
Genomic Characterization of Parengyodontium torokii sp. nov., a Biofilm-Forming Fungus Isolated from Mars 2020 Assembly Facility

 , ,
, ,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Isolation of Fungi
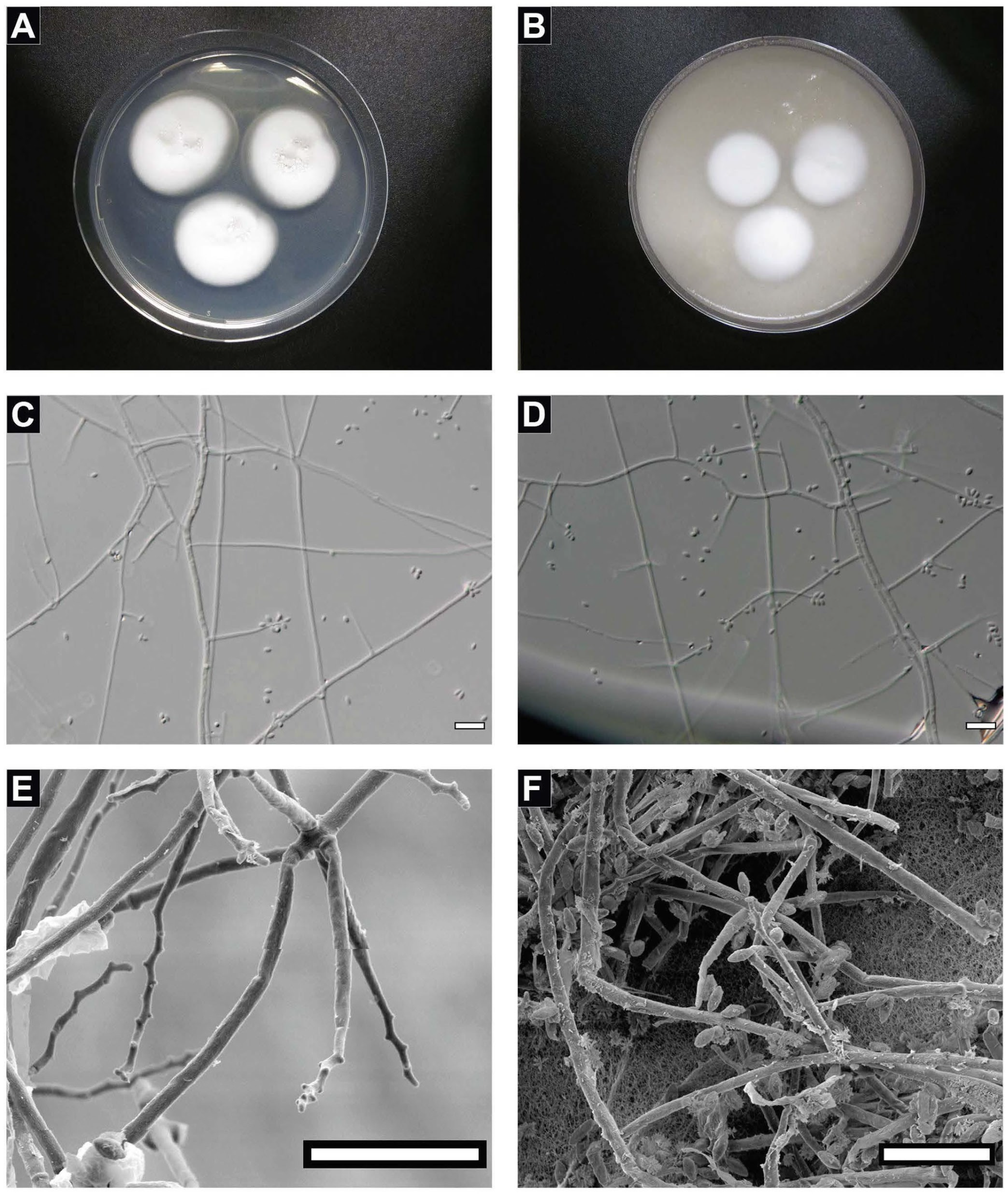
2.3. Morphological Analysis
2.4. Scanning Electron Microscopy
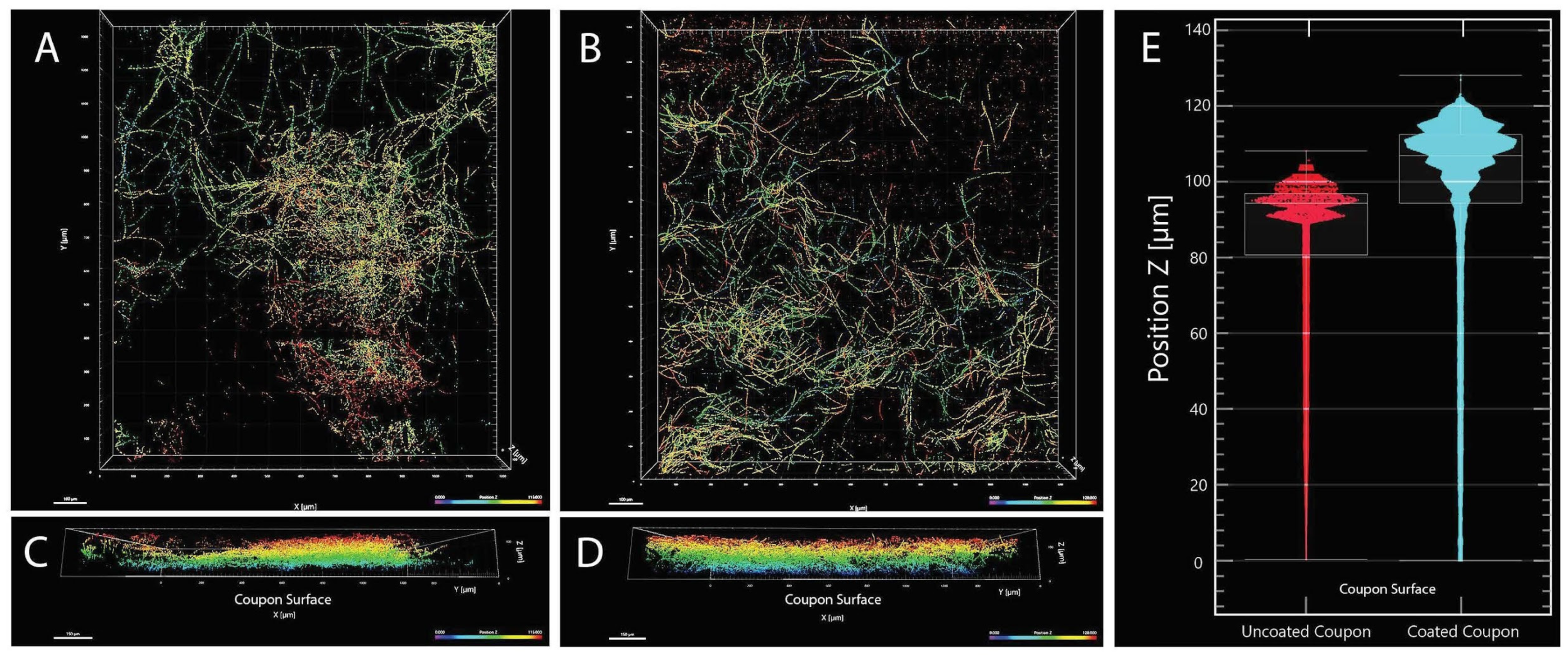
2.5. Biofilm Formation
2.6. Confocal Microscopy
2.7. ITS-Based Fungal Identification
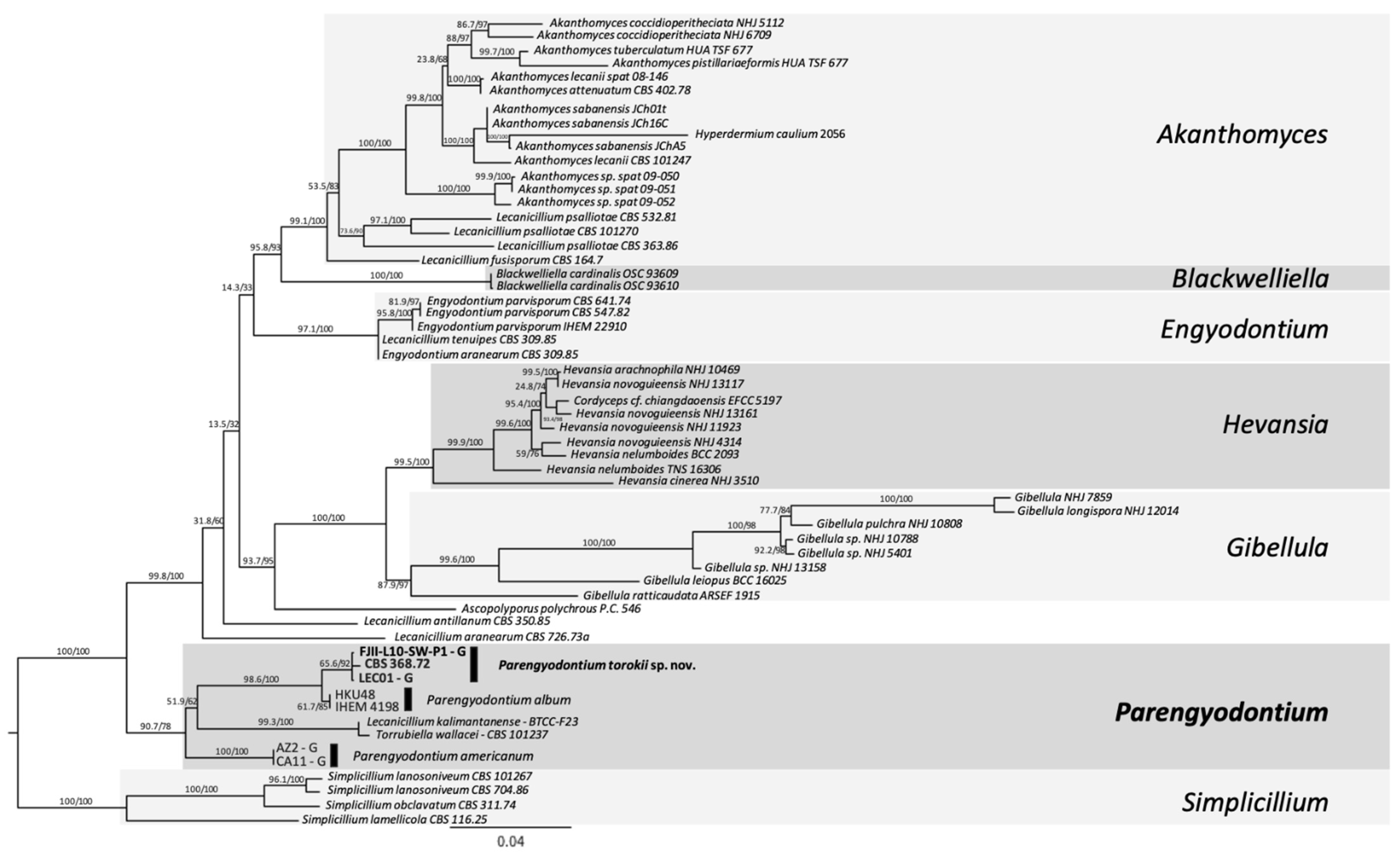
2.8. MLST-Based Phylogenetic Analyses
- (a)
- Three-gene MLST analyses. Sequences from ITS, 28S nrDNA, and β-tubulin genes were used in a dataset comprised of 22 fungi, including the outgroup. The outgroup selection was based on [16]. Multiple sequence alignments were generated using MAFFT default settings using PhyloSuite v.1.2.1 [32]. The alignments were trimmed to remove ambiguous characters using GBlocks [33,34]. For the concatenated dataset, PartitionFinder 2 [35] was used to select the best-fit model according to the Akaike Information Criterion corrected (AICc) [36]. The best-fitting substitution models according to AICc were: ITS and β-tubulin: GTR+I+G and LSU: TRN+I. ModelFinder [37] was used for the ITS dataset to select the best-fit model using the AICc criterion. The best-fit model according to AICc was TIM2+F+R2. The trimmed alignment was then used to construct a ML tree using IQ-TREE implemented in PhyloSuite. Ultrafast bootstrapping was done with 5000 replicates [38]. Nodes with UFBoot ≥90% are shown on the clades, but only nodes ≥95% were considered strongly supported. Bayesian inference phylogenies were inferred using MrBayes 3.2.6 [39] under partition model (2 parallel runs, 10 million generations), using PhyloSuite v. 2.1. Four independent chains of Metropolis-coupled MCMC were run for 10 million generations with trees sampled every 1000th generation, resulting in 10,000 trees. The first 25% of the trees were discarded as a burn-in parameter. The average standard deviation of split frequencies value approaching 0.001 was used to estimate that the two runs had converged closer to the stationary phase (10 million generations). Consensus trees were generated and viewed in PAUP* v.4.0a (build 166) [40]. Clades with a posterior probability (PP) ≥95% were considered significant and strongly supported.
- (b)
- Six-loci MLST analyses. Gene sequences utilized were: ITS region rRNA gene, D1/D2 domain of large subunit (LSU or 26S) rRNA gene, small subunit (SSU or 18S) rRNA gene, and housekeeping genes including two subunits of RNA polymerase II (RPB1 and RPB2) and the translation elongation factor 1-α (TEF1). These six-loci have already been established for differentiating Cordycipitaceae species [41]. Sequences of 58 fungal strains available were downloaded, and sequences were manually concatenated (representative sequences are available at [41]. The respective gene sequences that were available on NCBI for different Parengyodontium species (n = 8 isolates) were included in the phylogenetic analysis except for the Mars 2020 strain (FJII-L10-SW-P1), which was generated during this study. For MLST, sequences were aligned using MAFFT v7 [42], concatenated manually, trimmed using the ClipKit tool, smart-gap function [43] and a ML Tree was generated using the using IQTREE2 v2.0.6 [31,44]. The best substitution model was calculated using the ModelFinder algorithm [37] and 1000 ultrafast bootstraps [45] and SH-like approximate likelihood ratio test (aLRT) were used to test branch support [46]. Finally, the trees were visualized using the FigTree v 1.4.4 software (http://tree.bio.ed.ac.uk/software/figtree/, accessed on 12 November 2021).
2.9. Whole-Genome Sequencing Analyses
2.10. De Novo and Functional Genome Annotation
3. Comparative Analysis of Fungal Genomes
Metabolomics
4. Results
4.1. Taxonomy of the Strain FJII-L10-SW-P1
4.2. Biofilm Formation of the Strain FJII-L10-SW-P1
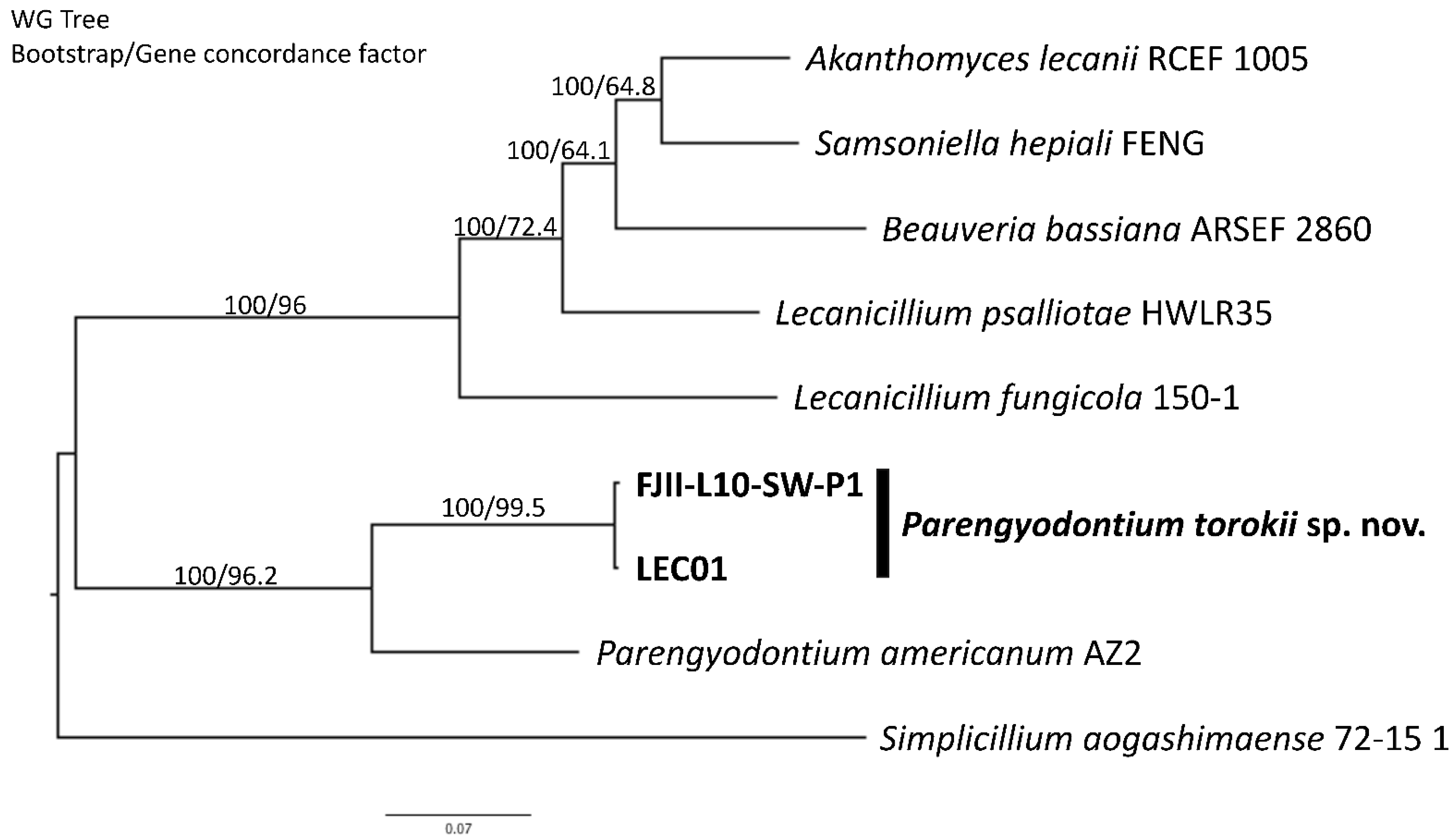
4.3. Phylogenetic Analyses of the Strain FJII-L10-SW-P1
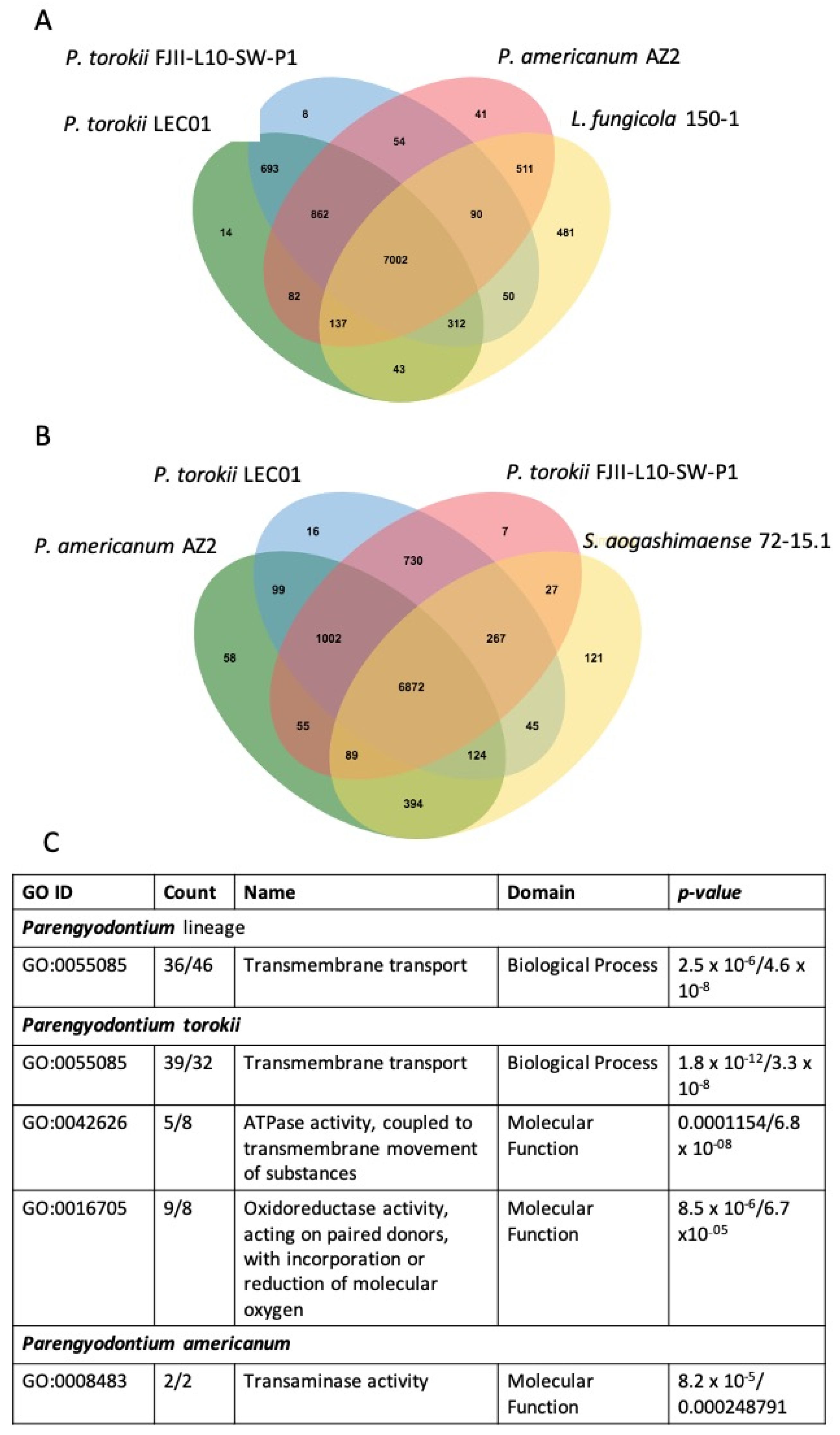
4.4. Whole-Genome Sequence Analyses
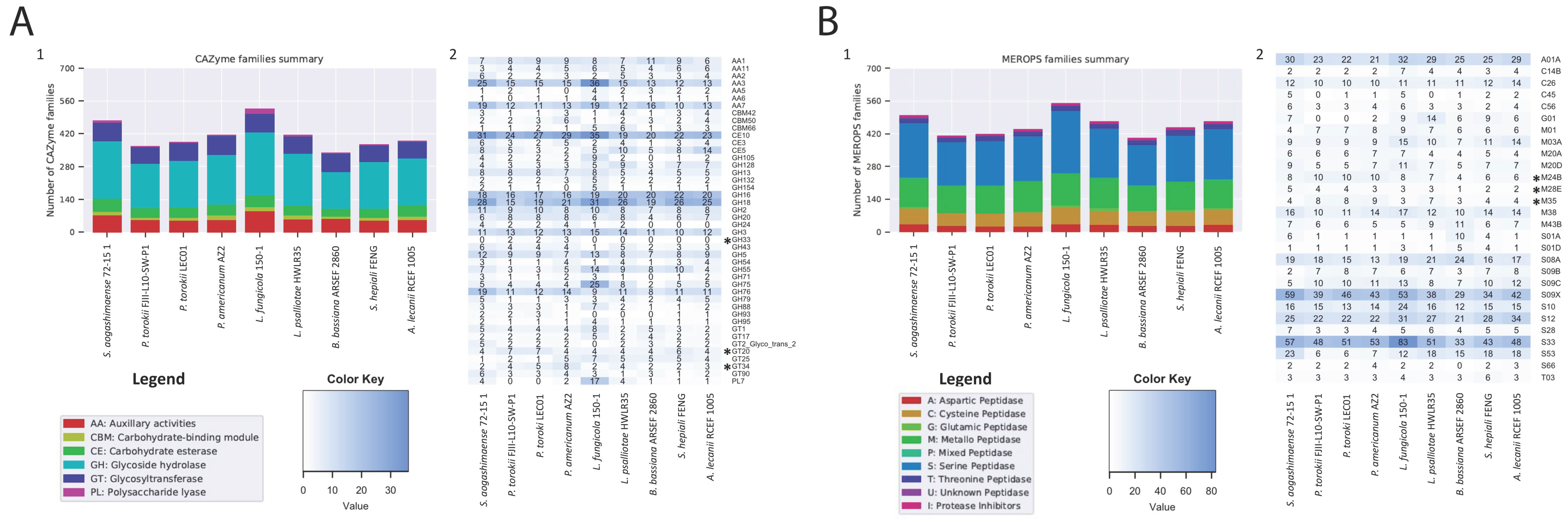
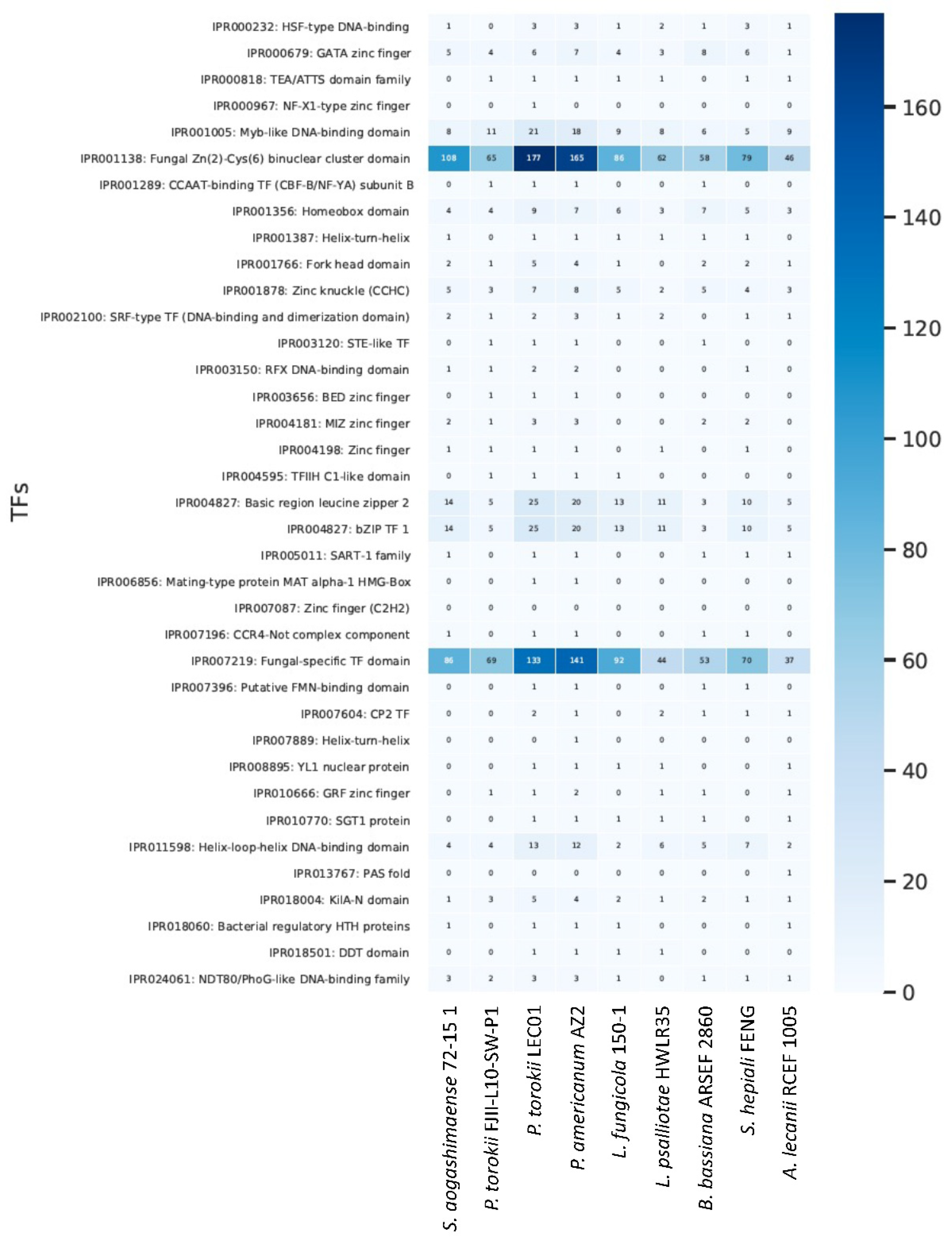
4.5. Genomic Features of the P. torokii FJII-L10-SW-P1 Strain
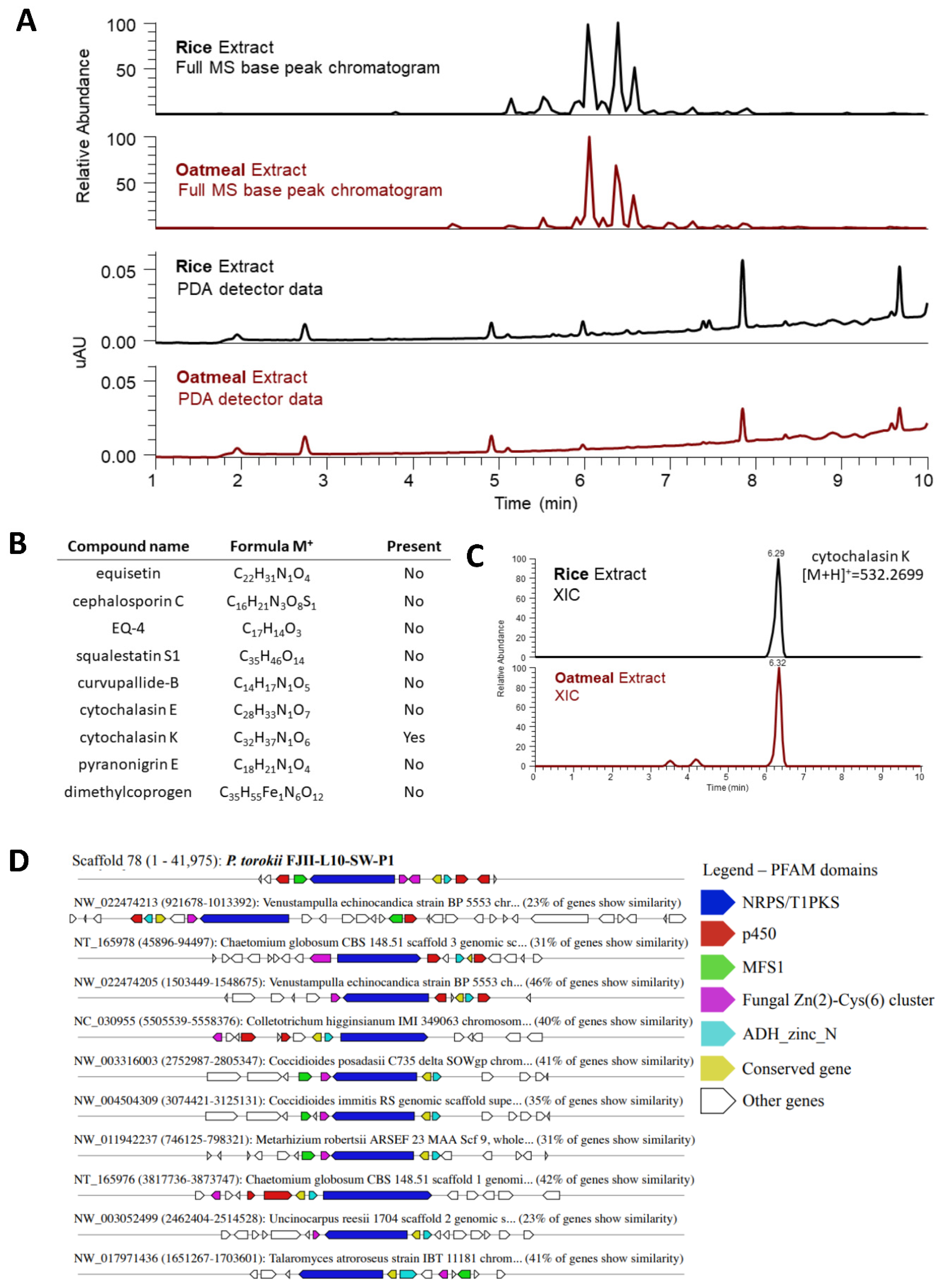
4.6. Metabolomic Profiling of P. torokii FJII-L10-SW-P1 Strain
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benardini, J.N., 3rd; La Duc, M.T.; Ballou, D.; Koukol, R. Implementing planetary protection on the atlas v fairing and ground systems used to launch the Mars Science Laboratory. Astrobiology 2014, 14, 33–41. [Google Scholar] [CrossRef]
- Benardini, J.N., 3rd; La Duc, M.T.; Beaudet, R.A.; Koukol, R. Implementing planetary protection measures on the Mars Science Laboratory. Astrobiology 2014, 14, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Vaishampayan, P.; Probst, A.J.; La Duc, M.T.; Bargoma, E.; Benardini, J.N.; Andersen, G.L.; Venkateswaran, K. New perspectives on viable microbial communities in low-biomass cleanroom environments. ISME J. 2013, 7, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Gorbushina, A. Microcolonial fungi: Survival potential of terrestrial vegetative structures. Astrobiology 2003, 3, 543–554. [Google Scholar] [CrossRef]
- Blachowicz, A.; Chiang, A.J.; Elsaesser, A.; Kalkum, M.; Ehrenfreund, P.; Stajich, J.E.; Torok, T.; Wang, C.C.C.; Venkateswaran, K. Proteomic and Metabolomic Characteristics of Extremophilic Fungi under Simulated Mars Conditions. Front. Microbiol. 2019, 10, 1013. [Google Scholar] [CrossRef] [PubMed]
- Mhatre, S.; Singh, N.K.; Wood, J.M.; Parker, C.W.; Pukall, R.; Verbarg, S.; Tindall, B.J.; Neumann-Schaal, M.; Venkateswaran, K. Description of Chloramphenicol Resistant Kineococcus rubinsiae sp. nov. Isolated from a Spacecraft Assembly Facility. Front. Microbiol. 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Radwan, O.; Gunasekera, T.S.; Ruiz, O.N. Draft Genome Sequence of Lecanicillium sp. Isolate LEC01, a Fungus Capable of Hydrocarbon Degradation. Microbiol. Resour. Announc. 2019, 8, e01744-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Q.; Wang, J.; Zhang, X.; Nong, X.; Xu, X.; Qi, S. Cytotoxic polyketides from the deep-sea-derived fungus Engyodontium album DFFSCS021. Mar. Drugs 2014, 12, 5902–5915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Yang, H.-Y.; You, X.-L.; Li, Y.-H. Diversity of endophytic fungi from roots of Panax ginseng and their saponin yield capacities. SpringerPlus 2013, 2, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kachuei, R.; Emami, M.; Naeimi, B.; Diba, K. Isolation of keratinophilic fungi from soil in Isfahan province, Iran. J. Mycol. Med. 2012, 22, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Simonovicova, A.; Godyova, M.; Kunert, J. Engyodontium album, a new species of microscopic fungi for Slovakia and its keratinolytic activity. Biol. Bratisl. 2004, 59, 17–18. [Google Scholar]
- Leplat, J.; Francois, A.; Bousta, F. Parengyodontium album, a frequently reported fungal species in the cultural heritage environment. Fungal Biol. Rev. 2020, 34, 126–135. [Google Scholar] [CrossRef]
- Vuillemin, P. Beauveria, nouveau genera de Verticilla-cees. Bull. Soc. Bot. Fr. 1912, 59, 7. [Google Scholar]
- Limber, D.P. A New Form Genus of the Moniliaceae. Mycologia 1940, 32, 23–30. [Google Scholar] [CrossRef]
- Gams, W.; de Hoog, G.S.; Samson, R.A.; Evans, H.C. The hyphomycete genus Engyodontium a link between Verticillium and Aphanocladium. Pers. Mol. Phylogeny Evol. Fungi 1984, 12, 135–147. [Google Scholar]
- Tsang, C.-C.; Chan, J.F.W.; Pong, W.-M.; Chen, J.H.K.; Ngan, A.H.Y.; Cheung, M.; Lai, C.K.C.; Tsang, D.N.C.; Lau, S.K.P.; Woo, P.C.Y. Cutaneous hyalohyphomycosis due to Parengyodontium album gen. et comb. nov. Med. Mycol. 2016, 54, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, M.M.; Muszewska, A.; Travis, J.; Moreno, L.F.; Ahmed, S.; Roe, C.; Mead, H.; Steczkiewicz, K.; Lemmer, D.; de Hoog, S.; et al. Genomic characterization of Parengyodontium americanum sp. nov. Fungal Genet Biol 2020, 138, 103351. [Google Scholar] [CrossRef]
- Liu, X.Z.; Wang, Q.M.; Theelen, B.; Groenewald, M.; Bai, F.Y.; Boekhout, T. Phylogeny of tremellomycetous yeasts and related dimorphic and filamentous basidiomycetes reconstructed from multiple gene sequence analyses. Stud. Mycol. 2015, 81, 1–26. [Google Scholar] [CrossRef] [Green Version]
- El-Elimat, T.; Figueroa, M.; Ehrmann, B.M.; Cech, N.B.; Pearce, C.J.; Oberlies, N.H. High-Resolution MS, MS/MS, and UV Database of Fungal Secondary Metabolites as a Dereplication Protocol for Bioactive Natural Products. J. Nat. Prod. 2013, 76, 1709–1716. [Google Scholar] [CrossRef] [Green Version]
- Paguigan, N.D.; El-Elimat, T.; Kao, D.; Raja, H.A.; Pearce, C.J.; Oberlies, N.H. Enhanced dereplication of fungal cultures via use of mass defect filtering. J. Antibiot. 2017, 70, 553–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, K.; Cooper, M.; La Duc, M.T.; Vaishampayan, P.; Stam, C.; Benardini, J.N.; Scalzi, G.; Moissl-Eichinger, C.; Venkateswaran, K. Evaluation of procedures for the collection, processing, and analysis of biomolecules from low-biomass surfaces. Appl. Environ. Microbiol. 2011, 77, 2943–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checinska Sielaff, A.; Urbaniak, C.; Mohan, G.B.M.; Stepanov, V.G.; Tran, Q.; Wood, J.M.; Minich, J.; McDonald, D.; Mayer, T.; Knight, R.; et al. Characterization of the total and viable bacterial and fungal communities associated with the International Space Station surfaces. Microbiome 2019, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, R.; Lundgren, P.; Malli-Mohan, G.B.; Urbaniak, C.; Benardini, J.N.; Venkateswaran, K. Comprehensive measurement of microbial burden in nutrient-deprived cleanrooms. In Proceedings of the 47th International Conference on Environmental Systems, ICES-2017-177, Charleston, SC, USA, 16–20 July 2017. [Google Scholar]
- Riddell, R.W. Permanent stained mycological preparations obtained by slide culture. Mycologia 1950, 42, 265–270. [Google Scholar] [CrossRef]
- Murray, J.; Muruko, T.; Gill, C.I.R.; Kearney, M.P.; Farren, D.; Scott, M.G.; McMullan, G.; Ternan, N.G. Evaluation of bactericidal and anti-biofilm properties of a novel surface-active organosilane biocide against healthcare associated pathogens and Pseudomonas aeruginosa biolfilm. PLoS ONE 2017, 12, e0182624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkateswaran, K.; Chung, S.; Allton, J.; Kern, R. Evaluation of various cleaning methods to remove Bacillus spores from spacecraft hardware materials. Astrobiology 2004, 4, 377–390. [Google Scholar] [PubMed] [Green Version]
- Lai, X.; Cao, L.; Tan, H.; Fang, S.; Huang, Y.; Zhou, S. Fungal communities from methane hydrate-bearing deep-sea marine sediments in South China Sea. ISME J 2007, 1, 756–762. [Google Scholar] [CrossRef]
- Taylor, D.L.; Bruns, T.D. Community structure of ectomycorrhizal fungi in a Pinus muricata forest: Minimal overlap between the mature forest and resistant propagule communities. Mol. Ecol. 1999, 8, 1837–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blachowicz, A.; Mayer, T.; Bashir, M.; Pieber, T.R.; León, P.D.; Venkateswaran, K. Human presence impacts fungal diversity of inflated lunar/Mars analog habitat. Microbiome 2017, 5, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Hoff, M.; Orf, S.; Riehm, B.; Darriba, D.; Stamatakis, A. Does the choice of nucleotide substitution models matter topologically? BMC Bioinform. 2016, 17, 143. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (* and Other Methods) Version 4; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Kepler, R.M.; Luangsa-ard, J.J.; Hywel-Jones, N.L.; Quandt, C.A.; Sung, G.-H.; Rehner, S.A.; Aime, M.C.; Henkel, T.W.; Sanjuan, T.; Zare, R.; et al. A phylogenetically-based nomenclature for Cordycipitaceae (Hypocreales). IMA Fungus 2017, 8, 335–353. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Steenwyk, J.L.; Buida, T.J., III; Li, Y.; Shen, X.-X.; Rokas, A. ClipKIT: A multiple sequence alignment trimming software for accurate phylogenomic inference. PLoS Biol. 2020, 18, e3001007. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Bijlani, S.; Singh, N.K.; Mason, C.E.; Wang, C.C.C.; Venkateswaran, K. Draft Genome Sequences of Tremellomycetes Strains Isolated from the International Space Station. Microbiol Resour Announc 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Stajich, J.; Palmer, J. stajichlab/AAFTF: v0.2.3 release with (v0.2.3). Zenodo 2019. Available online: https://zenodo.org/record/3437300 (accessed on 12 November 2021).
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Zenodo. nextgenusfs/funannotate: Funannotate v1.5.3. 2020. Available online: https://zenodo.org/record/2604804 (accessed on 12 November 2021).
- Frith, M.C. A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res. 2010, 39, e23. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.M.; Hubley, R.; Goubert, C.; Rosen, J.; Clark, A.G.; Feschotte, C.; Smit, A.F. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 2020, 117, 9451–9457. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Smit, A.; Hubley, R.; Green, P. RepeatMasker Open-4.0. 2013–2015. Available online: http://www.repeatmasker.org (accessed on 12 November 2021).
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. In Gene Prediction: Methods and Protocols; Kollmar, M., Ed.; Springer: New York, NY, USA, 2019; pp. 1–14. [Google Scholar]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2020, 49, D412–D419. [Google Scholar] [CrossRef]
- Blum, M.; Chang, H.-Y.; Chuguransky, S.; Grego, T.; Kandasaamy, S.; Mitchell, A.; Nuka, G.; Paysan-Lafosse, T.; Qureshi, M.; Raj, S.; et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 2020, 49, D344–D354. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics 2005, 21, i251–i257. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2013, 42, D503–D509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Hahn, M.W.; Lanfear, R. New Methods to Calculate Concordance Factors for Phylogenomic Datasets. Mol. Biol. Evol. 2020, 37, 2727–2733. [Google Scholar] [CrossRef]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Lu, M.; Ling, E.; Li, P.; Wang, C. A M35 family metalloprotease is required for fungal virulence against insects by inactivating host prophenoloxidases and beyond. Virulence 2020, 11, 222–237. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Huang, H.; Wu, B.; Xie, J.; Viljoen, A.; Wang, W.; Mostert, D.; Xie, Y.; Fu, G.; Xiang, D.; et al. The M35 Metalloprotease Effector FocM35_1 Is Required for Full Virulence of Fusarium oxysporum f. sp. cubense Tropical Race 4. Pathogens 2021, 10, 670. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Hawksworth, D.L.; Lücking, R. Fungal Diversity Revisited: 2.2 to 3.8 Million Species. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Visagie, C.M.; Houbraken, J.; Frisvad, J.C.; Hong, S.B.; Klaassen, C.H.; Perrone, G.; Seifert, K.A.; Varga, J.; Yaguchi, T.; Samson, R.A. Identification and nomenclature of the genus Penicillium. Stud. Mycol. 2014, 78, 343–371. [Google Scholar] [CrossRef] [Green Version]
- Lücking, R.; Aime, M.C.; Robbertse, B.; Miller, A.N.; Ariyawansa, H.A.; Aoki, T.; Cardinali, G.; Crous, P.W.; Druzhinina, I.S.; Geiser, D.M.; et al. Unambiguous identification of fungi: Where do we stand and how accurate and precise is fungal DNA barcoding? IMA Fungus 2020, 11, 14. [Google Scholar] [CrossRef]
- Lynch, A.S.; Robertson, G.T. Bacterial and fungal biofilm infections. Annu. Rev. Med. 2008, 59, 415–428. [Google Scholar] [CrossRef]
- Seneviratne, G.; Zavahir, J.S.; Bandara, W.M.M.S.; Weerasekara, M.L.M.A.W. Fungal-bacterial biofilms: Their development for novel biotechnological applications. World J. Microbiol. Biotechnol. 2007, 24, 739. [Google Scholar] [CrossRef]
- Khatoon, Z.; McTiernan, C.D.; Suuronen, E.J.; Mah, T.F.; Alarcon, E.I. Bacterial biofilm formation on implantable devices and approaches to its treatment and prevention. Heliyon 2018, 4, e01067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, K.A.; Kerkaert, J.D.; Cramer, R.A. Aspergillus fumigatus biofilms: Toward understanding how growth as a multicellular network increases antifungal resistance and disease progression. PLoS Pathog. 2021, 17, e1009794. [Google Scholar] [CrossRef] [PubMed]
- Burmølle, M.; Ren, D.; Bjarnsholt, T.; Sørensen, S.J. Interactions in multispecies biofilms: Do they actually matter? Trends Microbiol. 2014, 22, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, Y.; Wu, H.; Hóiby, N.; Molin, S.; Song, Z.-j. Current understanding of multi-species biofilms. Int. J. Oral Sci. 2011, 3, 74–81. [Google Scholar] [CrossRef]
- Varki, A. Sialic acids as ligands in recognition phenomena. FASEB J. 1997, 11, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Breton, C.; Šnajdrová, L.; Jeanneau, C.; Koča, J.; Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 2005, 16, 29R–37R. [Google Scholar] [CrossRef] [PubMed]
- Loussert, C.; Schmitt, C.; Prevost, M.-C.; Balloy, V.; Fadel, E.; Philippe, B.; Kauffmann-Lacroix, C.; Latgé, J.P.; Beauvais, A. In vivo biofilm composition of Aspergillus fumigatus. Cell. Microbiol. 2010, 12, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Chellappan, S.; Jasmin, C.; Basheer, S.M.; Elyas, K.K.; Bhat, S.G.; Chandrasekaran, M. Production, purification and partial characterization of a novel protease from marine Engyodontium album BTMFS10 under solid state fermentation. Process Biochem. 2006, 41, 956–961. [Google Scholar] [CrossRef]
- Gunkel, F.A.; Gassen, H.G. Proteinase K from Tritirachium album Limber. Characterization of the chromosomal gene and expression of the cDNA in Escherichia coli. Eur. J. Biochem. 1989, 179, 185–194. [Google Scholar] [CrossRef]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2011, 41, 271–290. [Google Scholar] [CrossRef] [Green Version]
- Kitchener, R.L.; Grunden, A.M. Prolidase function in proline metabolism and its medical and biotechnological applications. J. Appl. Microbiol. 2012, 113, 233–247. [Google Scholar] [CrossRef]
- Monod, M.; Léchenne, B.; Jousson, O.; Grand, D.; Zaugg, C.; Stöcklin, R.; Grouzmann, E. Aminopeptidases and dipeptidyl-peptidases secreted by the dermatophyte Trichophyton rubrum. Microbiol. 2005, 151, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Yang, S.; Lee, B.H. Bioinformatic mapping of a more precise Aspergillus niger degradome. Sci. Rep. 2021, 11, 693. [Google Scholar] [CrossRef]
- Trendowski, M. Using cytochalasins to improve current chemotherapeutic approaches. Anticancer Agents Med. Chem. 2015, 15, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raguz, S.; Yagüe, E. Resistance to chemotherapy: New treatments and novel insights into an old problem. Br. J. Cancer 2008, 99, 387–391. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parengyodontium toroki FJII-L10-SW-P1 | Parengyodontium toroki LEC01 | Parengyodontium americanum AZ2 | Akanthomyces lecanii RCEF 1005 | Simplicillium aogashimaense 72-15 1 | Lecanicillium fungicola 150-1 | Lecanicillium psalliotae HWLR35 | Beauveria bassiana ARSEF 2860 | Samsoniella hepiali FENG | |
|---|---|---|---|---|---|---|---|---|---|
| Assembly | |||||||||
| # of contigs | 440 | 352 | 295 | 131 | 20 | 782 | 197 | 239 | 222 |
| Genome size | 30,424,506 | 31,084,693 | 32,962,623 | 35,580,375 | 29,244,117 | 44,547,425 | 36,133,949 | 33,693,821 | 34,650,604 |
| Largest contig | 718,708 | 1,200,404 | 821,300 | 5,461,016 | 4,930,463 | 493,648 | 4,365,396 | 2,084,429 | 1,229,925 |
| Repetitive DNA (%) | 2.83% | 7.31% | 9.98% | 11.29% | 7.38% | 8.44% | 11.34% | 11.78% | 11.91% |
| GC (%) | 50.45 | 50.28 | 52.88 | 53.10 | 49.01 | 49.87 | 52.73 | 51.36 | 53.89 |
| N50 | 122,374 | 310,369 | 345,622 | 3,613,853 | 3,162,613 | 154,124 | 2,330,369 | 724,305 | 576,310 |
| L50 | 70 | 27 | 31 | 4 | 4 | 92 | 6 | 13 | 20 |
| Annotation | |||||||||
| tRNA | 70 | 77 | 95 | 115 | 85 | 144 | 121 | 111 | 115 |
| intron | 16,303 | 16,562 | 16,709 | 14,195 | 15,835 | 19,694 | 13,365 | 15,912 | 13,429 |
| Exons | 25,899 | 26,363 | 27,008 | 24,306 | 25,962 | 32,964 | 23,566 | 25,710 | 23,649 |
| average exon length | 478 | 482 | 472 | 494 | 483 | 476 | 502 | 476 | 518 |
| mRNA | 9596 | 9801 | 10,299 | 10,111 | 10,127 | 13,270 | 10,201 | 9798 | 10,220 |
| CDS | 9596 | 9801 | 10,299 | 10,111 | 10,127 | 13,270 | 10,201 | 9798 | 10,220 |
| gene | 9666 | 9878 | 10,394 | 10,226 | 10,212 | 13,414 | 10,322 | 9909 | 10,335 |
| average gene length | 1658 | 1642 | 1570 | 1565 | 1579 | 1536 | 1521 | 1627 | 1560 |
| average protein length | 496 | 501 | 479 | 484 | 489 | 470 | 472 | 498 | 482 |
| Functional | |||||||||
| go_terms | 2913 | 5980 | 6243 | 1832 | 3109 | 2750 | 2306 | 2301 | 2947 |
| interproscan | 3915 | 8073 | 8454 | 2523 | 4184 | 3879 | 3206 | 3118 | 4061 |
| eggnog | 9330 | 9455 | 9892 | 9736 | 9780 | 12,457 | 9749 | 9461 | 9793 |
| pfam | 6961 | 7187 | 7476 | 7269 | 7530 | 9092 | 7160 | 7001 | 7141 |
| cazyme | 355 | 370 | 399 | 380 | 459 | 507 | 403 | 331 | 364 |
| merops | 412 | 418 | 438 | 472 | 498 | 549 | 471 | 402 | 447 |
| busco | 3685 | 3742 | 3739 | 3661 | 3748 | 3755 | 3596 | 3747 | 3573 |
| secretion | 826 | 899 | 995 | 1060 | 1144 | 1403 | 1144 | 1008 | 1009 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, C.W.; Teixeira, M.d.M.; Singh, N.K.; Raja, H.A.; Cank, K.B.; Spigolon, G.; Oberlies, N.H.; Barker, B.M.; Stajich, J.E.; Mason, C.E.; et al. Genomic Characterization of Parengyodontium torokii sp. nov., a Biofilm-Forming Fungus Isolated from Mars 2020 Assembly Facility. J. Fungi 2022, 8, 66. https://doi.org/10.3390/jof8010066
Parker CW, Teixeira MdM, Singh NK, Raja HA, Cank KB, Spigolon G, Oberlies NH, Barker BM, Stajich JE, Mason CE, et al. Genomic Characterization of Parengyodontium torokii sp. nov., a Biofilm-Forming Fungus Isolated from Mars 2020 Assembly Facility. Journal of Fungi. 2022; 8(1):66. https://doi.org/10.3390/jof8010066
Chicago/Turabian StyleParker, Ceth W., Marcus de Melo Teixeira, Nitin K. Singh, Huzefa A. Raja, Kristof B. Cank, Giada Spigolon, Nicholas H. Oberlies, Bridget M. Barker, Jason E. Stajich, Christopher E. Mason, and et al. 2022. "Genomic Characterization of Parengyodontium torokii sp. nov., a Biofilm-Forming Fungus Isolated from Mars 2020 Assembly Facility" Journal of Fungi 8, no. 1: 66. https://doi.org/10.3390/jof8010066
APA StyleParker, C. W., Teixeira, M. d. M., Singh, N. K., Raja, H. A., Cank, K. B., Spigolon, G., Oberlies, N. H., Barker, B. M., Stajich, J. E., Mason, C. E., & Venkateswaran, K. (2022). Genomic Characterization of Parengyodontium torokii sp. nov., a Biofilm-Forming Fungus Isolated from Mars 2020 Assembly Facility. Journal of Fungi, 8(1), 66. https://doi.org/10.3390/jof8010066