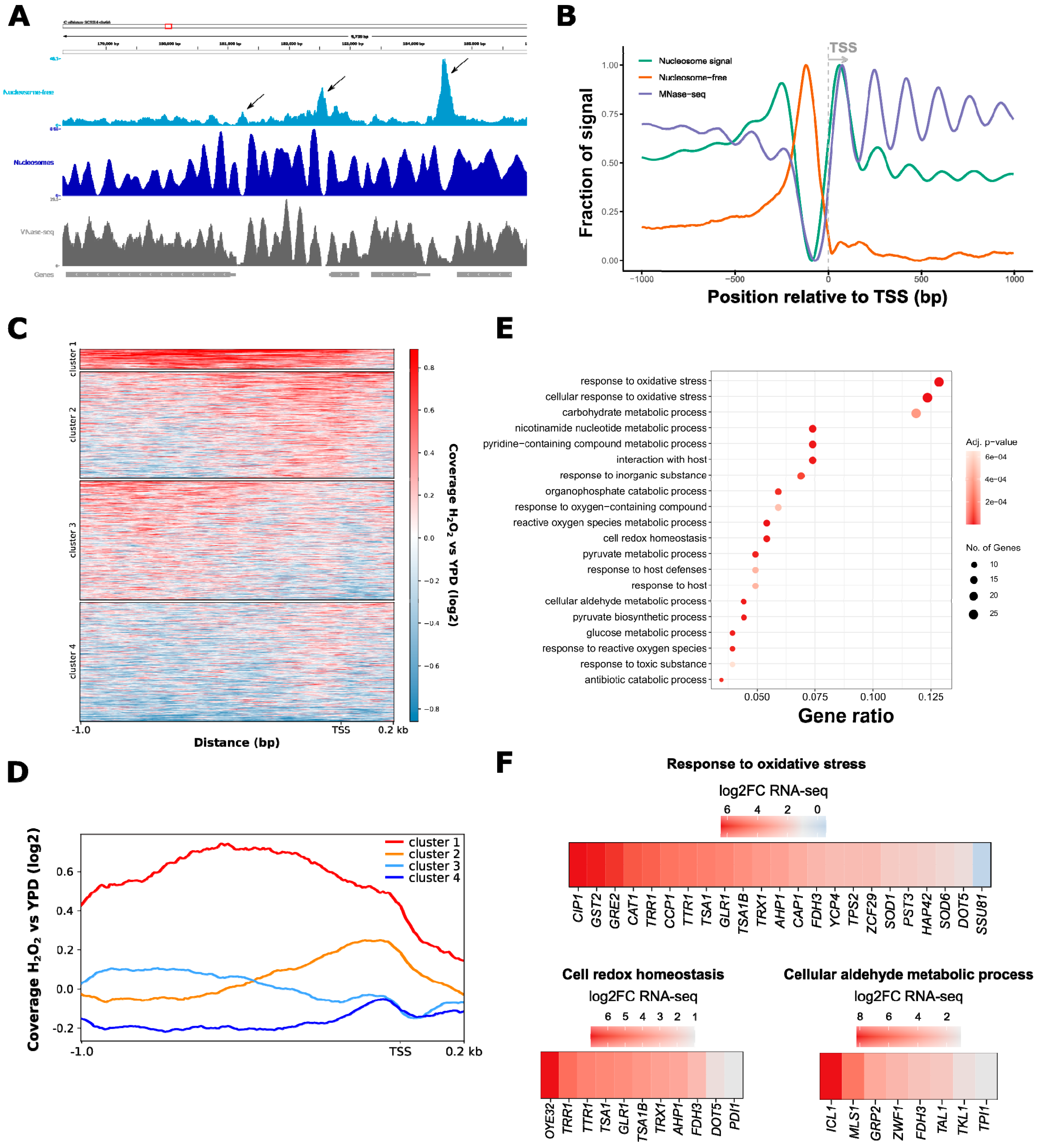
3.1. ATAC-Seq in C. albicans Reflects Nucleosomal Organization Genome-Wide
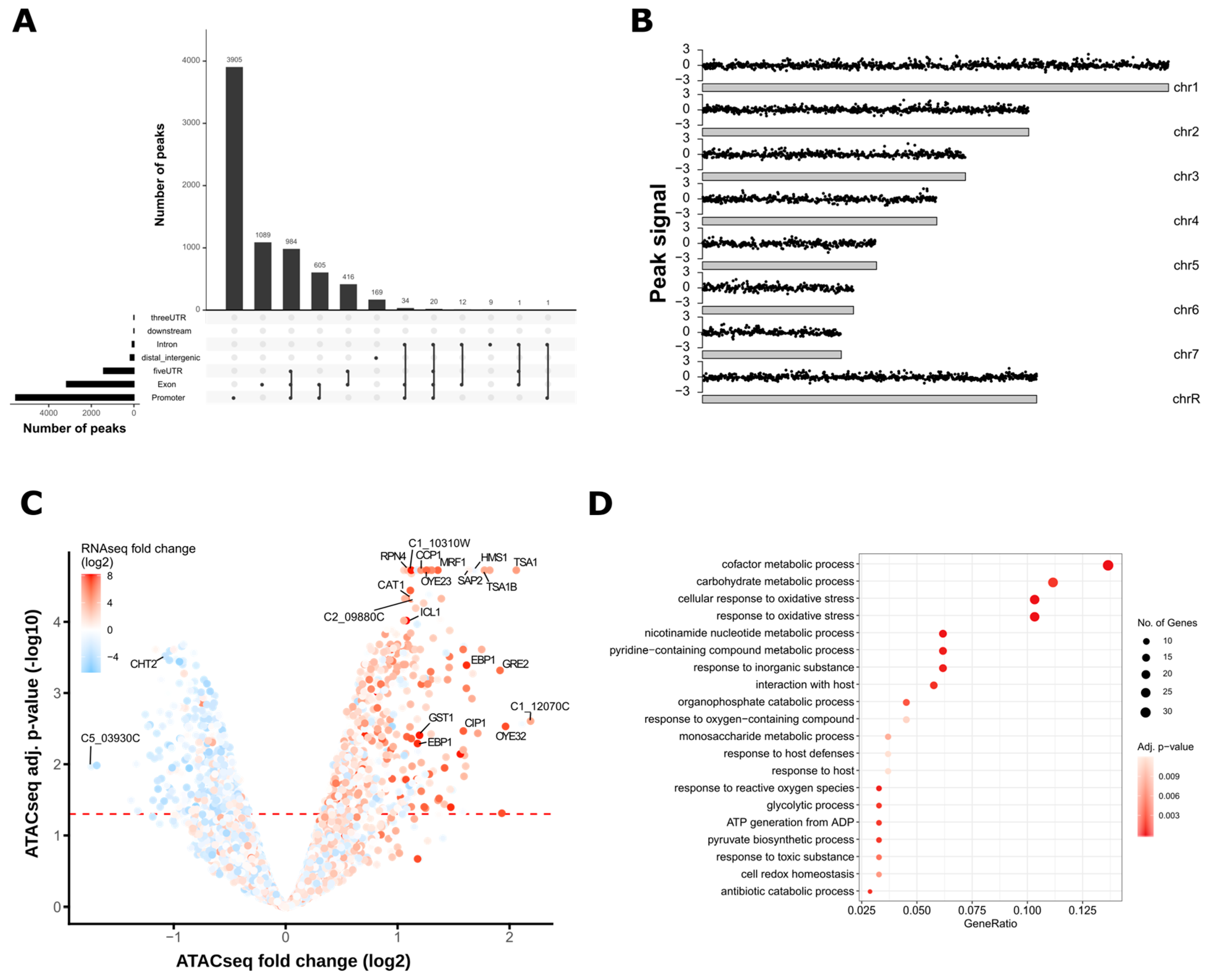
3.2. ATAC-Seq Detects Genome-Wide Changes in Chromatin Accessibility after Oxidative Stress
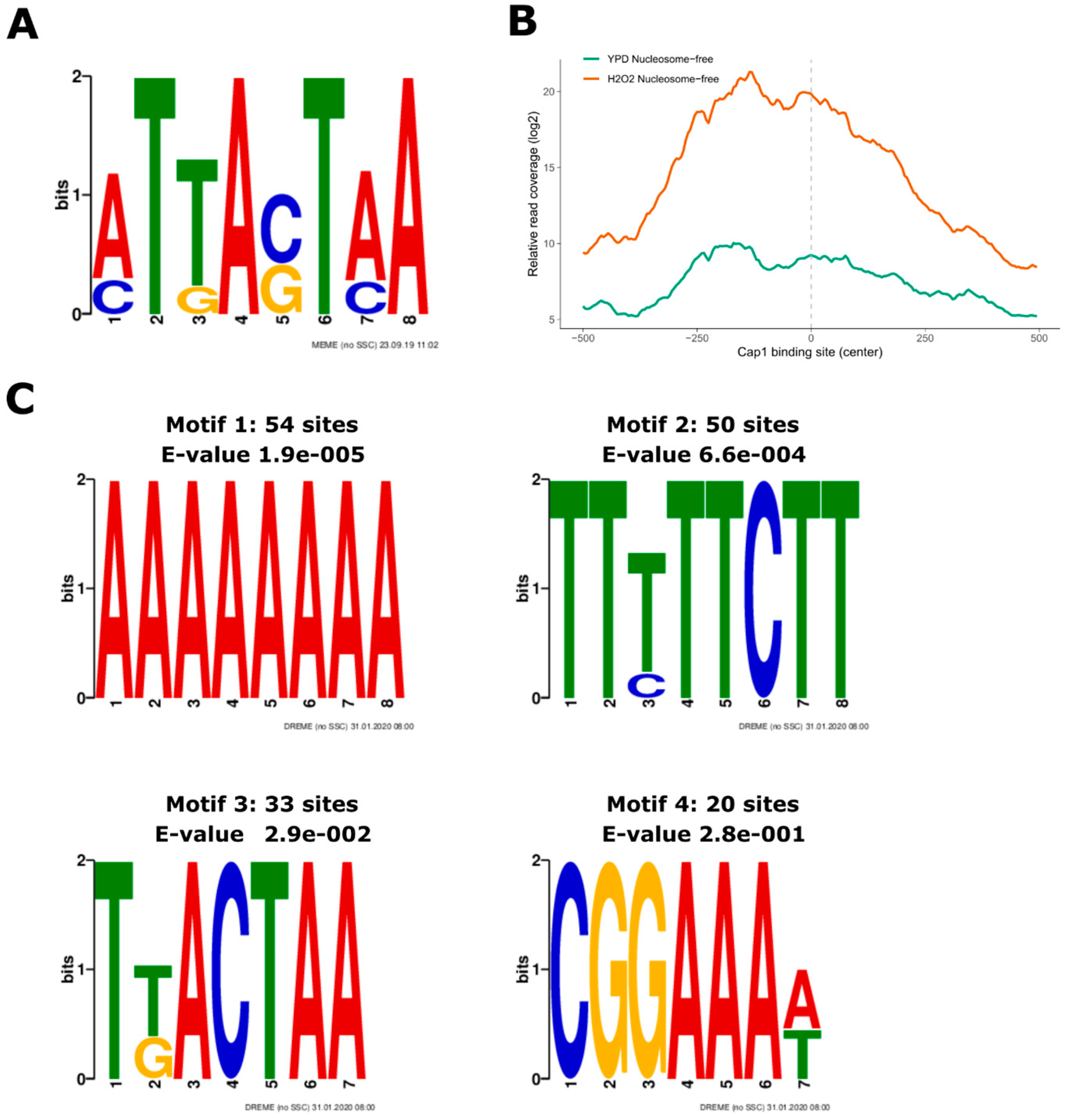
3.3. Oxidative Stress-Responsive ATAC-Seq Peaks Are Enriched for Cap1 Binding Sites
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rai, M.N.; Balusu, S.; Gorityala, N.; Dandu, L.; Kaur, R. Functional genomic analysis of Candida glabrata-macrophage interaction: Role of chromatin remodeling in virulence. PLoS Pathog. 2012, 8, e1002863. [Google Scholar] [CrossRef] [Green Version]
- Juárez-Reyes, A.; Castaño, I. Chromatin architecture and virulence-related gene expression in eukaryotic microbial pathogens. Curr. Genet. 2019, 65, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Dantas, A.; Lee, K.K.; Raziunaite, I.; Schaefer, K.; Wagener, J.; Yadav, B.; Gow, N.A. Cell biology of Candida albicans-host interactions. Curr. Opin. Microbiol. 2016, 34, 111–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Maury, L.; Marguerat, S.; Bähler, J. Tuning gene expression to changing environments: From rapid responses to evolutionary adaptation. Nat. Rev. Genet. 2008, 9, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Cases, I.; de Lorenzo, V.; Ouzounis, C.A. Transcription regulation and environmental adaptation in bacteria. Trends Microbiol. 2003, 11, 248–253. [Google Scholar] [CrossRef]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell. Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef]
- Voss, T.C.; Hager, G.L. Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet. 2014, 15, 69–81. [Google Scholar] [CrossRef]
- Brown, A.J.P.; Budge, S.; Kaloriti, D.; Tillmann, A.; Jacobsen, M.D.; Yin, Z.; Ene, I.V.; Bohovych, I.; Sandai, D.; Kastora, S.; et al. Stress adaptation in a pathogenic fungus. J. Exp. Biol. 2014, 217, 144–155. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef]
- Wilson, D.; Thewes, S.; Zakikhany, K.; Fradin, C.; Albrecht, A.; Almeida, R.; Brunke, S.; Grosse, K.; Martin, R.; Mayer, F.; et al. Identifying infection-associated genes of Candida albicans in the postgenomic era. FEMS Yeast Res. 2009, 9, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Mayer, F.L.; Wilson, D.; Hube, B. Candida albicans pathogenicity mechanisms. Virulence 2013, 4, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, S.M.; French, S.; Kohn, L.A.; Chen, V.; Johnson, A.D. Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat Genet. 2010, 42, 590–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, S.M.; Gianetti, B.A.; Witchley, J.N. Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat. Rev. Microbiol. 2017, 15, 96–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enjalbert, B.; Nantel, A.; Whiteway, M. Stress-induced gene expression in Candida albicans: Absence of a general stress response. Mol. Biol. Cell. 2002, 14, 1460–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivarathri, R.; Tscherner, M.; Zwolanek, F.; Singh, N.K.; Chauhan, N.; Kuchler, K. The fungal histone acetyl transferase Gcn5 controls virulence of the human pathogen Candida albicans through multiple pathways. Sci. Rep. 2019, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Tscherner, M.; Zwolanek, F.; Jenull, S.; Sedlazeck, F.J.; Petryshyn, A.; Frohner, I.E.; Mavrianos, J.; Chauhan, N.; von Haeseler, A.; Kuchler, K. The Candida albicans histone acetyltransferase Hat1 regulates stress resistance and virulence via distinct chromatin assembly pathways. PLoS Pathog. 2015, 11, e1005218. [Google Scholar] [CrossRef] [Green Version]
- Kuchler, K.; Jenull, S.; Shivarathri, R.; Chauhan, N. Fungal KATs/KDACs: A new highway to better antifungal drugs? PLoS Pathog. 2016, 12, e1005938. [Google Scholar] [CrossRef]
- Da Rosa, J.L.; Boyartchuk, V.L.; Zhu, L.J.; Kaufman, P.D. Histone acetyltransferase Rtt109 is required for Candida albicans pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 1594–1599. [Google Scholar] [CrossRef] [Green Version]
- Wurtele, H.; Tsao, S.; Lépine, G.; Mullick, A.; Tremblay, J.; Drogaris, P.; Lee, E.H.; Thibault, P.; Verreault, A.; Raymond, M. Modulation of histone H3 lysine 56 acetylation as an antifungal therapeutic strategy. Nat Med. 2010, 16, 774–780. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Bardet, A.F.; Nobile, C.J.; Petryshyn, A.; Glaser, W.; Schöck, U.; Stark, A.; Kuchler, K. A histone deacetylase adjusts transcription kinetics at coding sequences during Candida albicans morphogenesis. PLoS Genet. 2012, 8, e1003118. [Google Scholar] [CrossRef] [Green Version]
- Garnaud, C.; Champleboux, M.; Maubon, D.; Cornet, M.; Govin, J. Histone deacetylases and their inhibition in Candida species. Front. Microbiol. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, N.; Wright, G.D.; Cowen, L.E. Antifungal drugs: The current armamentarium and development of new agents. In The Fungal Kingdom; ASM Press: Washington, DC, USA, 2016; pp. 903–922. [Google Scholar]
- Lockhart, S.R. Candida auris and multidrug resistance: Defining the new normal. Fungal Genet. Biol. 2019, 131, 103243. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.F.; Gade, L.; Chow, N.A.; Loparev, V.N.; Juieng, P.; Berkow, E.L.; Farrer, R.A.; Litvintseva, A.P.; Cuomo, C.A. Genomic insights into multidrug-resistance, mating and virulence in Candida auris and related emerging species. Nat. Commun. 2018, 9, 5346. [Google Scholar] [CrossRef] [Green Version]
- Kean, R.; Brown, J.; Gulmez, D.; Ware, A.; Ramage, G. Candida auris: A decade of understanding of an enigmatic pathogenic yeast. J. Fungi 2020, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, W.K.M.; Pugh, B.F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell. Biol. 2017, 18, 548–562. [Google Scholar] [CrossRef]
- Brogaard, K.; Xi, L.; Wang, J.-P.; Widom, J. A map of nucleosome positions in yeast at base-pair resolution. Nature 2012, 486, 496–501. [Google Scholar] [CrossRef]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Schep, A.N.; Buenrostro, J.D.; Denny, S.K.; Schwartz, K.; Sherlock, G.; Greenleaf, W.J. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res. 2015, 25, 1757–1770. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Hofmeister, B.T.; Vollmers, C.; DuBois, R.M.; Schmitz, R.J. Combining ATAC-seq with nuclei sorting for discovery of cis-regulatory regions in plant genomes. Nucleic Acids Res. 2017, 45, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huang, B.; Chen, H.; Yin, Q.; Liu, Y.; Xiang, Y.; Zhang, B.; Liu, B.; Wang, Q.; Xia, W.; et al. The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 2016, 534, 652–657. [Google Scholar] [CrossRef]
- Maezawa, S.; Yukawa, M.; Alavattam, K.G.; Barski, A.; Namekawa, S.H. Dynamic reorganization of open chromatin underlies diverse transcriptomes during spermatogenesis. Nucleic Acids Res. 2017, 46, 593–608. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Wang, M.; Wei, X.; Wu, L.; Xu, J.; Dai, X.; Xia, J.; Cheng, M.; Yuan, Y.; Zhang, P.; et al. An ATAC-seq atlas of chromatin accessibility in mouse tissues. Sci. Data 2019, 6, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Lareau, C.A.; Duarte, F.M.; Chew, J.G.; Kartha, V.K.; Burkett, Z.D.; Kohlway, A.S.; Pokholok, D.; Aryee, M.J.; Steemers, F.J.; Lebofsky, R.; et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 2019, 37, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Toenhake, C.G.; Fraschka, S.A.K.; Vijayabaskar, M.S.; Westhead, D.R.; van Heeringen, S.J.; Bártfai, R. Chromatin accessibility-based characterization of the gene regulatory network underlying plasmodium falciparum blood-stage development. Cell Host Microbe 2018, 23, 557–569. [Google Scholar] [CrossRef] [Green Version]
- Rogerson, C.; Britton, E.; Withey, S.; Hanley, N.; Ang, Y.S.; Sharrocks, A.D. Identification of a primitive intestinal transcription factor network shared between esophageal adenocarcinoma and its precancerous precursor state. Genome Res. 2019, 29, 723–736. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Schulz, M.H.; Look, T.; Begemann, M.; Zenke, M.; Costa, I.G. Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 2019, 20, 45. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Leng, L.; Liu, C.; Lu, C.; Yuan, Y.; Wu, L.; Gong, F.; Zhang, S.; Wei, X.; Wang, M.; et al. An integrated chromatin accessibility and transcriptome landscape of human pre-implantation embryos. Nat. Commun. 2019, 10, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enjalbert, B.; MacCallum, D.M.; Odds, F.C.; Brown, A.J.P. Niche-specific activation of the oxidative stress response by the pathogenic fungus Candida albicans. Infect. Immun. 2007, 75, 2143–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaloriti, D.; Jacobsen, M.; Yin, Z.; Patterson, M.; Tillmann, A.; Smith, D.A.; Cook, E.; You, T.; Grimm, M.J.; Bohovych, I.; et al. Mechanisms underlying the exquisite sensitivity of Candida albicans to combinatorial cationic and oxidative stress that enhances the potent fungicidal activity of phagocytes. MBio 2014, 5, e01334-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Znaidi, S.; Barker, K.S.; Weber, S.; Alarco, A.-M.; Liu, T.T.; Boucher, G.; Rogers, P.D.; Raymond, M. Identification of the Candida albicans Cap1p regulon. Eukaryot. Cell 2009, 8, 806–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillum, A.M.; Tsay, E.Y.H.; Kirsch, D.R. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. MGG Mol. Gen. Genet. 1984, 198, 179–182. [Google Scholar] [CrossRef]
- Goryshin, I.Y.; Miller, J.A.; Kil, Y.V.; Lanzov, V.A.; Reznikoff, W.S. Tn5/IS50 target recognition. Proc. Natl. Acad. Sci. USA 1998, 95, 10716–10721. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, C.S.; Winston, F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformaion of Escherichia coli. Gene 1987, 57, 267–272. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Sedlazeck, F.J.; Rescheneder, P.; von Haeseler, A. NextGenMap: Fast and accurate read mapping in highly polymorphic genomes. Bioinformatics 2013, 29, 2790–2791. [Google Scholar] [CrossRef] [Green Version]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for computing and annotating genomic ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef] [PubMed]
- Tsankov, A.M.; Thompson, D.A.; Socha, A.; Regev, A.; Rando, O.J. The role of nucleosome positioning in the evolution of gene regulation. PLoS Biol. 2010, 8, e1000414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMISC: J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2014, 31, 166–169. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; He, Q.-Y. ChIPseeker: An R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 2015, 31, 2382–2383. [Google Scholar] [CrossRef] [Green Version]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37 (Suppl. S2), W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, T.L. DREME: Motif discovery in transcription factor ChIP-seq data. Bioinformatics 2011, 27, 1653–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Adey, A.; Morrison, H.G.; Asan Xun, X.; Kitzman, J.O.; Turner, E.H.; Stackhouse, B.; MacKenzie, A.P.; Caruccio, N.C.; Zhang, X.; Shendure, J. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol. 2010, 11, R119. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Morozov, A.V. Gene regulation by nucleosome positioning. Trends Genet. 2010, 26, 476–483. [Google Scholar] [CrossRef]
- Cottier, F.; Tan, A.S.M.; Chen, J.; Lum, J.; Zolezzi, F.; Poidinger, M.; Pavelka, N. The transcriptional stress response of Candida albicans to weak organic acids. G3 Genes Genomes Genet. 2015, 5, 497–505. [Google Scholar]
- Lorenz, M.C.; Bender, J.A.; Fink, G.R. Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryot. Cell. 2004, 3, 1076–1087. [Google Scholar] [CrossRef] [Green Version]
- Enjalbert, B.; Smith, D.A.; Cornell, M.J.; Alam, I.; Nicholls, S.; Brown, A.J.P.; Quinn, J. Role of the Hog1 stress-activated protein kinase in the global transcriptional response to stress in the fungal pathogen Candida albicans. Mol. Biol. Cell. 2006, 17, 1018–1032. [Google Scholar] [CrossRef] [Green Version]
- Gasch, A.P.; Spellman, P.T.; Kao, C.M.; Carmel-Harel, O.; Eisen, M.B.; Storz, G.; Botstein, D.; Brown, P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000, 11, 4241–4257. [Google Scholar] [CrossRef]
- Albert, B.; Kos-Braun, I.C.; Henras, A.K.; Dez, C.; Rueda, M.P.; Zhang, X.; Gadal, O.; Kos, M.; Shore, D. A ribosome assembly stress response regulates transcription to maintain proteome homeostasis. Elife 2019, 8, e45002. [Google Scholar] [CrossRef]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.M.; Ernst, J.; Wilder, S.P.; Kundaje, A.; Harris, R.S.; Libbrecht, M.; Giardine, B.; Ellenbogen, P.M.; Bilmes, J.A.; Birney, E.; et al. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res. 2013, 41, 827–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaton, A.M.; Gómez-Rodríguez, M.; Mieczkowski, J.; Tolstorukov, M.Y.; Kundu, S.; Sadreyev, R.I.; Jansen, L.E.; Kingston, R.E. Enhancer regions show high histone H3.3 turnover that changes during differentiation. Elife 2016, 5, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, D.G.; Soifer, I.; Wranik, B.J.; Botstein, D.; Scott McIsaac, R. Simultaneous profiling of DNA accessibility and gene expression dynamics with ATAC-seq and RNA-seq. Methods Mol. Biol. 2018, 1819, 317–333. [Google Scholar]
- Wysong, D.R.; Christin, L.; Sugar, A.M.; Robbins, P.W.; Diamond, R.D. Cloning and sequencing of a Candida albicans catalase gene and effects of disruption of this gene. Infect. Immun. 1998, 66, 1953–1961. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cao, Y.Y.; Jia, X.M.; Cao, Y.B.; Gao, P.H.; Fu, X.P.; Ying, K.; Chen, W.S.; Jiang, Y.Y. Cap1p is involved in multiple pathways of oxidative stress response in Candida albicans. Free Radic. Biol. Med. 2006, 40, 1201–1209. [Google Scholar] [CrossRef]
- Piekarska, K.; Hardy, G.; Mol, E.; van den Burg, J.; Strijbis, K.; van Roermund, C.; van den Berg, M.; Distel, B. The activity of the glyoxylate cycle in peroxisomes of Candida albicans depends on a functional beta-oxidation pathway: Evidence for reduced metabolite transport across the peroxisomal membrane. Microbiology 2008, 154, 3061–3072. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef]
- Urban, C.; Xiong, X.; Sohn, K.; Schröppel, K.; Brunner, H.; Rupp, S. The moonlighting protein Tsa1p is implicated in oxidative stress response and in cell wall biogenesis in Candida albicans. Mol. Microbiol. 2005, 57, 1318–1341. [Google Scholar] [CrossRef]
- Lee, K.; Kim, S.C.; Jung, I.; Kim, K.; Seo, J.; Lee, H.-S.; Bogu, G.K.; Kim, D.; Lee, S.; Lee, B.; et al. Genetic landscape of open chromatin in yeast. PLoS Genet. 2013, 9, e1003229. [Google Scholar] [CrossRef]
- Hernday, A.D.; Lohse, M.B.; Fordyce, P.M.; Nobile, C.J.; Derisi, J.L.; Johnson, A.D. Structure of the transcriptional network controlling white-opaque switching in Candida albicans. Mol. Microbiol. 2013, 90, 22–35. [Google Scholar]
- Nobile, C.J.; Fox, E.P.; Nett, J.E.; Sorrells, T.R.; Mitrovich, Q.M.; Hernday, A.D.; Tuch, B.B.; Andes, D.R.; Johnson, A.D. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 2012, 148, 126–138. [Google Scholar] [CrossRef] [Green Version]
- Segal, E.; Widom, J. Poly(dA:dT) tracts: Major determinants of nucleosome organization. Curr. Opin. Struct. Biol. 2009, 19, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Rhee, H.S.; Pugh, B.F. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature 2012, 483, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.R.; Quach, B.; Furey, T.S. Correcting nucleotide-specific biases in high-throughput sequencing data. BMC Bioinformatics 2017, 18, 357. [Google Scholar] [CrossRef]
- Martins, A.L.; Walavalkar, N.M.; Anderson, W.D.; Zang, C.; Guertin, M.J. Universal correction of enzymatic sequence bias reveals molecular signatures of protein/DNA interactions. Nucleic Acids Res. 2017, 46, e9. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jenull, S.; Tscherner, M.; Mair, T.; Kuchler, K. ATAC-Seq Identifies Chromatin Landscapes Linked to the Regulation of Oxidative Stress in the Human Fungal Pathogen Candida albicans. J. Fungi 2020, 6, 182. https://doi.org/10.3390/jof6030182
Jenull S, Tscherner M, Mair T, Kuchler K. ATAC-Seq Identifies Chromatin Landscapes Linked to the Regulation of Oxidative Stress in the Human Fungal Pathogen Candida albicans. Journal of Fungi. 2020; 6(3):182. https://doi.org/10.3390/jof6030182
Chicago/Turabian StyleJenull, Sabrina, Michael Tscherner, Theresia Mair, and Karl Kuchler. 2020. "ATAC-Seq Identifies Chromatin Landscapes Linked to the Regulation of Oxidative Stress in the Human Fungal Pathogen Candida albicans" Journal of Fungi 6, no. 3: 182. https://doi.org/10.3390/jof6030182
APA StyleJenull, S., Tscherner, M., Mair, T., & Kuchler, K. (2020). ATAC-Seq Identifies Chromatin Landscapes Linked to the Regulation of Oxidative Stress in the Human Fungal Pathogen Candida albicans. Journal of Fungi, 6(3), 182. https://doi.org/10.3390/jof6030182