
Hidden Fungi: Combining Culture-Dependent and -Independent DNA Barcoding Reveals Inter-Plant Variation in Species Richness of Endophytic Root Fungi in Elymus repens


Abstract
:1. Introduction
2. Materials and Methods
2.1. Elymus Repens Sampling
2.2. Root Surface Sterilisation and Endophyte Culturing
2.3. High-Throughput Amplicon Sequencing
2.4. Fungal DNA Extraction, Amplification and Sanger Sequencing
2.5. Endophyte Identification
2.6. Sequence Processing and Community Analyses
3. Results
3.1. OTU Richness Described by Direct Amplicon Sequencing
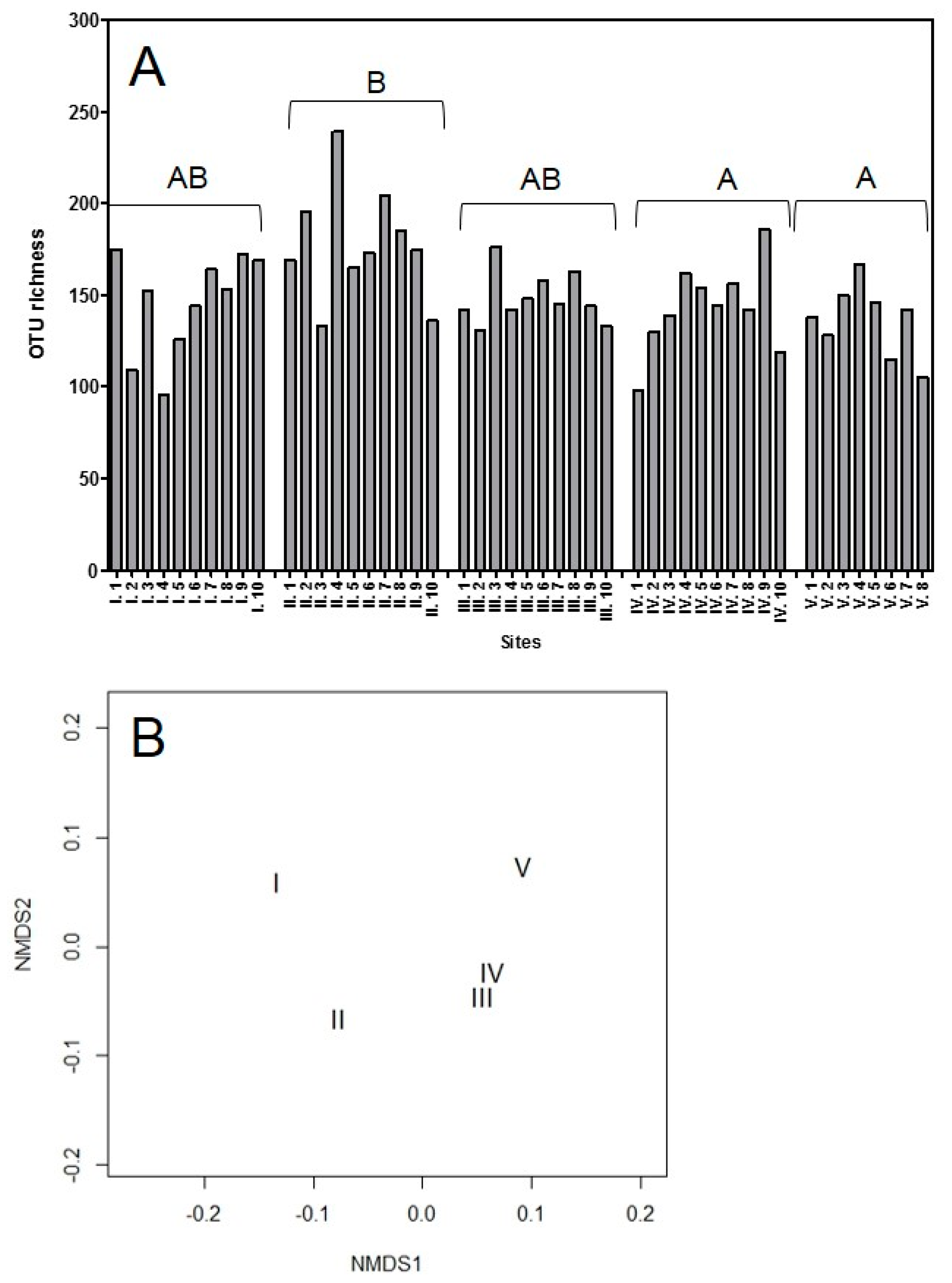
3.1.1. The OTU Richness and Community Structure of All Sites
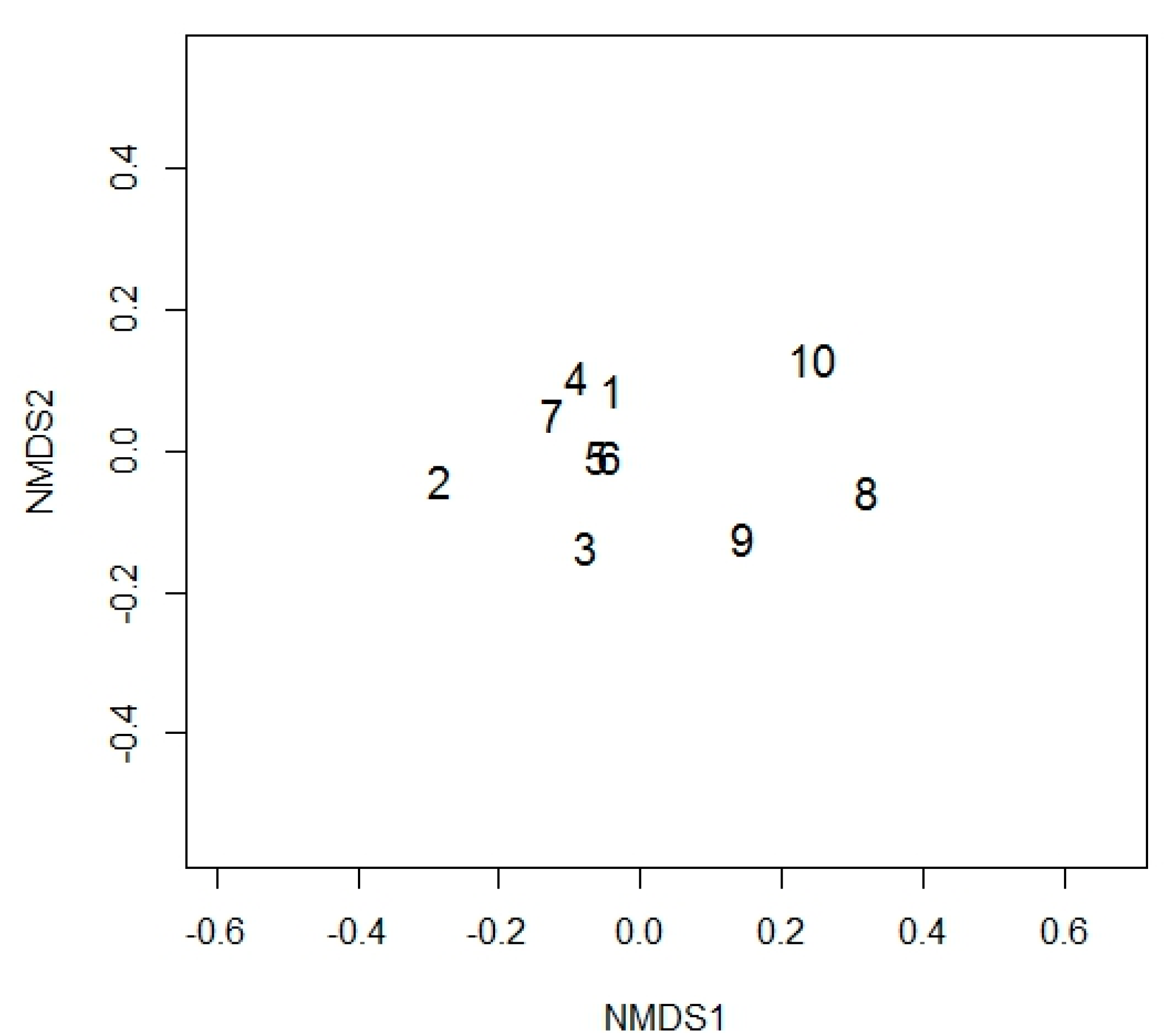
3.1.2. The OTU Richness and Community Structure of Site III
3.2. Cultured OTU Richness from Site III
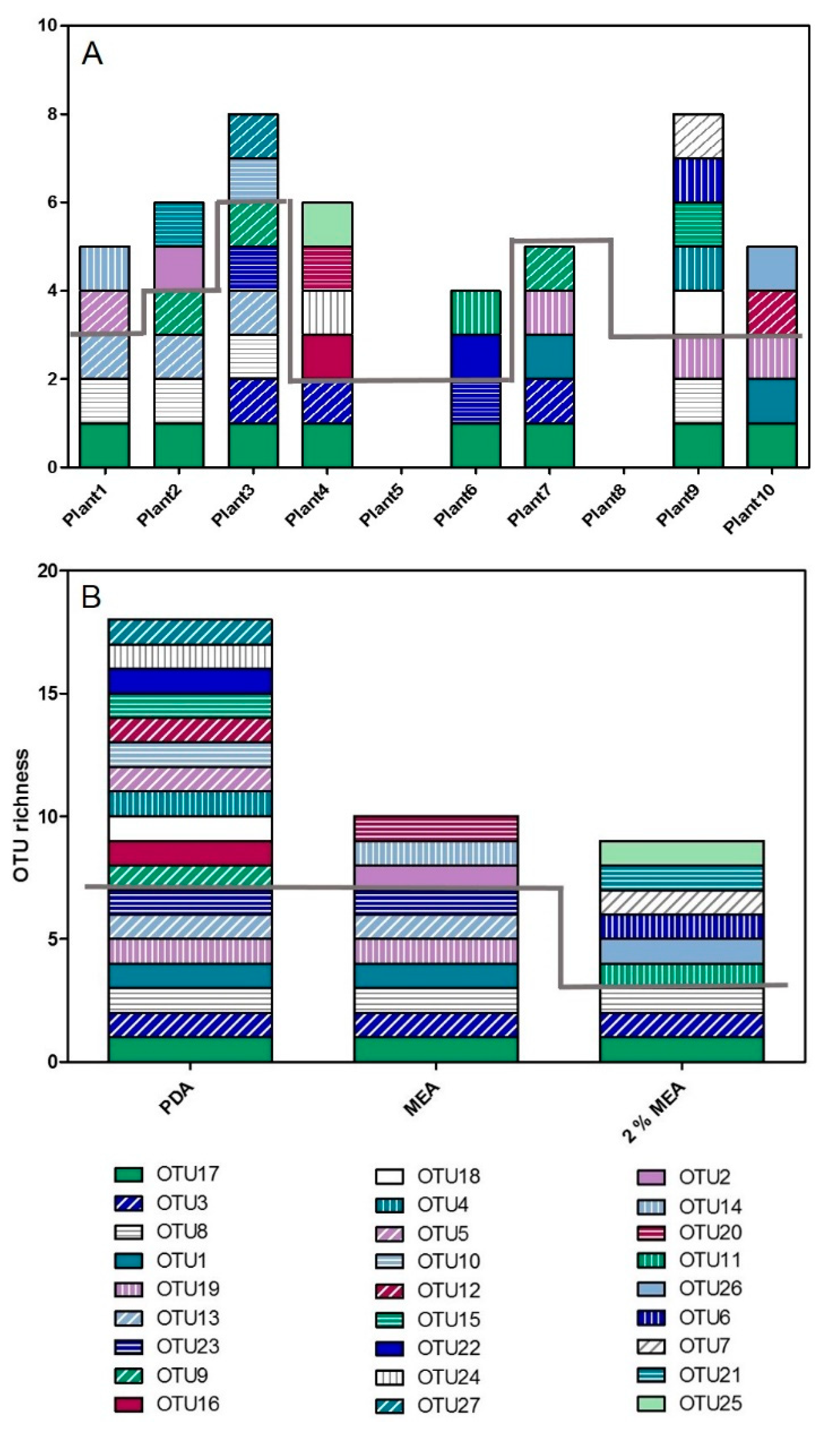
3.2.1. Inter-Plant Variation in OTU Richness
3.2.2. Media Influence on OTU Richness
4. Discussion
4.1. Endophyte Community Described by Direct Amplicon Sequencing
4.2. Comparison Between the Cultured and Directly Sequenced Community of Site III
4.3. The Influence of Media on OTU Richness
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vorholt, J.A. Microbial life in the phyllosphere. Nat. Rev. Genet. 2012, 10, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe 2015, 17, 392–403. [Google Scholar] [CrossRef] [Green Version]
- Torres-Cortés, G.; Bonneau, S.; Bouchez, O.; Genthon, C.; Briand, M.; Jacques, M.-A.; Barret, M. Functional microbial features driving community assembly during seed germination and emergence. Front. Plant Sci. 2018, 9, 902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bary, A. Vergleichende Morphologie und Biologie der Pilze, Mycetozoen und Bacterien; Wilhelm Engelmann: Leipzig, Germany, 1884. [Google Scholar]
- Suz, L.M.; Sarasan, V.; Wearn, J.A.; Bidartondo, M.I.; Hodkinson, T.R.; Kowal, J.; Murphy, B.R.; Rodriguez, R.J.; Gange, A. Positive plant-fungal interactions. In State of the World’s Fungi; Willis, K.J., Ed.; Royal Botanic Garden Kew: Richmond, UK, 2018; pp. 32–39. [Google Scholar]
- Clay, K.; Holah, J. Fungal endophyte symbiosis and plant diversity in successional fields. Science 1999, 285, 1742–1744. [Google Scholar] [CrossRef] [PubMed]
- Omacini, M.; Chaneton, E.J.; Ghersa, C.M.; Müller, C.B. Symbiotic fungal endophytes control insect host–parasite interaction webs. Nat. Cell Biol. 2001, 409, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Naveed, M.; Mitter, B.; Reichenauer, T.G.; Wieczorek, K.; Sessitsch, A. Increased drought stress resilience of maize through endophytic colonization by Burkholderia phytofirmans PsJN and Enterobacter sp. FD17. Environ. Exp. Bot. 2014, 97, 30–39. [Google Scholar] [CrossRef]
- Hubbard, M.; Germida, J.; Vujanovic, V. Fungal endophytes enhance wheat heat and drought tolerance in terms of grain yield and second-generation seed viability. J. Appl. Microbiol. 2014, 116, 109–122. [Google Scholar] [CrossRef]
- Rodriguez, R.J.; Henson, J.; Van Volkenburgh, E.; Hoy, M.; Wright, L.; Beckwith, F.; Kim, Y.-O.; Redman, R.S. Stress tolerance in plants via habitat-adapted symbiosis. ISME J. 2008, 2, 404–416. [Google Scholar] [CrossRef] [Green Version]
- Taghinasab, M.; Imani, J.; Steffens, D.; Glaeser, S.P.; Kogel, K.-H. The root endophytes Trametes versicolor and Piriformospora indica increase grain yield and P content in wheat. Plant Soil 2018, 426, 339–348. [Google Scholar] [CrossRef]
- Wicaksono, W.A.; Jones, E.E.; Monk, J.; Ridgway, H.J. Using bacterial endophytes from a New Zealand native medicinal plant for control of grapevine trunk diseases. Biol. Control. 2017, 114, 65–72. [Google Scholar] [CrossRef]
- Deshmukh, S.D.; Kogel, K.-H. Piriformospora indica protects barley from root rot caused by Fusarium graminearum. J. Plant Dis. Prot. 2007, 114, 263–268. [Google Scholar] [CrossRef]
- di Menna, M.E.; Finch, S.C.; Popay, A.J.; Smith, B.L. A review of the Neotyphodium lolii/Lolium perenne symbiosis and its associated effects on animal and plant health, with particular emphasis on ryegrass staggers. N. Z. Vet. J. 2012, 60, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-N.; Bashyal, B.P.; Wijeratne, E.M.K.; U’Ren, J.M.; Liu, M.X.; Gunatilaka, M.K.; Arnold, A.E.; Gunatilaka, A.A.L. Smardaesidins A–G, Isopimarane and 20-nor-Isopimarane Diterpenoids from Smardaea sp., a fungal endophyte of the moss Ceratodon purpureus(1). J. Nat. Prod. 2011, 74, 2052–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallmann, J.; Berg, G.; Schulz, B. Isolation procedures for endophytic microorganisms. In Microbial Root Endophytes; Schulz, B.J.E., Boyle, C.J.C., Sieber, T.N., Eds.; Springer Science and Business Media: Berlin, Germany, 2007; pp. 299–319. [Google Scholar]
- Guo, L.D.; Hyde, K.D.; Liew, E.C.Y. Identification of endophytic fungi from Livistona chinensis based on morphology and rDNA sequences. New Phytol. 2000, 147, 617–630. [Google Scholar] [CrossRef]
- Verma, V.C.; Gond, S.K.; Kumar, A.; Kharwar, R.N.; Boulanger, L.-A.; Strobel, G.A. Endophytic fungal flora from roots and fruits of an Indian neem plant Azadirachta indica A. Juss., and impact of culture media on their isolation. Indian J. Microbiol. 2011, 51, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Elvira-Recuenco, M.; van Vuurde, J.W. Natural incidence of endophytic bacteria in pea cultivars under field conditions. Can. J. Microbiol. 2000, 46, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Bills, G.F.; Polishook, J.D. Recovery of endophytic fungi from Chamaecyparis thyoides. Sydowia 1992, 44, 1–12. [Google Scholar]
- Khidir, H.; Eudy, D.; Porras-Alfaro, A.; Herrera, J.; Natvig, D.; Sinsabaugh, R. A general suite of fungal endophytes dominate the roots of two dominant grasses in a semiarid grassland. J. Arid. Environ. 2010, 74, 35–42. [Google Scholar] [CrossRef]
- Luo, J.; Walsh, E.; Miller, S.; Blystone, D.; Dighton, J.; Zhang, N. Root endophytic fungal communities associated with pitch pine, switchgrass, and rosette grass in the pine barrens ecosystem. Fungal Biol. 2017, 121, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Hammami, H.; Baptista, P.; Martins, F.; Gomes, T.; Abdelly, C.; Mahmoud, O.M.-B. Impact of a natural soil salinity gradient on fungal endophytes in wild barley (Hordeum maritimum With.). World J. Microbiol. Biotechnol. 2016, 32, 184. [Google Scholar] [CrossRef] [Green Version]
- Potshangbam, M.; Devi, S.I.; Sahoo, D.; Strobel, G.A. Functional characterization of endophytic fungal community associated with Oryza sativa L. and Zea mays L. Front. Microbiol. 2017, 8, 325. [Google Scholar] [CrossRef] [PubMed]
- Tejesvi, M.V.; Ruotsalainen, A.L.; Markkola, A.M.; Pirttilä, A.M. Root endophytes along a primary succession gradient in northern Finland. Fungal Divers. 2010, 41, 125–134. [Google Scholar] [CrossRef]
- Comby, M.; Lacoste, S.; Baillieul, F.; Profizi, C.; Dupont, J. Spatial and temporal variation of cultivable communities of co-occurring endophytes and pathogens in wheat. Front. Microbiol. 2016, 7, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Souza, R.S.C.; Okura, V.K.; Armanhi, J.; Jorrín, B.; Lozano, N.; Da Silva, M.J.; Gonzalez-Guerrero, M.; De Araújo, L.M.; Verza, N.C.; Bagheri, H.C.; et al. Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kavanagh, V.B.; Hall, L.M.; Hall, J.C. Potential hybridization of genetically engineered Triticale with wild and weedy relatives in Canada. Crop. Sci. 2010, 50, 1128–1140. [Google Scholar] [CrossRef]
- Mason-Gamer, R.J. Allohexaploidy, introgression, and the complex phylogenetic history of Elymus repens (Poaceae). Mol. Phylogenetics Evol. 2008, 47, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Ihrmark, K.; Bödeker, I.T.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandström-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region-evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Taylor, J. Amplicon and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols–A Guide to Methods and Application; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press Inc Elsevier Science: New York. NY, USA; pp. 315–322.
- Stielow, J.; Lévesque, C.; Seifert, K.; Meyer, W.; Irinyi, L.; Smits, D.; Renfurm, R.; Verkley, G.; Groenewald, M.; Chaduli, D.; et al. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia-Mol. Phylogeny Evol. Fungi 2015, 35, 242–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehner, S.A.; Buckley, E.; Craven, K.; Peterson, P.; Windham, D.; Mitchell, T.; Martin, S. A Beauveria phylogeny inferred from nuclear ITS and EF1-alpha sequences: Evidence for cryptic diversification and links to Cordyceps teleomorphs. Mycologia 2005, 97, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Blaxter, M.; Mann, J.D.; Chapman, T.; Thomas, F.; Whitton, C.; Floyd, R.; Abebe, E. Defining operational taxonomic units using DNA barcode data. Philos. Trans. R. Soc. B Biol. Sci. 2005, 360, 1935–1943. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.-H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glöckner, K.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2018, 47, D259–D264. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bálint, M.; Bahram, M.; Eren, A.M.; Faust, K.; Fuhrman, J.A.; Lindahl, B.; O’Hara, R.B.; Öpik, M.; Sogin, M.L.; Unterseher, M.; et al. Millions of reads, thousands of taxa: Microbial community structure and associations analyzed via marker genes. FEMS Microbiol. Rev. 2016, 40, 686–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauserud, H.; Kumar, S.; Brysting, A.K.; Nordén, J.; Carlsen, T. High consistency between replicate 454 pyrosequencing analyses of ectomycorrhizal plant root samples. Mycorrhiza 2012, 22, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J. Vegan: Ecological diversity. R Proj. 2016, 368, 1–12. [Google Scholar]
- Birch, P.R.J.; Whisson, S.C. Phytophthora infestans enters the genomics era. Mol. Plant Pathol. 2001, 2, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Than, D.J.; Hughes, K.J.D.; Boonhan, N.; Tomlinson, J.A.; Woodhall, J.W.; Bellgard, S.E. A TaqMan real-time PCR assay for the detection of Phytophthora ‘taxon Agathis’ in soil, pathogen of Kauri in New Zealand. For. Pathol. 2013, 43, 324–330. [Google Scholar] [CrossRef]
- Cavalier-Smith, T.; Chao, E.E.-Y. Phylogeny and classification of phylum Cercozoa (Protozoa). Protist 2003, 154, 341–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flues, S.; Blokker, M.; Dumack, K.; Bonkowski, M. Diversity of Cercomonad species in the phyllosphere and rhizosphere of different plant species with a description of Neocercomonas epiphylla (Cercozoa, Rhizaria) a leaf-associated Protist. J. Eukaryot. Microbiol. 2018, 65, 587–599. [Google Scholar] [CrossRef]
- Niwa, R.; Kawahara, A.; Murakami, H.; Tanaka, S.; Ezawa, T. Complete sructure of nuclear rDNA of the obligate plant parasite Plasmodiophora brassicae: Intraspecific polymorphisms in the exon and group I intron of the large subunit rDNA. Protist 2011, 162, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Jayawardena, R.S.; Purahong, W.; Zhang, W.; Wubet, T.; Li, X.; Liu, M.; Zhao, W.; Hyde, K.D.; Liu, J.; Yan, J. Biodiversity of fungi on Vitis vinifera L. revealed by traditional and high-resolution culture-independent approaches. Fungal Divers. 2018, 90, 1–84. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.-L.; Zhang, C.-L.; Lin, F.-C.; Kubicek, C.P. Identity, diversity, and molecular phylogeny of the endophytic mycobiota in the roots of rare wild rice (Oryza granulata) from a nature reserve in Yunnan, China. Appl. Environ. Microbiol. 2010, 76, 1642–1652. [Google Scholar] [CrossRef] [Green Version]
- Dissanayake, A.J.; Purahong, W.; Wubet, T.; Hyde, K.D.; Zhang, W.; Xu, H.; Zhang, G.; Fu, C.; Liu, M.; Xing, Q.; et al. Direct comparison of culture-dependent and culture-independent molecular approaches reveal the diversity of fungal endophytic communities in stems of grapevine (Vitis vinifera). Fungal Divers. 2018, 90, 85–107. [Google Scholar] [CrossRef]
- Nilsson, H.; Kristiansson, E.; Ryberg, M.; Hallenberg, N.; Larsson, K.-H. Intraspecific ITS variability in the kingdom fungi as expressed in the international sequence databases and its implications for molecular species identification. Evol. Bioinform. 2008, 4, EBO.S653–201. [Google Scholar] [CrossRef]
- Hughes, K.W.; Petersen, R.H.; Lickey, E.B. Using heterozygosity to estimate a percentage DNA sequence similarity for environmental species’ delimitation across basidiomycete fungi. New Phytol. 2009, 182, 795–798. [Google Scholar] [CrossRef]
- Gazis, R.; Rehner, S.A.; Chaverri, P. Species delimitation in fungal endophyte diversity studies and its implications in ecological and biogeographic inferences. Mol. Ecol. 2011, 20, 3001–3013. [Google Scholar] [CrossRef]
- Merc. Malt Extract Agar Modified, Vegitone and Potato Dextrose Agar. Malt Extract Agar Modified, Vegitone and Potato Dextrose Agar 2019. Available online: https://www.sigmaaldrich.com/catalog/product/sial/38954?lang=en®ion=IE (accessed on 1 February 2019).
- Pelaez, F.; Collado, J.; Arenal, F.; Basílio, A.; Cabello, A.; Matas, M.D.; Garcia, J.; Del Val, A.G.; Gonzalez, V.; Gorrochategui, J.; et al. Endophytic fungi from plants living on gypsum soils as a source of secondary metabolites with antimicrobial activity. Mycol. Res. 1998, 102, 755–761. [Google Scholar] [CrossRef]
- Herrera, J.; Khidir, H.H.; Eudy, D.M.; Porras-Alfaro, A.; Natvig, D.O.; Sinsabaugh, R.L. Shifting fungal endophyte communities colonize Bouteloua gracilis: Effect of host tissue and geographical distribution. Mycologia 2010, 102, 1012–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, Y.-S.; Weller, D.M. Take-all of wheat and natural disease suppression: A review. Plant Pathol. J. 2013, 29, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Márquez, S.; Bills, G.F.; Zabalgogeazcoa, I. The endophytic mycobiota of the grass Dactylis glomerata. Fungal Divers. 2007, 27, 171–195. [Google Scholar]
- Fisher, P.J.; Petrini, O. Fungal saprobes and pathogens as endophytes of rice (Oryza sativa L.). New Phytol. 1992, 120, 137–143. [Google Scholar] [CrossRef]
- Høyer, A.K.; Jørgensen, H.J.L.; Jensen, B.; Murphy, B.R.; Hodkinson, T.R.; Doohan, F.M.; Saunders, M.J. Emerging methods for biological control of barley diseases including the role of endophytes. In Endophytes for a Growing World; Hodkinson, T.R., Doohan, F.M., Saunders, M.J., Murphy, B.R., Eds.; Cambridge University Press: Cambridge, UK, 2019; pp. 93–119. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Kingdom | Division | Class | Number of Plants | Site | ||||
|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | V | ||||
| Chromista | – | – | 2 | × | ||||
| Fungi | Ascomycota | Archaeorhizomycetes | 12 | × | × | × | × | × |
| Dothideomycetes | 48 | × | × | × | × | × | ||
| Eurotiomycetes | 48 | × | × | × | × | × | ||
| Lecanoromycetes | 13 | × | × | × | × | × | ||
| Leotiomycetes | 48 | × | × | × | × | × | ||
| Orbiliomycetes | 16 | × | × | × | × | × | ||
| Pezizomycetes | 34 | × | × | × | × | × | ||
| Saccharomycetes | 43 | × | × | × | × | × | ||
| Sordariomycetes | 48 | × | × | × | × | × | ||
| Taphrinomycetes | 1 | × | ||||||
| Xylonomycetes | 2 | × | ||||||
| Basidiomycota | Agaricomycetes | 48 | × | × | × | × | × | |
| Agaricostilbomycetes | 1 | × | ||||||
| Exobasidiomycetes | 1 | × | ||||||
| Malasseziomycetes | 32 | × | × | × | × | × | ||
| Microbotryomycetes | 26 | × | × | × | × | × | ||
| Pucciniomycetes | 4 | × | ||||||
| Tremellomycetes | 47 | × | × | × | × | × | ||
| Tritirachiomycetes | 1 | × | ||||||
| Ustilaginomycetes | 17 | × | × | × | × | × | ||
| Wallemiomycetes | 7 | × | × | × | × | |||
| Chytridiomycota | Rhizophydiomycetes | 1 | × | |||||
| Spizellomycetes | 4 | × | × | × | × | |||
| Glomeromycotina | Archaeosporomycetes | 6 | × | × | ||||
| Glomeromycetes | 48 | × | × | × | × | × | ||
| Paraglomeromycetes | 12 | × | × | × | × | × | ||
| Mortierellomycota | Mortierellomycetes | 48 | × | × | × | × | × | |
| Mucoromycota | Endogonomycetes | 13 | × | × | × | × | × | |
| Mucoromycetes | 32 | × | × | × | × | × | ||
| Olpidiomycota | Olpidiomycetes | 3 | × | × | ||||
| Rozellomycota | – | 5 | × | × | ||||
| Rhizaria | Cercozoa | – | 2 | × | × | |||
| Class | Species | Class | Species |
|---|---|---|---|
| – | Fungi sp. 1 | Leotiomycetes | Articulospora sp. |
| – | Fungi sp. 2 | Glarea sp. | |
| – | Ascomycota sp. | Hymenoscyphus sp. | |
| Dothideomycetes | Dothideomycetes sp. | Tetracladium sp. | |
| Capnodiales sp. | Tetracladium marchalianum | ||
| Pleosporales sp. | Tricladium splendens | ||
| Xenopyrenochaetopsis pratorum | Rhexocercosporidium panacis | ||
| Didymellaceae sp. | Microscypha sp. | ||
| Neoascochyta graminicola | Sordariomycetes | Sordariomycetes sp. | |
| Didymosphaeriaceae sp. | Codinaea acaciae | ||
| Stagonospora pseudovitensis | Pseudolachnella sp. | ||
| Melanommataceae sp. | Gibellulopsis nigrescens | ||
| Periconia sp. | Dactylonectria macrodidyma | ||
| Ophiosphaerella sp. | Gaeumannomyces graminis | ||
| Phaeosphaeria triglochinicola | Slopeiomyces cylindrosporus | ||
| Phaeosphaeriaceae sp. | Myrmecridium sp. | ||
| Alternaria sp. | Pleotrichocladium opacum | ||
| Alternaria hordeicola | Schizothecium glutinans | ||
| Drechslera sp. | Microdochium sp. | ||
| Eurotiomycetes | Exophiala sp. | Microdochium bolleyi | |
| Aspergillus sydowii | Microdochium phragmitis | ||
| Leotiomycetes | Helotiales sp. | Agaricomycetes | Agaricomycetes sp. |
| Helotiaceae sp. 1 | Glomeromycetes | Glomeraceae sp. | |
| Helotiaceae sp. 2 | Mortierellomycetes | Mortierella exigua |
| OTU | Sequence(s) | Class | Identification |
|---|---|---|---|
| 1 | 4 | Dothideomycetes | Clohesyomyces sp. |
| 2 | 1 | Dothideomycetes sp. 1 | |
| 3 | 8 | Dothideomycetes sp. 2 | |
| 4 | 1 | Dothideomycetes sp. 3 | |
| 5 | 1 | Dothideomycetes sp. 4 | |
| 6 | 1 | Dothideomycetes sp. 5 | |
| 7 | 1 | Dothideomycetes sp. 6 | |
| 8 | 15 | Dothideomycetes sp. 7 | |
| 9 | 6 | Epicoccum nigrum | |
| 10 | 1 | Epicoccum sp. | |
| 11 | 2 | Ophiosphaerella korrea | |
| 12 | 1 | Ophiosphaerella sp. 1 | |
| 13 | 4 | Periconia sp. 1 | |
| 14 | 1 | Periconia sp. 2 | |
| 15 | 1 | Pleosporaceae sp. | |
| 16 | 2 | Leotiomycetes | Glarea sp. |
| 17 | 34 | Leptodontidium sp. | |
| 18 | 2 | Pezizomycetes | Pyronema domesticum |
| 19 | 7 | Sordariomycetes | Chaetosphaeriaceae sp. |
| 20 | 1 | Diaporthe sp. | |
| 21 | 2 | Falciphora sp. | |
| 22 | 1 | Gaeumannomyces graminis | |
| 23 | 2 | Lasiosphaeriaceae sp. | |
| 24 | 1 | Sordariomycetes sp. 1 | |
| 25 | 1 | Sordariomycetes sp. 2 | |
| 26 | 2 | Sordariomycetes sp. 3 | |
| 27 | 1 | Xylariaceae sp. | |
| 28–54 | 27 | Fungus sp. 28–54 | |
| 55–66 | 12 | Fungus sp. 55–66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Høyer, A.K.; Hodkinson, T.R. Hidden Fungi: Combining Culture-Dependent and -Independent DNA Barcoding Reveals Inter-Plant Variation in Species Richness of Endophytic Root Fungi in Elymus repens. J. Fungi 2021, 7, 466. https://doi.org/10.3390/jof7060466
Høyer AK, Hodkinson TR. Hidden Fungi: Combining Culture-Dependent and -Independent DNA Barcoding Reveals Inter-Plant Variation in Species Richness of Endophytic Root Fungi in Elymus repens. Journal of Fungi. 2021; 7(6):466. https://doi.org/10.3390/jof7060466
Chicago/Turabian StyleHøyer, Anna K., and Trevor R. Hodkinson. 2021. "Hidden Fungi: Combining Culture-Dependent and -Independent DNA Barcoding Reveals Inter-Plant Variation in Species Richness of Endophytic Root Fungi in Elymus repens" Journal of Fungi 7, no. 6: 466. https://doi.org/10.3390/jof7060466
APA StyleHøyer, A. K., & Hodkinson, T. R. (2021). Hidden Fungi: Combining Culture-Dependent and -Independent DNA Barcoding Reveals Inter-Plant Variation in Species Richness of Endophytic Root Fungi in Elymus repens. Journal of Fungi, 7(6), 466. https://doi.org/10.3390/jof7060466