
Metagenomics Next Generation Sequencing (mNGS): An Exciting Tool for Early and Accurate Diagnostic of Fungal Pathogens in Plants

 , , and
, , and
Abstract
:1. Introduction
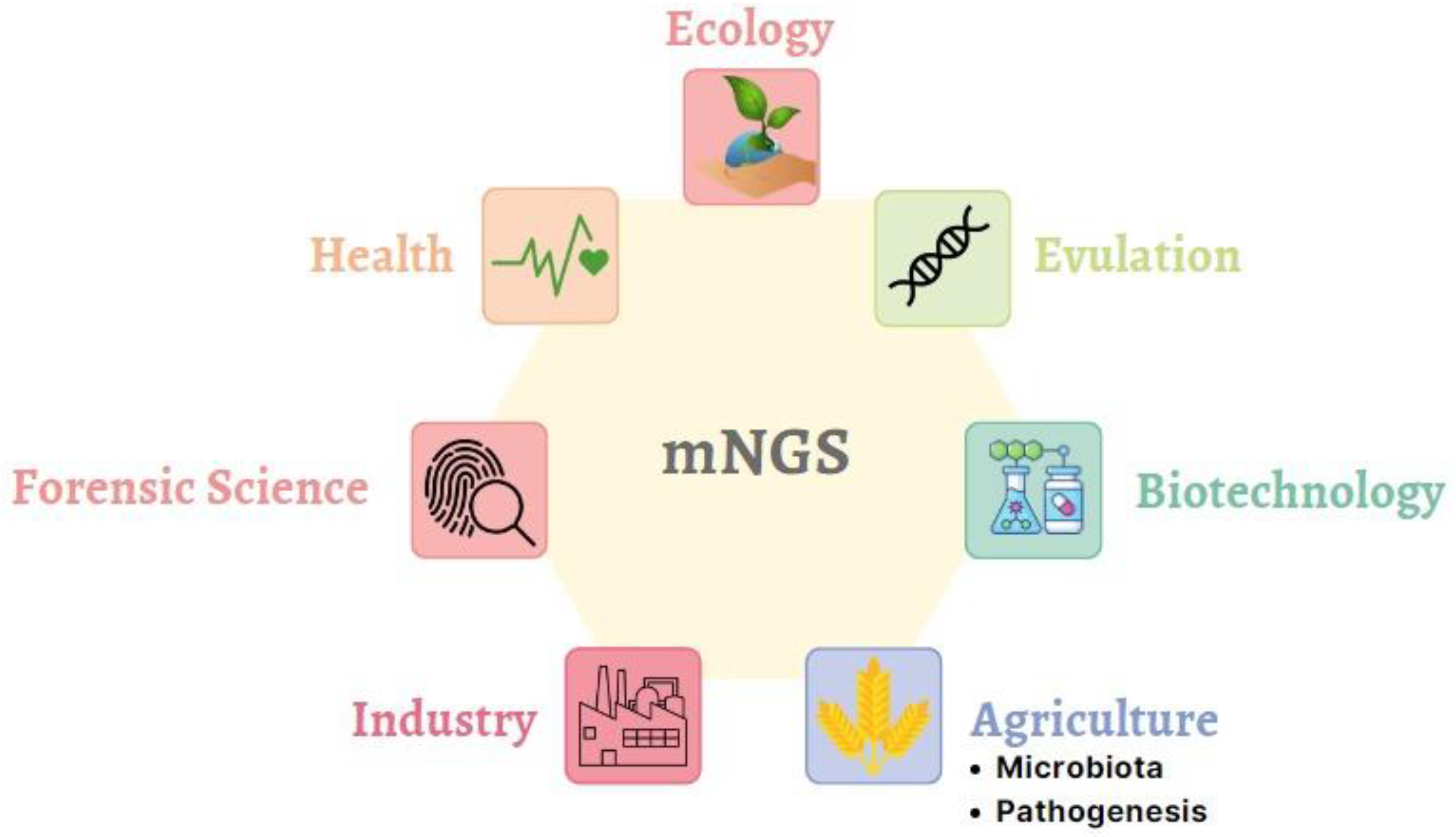
2. Multiple Real-World Applications for mNGS
3. mNGS Methodology for Detecting Fungal Pathogens in Plants
3.1. Wet Lab Applications
3.2. Preparation of Library
3.3. Sequencing
3.4. Bioinformatics Data Analysis
4. Successful Applications of mNGS in Fungal Plant Pathogen Detection
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woese, C.R. The problem of evolving a genetic code. Bioscience 1970, 20, 471–485. [Google Scholar] [CrossRef]
- Walters, W.A.; Knight, R. Technology and techniques for microbial ecology via DNA sequencing. Ann. Am. Thorac. Soc. 2014, 11 (Suppl. S1), S16–S20. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Gettu, N.; Swain, B.; Kumari, K.; Ojha, N.; Gunthe, S.S. Bioaerosol impact on crop health over India due to emerging fungal diseases (EFDs): An important missing link. Environ. Sci. Pollut. Res. 2020, 27, 12802–12829. [Google Scholar] [CrossRef]
- Ller, R.R.; Montoya, V.; Gardy, J.L.; Patrick, D.M.; Tang, P. Metagenomics for pathogen detection in public health. Genome Med. 2013, 5, 1–14. [Google Scholar]
- Hugenholtz, P.; Tyson, G.W. Metagenomics. Nature 2008, 455, 481–483. [Google Scholar] [CrossRef]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical metagenomic next-generation sequencing for pathogen detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Lipkin, W.I. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 2008, 358, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Li, Z.; Li, R.; Tan, P.; Zhang, R.; Li, J. mNGS in clinical microbiology laboratories: On the road to maturity. Crit. Rev. Microbiol. 2019, 45, 668–685. [Google Scholar] [CrossRef]
- Gu, W.; Deng, X.; Lee, M.; Sucu, Y.D.; Arevalo, S.; Stryke, D.; Federman, S.; Gopez, A.; Reyes, K.; Zorn, K.; et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat. Med. 2021, 27, 115–124. [Google Scholar] [CrossRef]
- Li, H.; Gao, H.; Meng, H.; Wang, Q.; Li, S.; Chen, H.; Li, Y.; Wang, H. Detection of pulmonary infectious pathogens from lung biopsy tissues by metagenomic next-generation sequencing. Front. Cell. Infect. Microbiol. 2018, 8, 205. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.-Y.; Wang, H.-Y.; Zhou, Y.; Zhang, H.-C.; Zhu, Y.-M.; Zhou, X.; Ying, Y.; Cui, P.; Wu, H.-L.; Zhang, W.-H.; et al. Improving pulmonary infection diagnosis with metagenomic next generation sequencing. Front. Cell. Infect. Microbiol. 2021, 10, 567615. [Google Scholar] [CrossRef]
- Wang, S.; Chen, Y.; Wang, D.; Wu, Y.; Zhao, D.; Zhang, J.; Xie, H.; Gong, Y.; Sun, R.; Nie, X.; et al. The feasibility of metagenomic next-generation sequencing to identify pathogens causing tuberculous meningitis in cerebrospinal fluid. Front. Microbiol. 2019, 10, 1993. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Huang, Z.; Li, W.; Fang, X.; Zhang, W. Can metagenomic next-generation sequencing identify the pathogens responsible for culture-negative prosthetic joint infection? BMC Infect. Dis. 2020, 20, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome projects. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Gevers, D.; Knight, R.; Petrosino, J.F.; Huang, K.; McGuire, A.L.; Birren, B.W.; Nelson, K.E.; White, O.; Methé, B.A.; Huttenhower, C. The Human Microbiome Project: A community resource for the healthy human microbiome. PLoS Biol. 2012, 10, e1001377. [Google Scholar] [CrossRef] [Green Version]
- Habtom, H.; Pasternak, Z.; Matan, O.; Azulay, C.; Gafny, R.; Jurkevitch, E. Applying microbial biogeography in soil forensics. Forensic Sci. Int. Genet. 2019, 38, 195–203. [Google Scholar] [CrossRef]
- Keet, J.H.; Ellis, A.G.; Hui, C.; Le Roux, J.J. Strong spatial and temporal turnover of soil bacterial communities in South Africa’s hyperdiverse fynbos biome. Soil Biol. Biochem. 2019, 136, 107541. [Google Scholar] [CrossRef]
- Wang, S.; Song, F.; Wang, Y.; Huang, Y.; Xie, B.; Luo, H. High resolution melting analysis (HRM) based on 16SrRNA as a tool for personal identification with the human oral microbiome. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 161–163. [Google Scholar] [CrossRef]
- Schmedes, S.E.; Woerner, A.E.; Novroski, N.M.; Wendt, F.R.; King, J.L.; Stephens, K.M.; Budowle, B. Targeted sequencing of clade-specific markers from skin microbiomes for forensic human identification. Forensic Sci. Int. Genet. 2018, 32, 50–61. [Google Scholar] [CrossRef]
- Bell, C.R.; Wilkinson, J.E.; Robertson, B.K.; Javan, G.T. Sex-related differences in the thanatomicrobiome in postmortem heart samples using bacterial gene regions V1-2 and V4. Lett. Appl. Microbiol. 2018, 67, 144–153. [Google Scholar] [CrossRef]
- Zhou, W.; Bian, Y. Thanatomicrobiome composition profiling as a tool for forensic investigation. Forensic Sci. Res. 2018, 3, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.M.; Pasternak, Z.; Mason, C.E.; Elhaik, E. Forensic applications of microbiomics: A review. Front. Microbiol. 2021, 11, 3455. [Google Scholar] [CrossRef]
- Zhang, Y.; Pechal, J.L.; Schmidt, C.J.; Jordan, H.R.; Wang, W.W.; Benbow, M.E.; Sze, S.-H.; Tarone, A. Machine learning performance in a microbial molecular autopsy context: A cross-sectional postmortem human population study. PLoS ONE 2019, 14, e0213829. [Google Scholar] [CrossRef]
- Marella, G.L.; Feola, A.; Marsella, L.T.; Mauriello, S.; Giugliano, P.; Arcudi, G. Diagnosis of drowning, an everlasting challenge in forensic medicine: Review of the literature and proposal of a diagnostic algorithm. Acta Med. Int. 2019, 35, 900–919. [Google Scholar]
- Metcalf, J.L.; Parfrey, L.W.; Gonzalez, A.; Lauber, C.L.; Knights, D.; Ackermann, G.; Humphrey, G.C.; Gebert, M.J.; Van Treuren, W.; Berg-Lyons, D.; et al. A microbial clock provides an accurate estimate of the postmortem interval in a mouse model system. Elife 2013, 2, e01104. [Google Scholar] [CrossRef]
- Pechal, J.L.; Crippen, T.L.; Benbow, M.E.; Tarone, A.M.; Dowd, S.; Tomberlin, J.K. The potential use of bacterial community succession in forensics as described by high throughput metagenomic sequencing. Int. J. Leg. Med. 2014, 128, 193–205. [Google Scholar] [CrossRef]
- Lorenz, P.; Eck, J. Metagenomics and industrial applications. Nat. Rev. Microbiol. 2005, 3, 510–516. [Google Scholar] [CrossRef]
- Tran, D.M.; Nguyen, T.H.; Huynh, T.U.; Do, T.O.; Nguyen, Q.V.; Nguyen, A.D. Analysis of endophytic microbiome dataset from roots of black pepper (Piper nigrum L.) cultivated in the Central Highlands region, Vietnam using 16S rRNA gene metagenomic next-generation sequencing. Data Brief 2022, 42, 108108. [Google Scholar] [CrossRef]
- Hussain, F.; Usman, F. Fungal biotic stresses in plants and its control strategy Abiotic Biot. Stress Plants 2019. [Google Scholar]
- Burdon, J.J.; Zhan, J. Climate change and disease in plant communities. PLoS Biol. 2020, 18, e3000949. [Google Scholar] [CrossRef]
- Mzava, O.; Cheng, A.P.; Chang, A.; Smalling, S.; Djomnang, L.-A.K.; Lenz, J.S.; Longman, R.; Steadman, A.; Gómez-Escobar, L.G.; Schenck, E.J.; et al. A metagenomic DNA sequencing assay that is robust against environmental DNA contamination. Nat. Commun. 2022, 13, 1–10. [Google Scholar] [CrossRef]
- Ruppert, K.M.; Kline, R.J.; Rahman, M.S. Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A systematic review in methods, monitoring, and applications of global eDNA. Glob. Ecol. Conserv. 2019, 17, e00547. [Google Scholar] [CrossRef]
- Chopra, R.S.; Chopra, C.; Sharma, N.R. (Eds.) Metagenomics: Techniques, Applications, Challenges and Opportunities; Springer: Singapore, 2020. [Google Scholar]
- Daniel, R. The metagenomics of soil. Nat. Rev. Microbiol. 2005, 3, 470–478. [Google Scholar] [CrossRef]
- Jiang, H.; Xing, Z.; Liu, X.; Chai, Q.; Xin, Z.; Zhu, C.; Lin, R.; Deng, X.; Cui, D.; Gao, H.; et al. Comparison and development of a metagenomic next-generation sequencing protocol for combined detection of DNA and RNA pathogens in cerebrospinal fluid. BMC Infect. Dis. 2022, 22, 1–10. [Google Scholar] [CrossRef]
- Cuscó, A.; Catozzi, C.; Viñes, J.; Sanchez, A.; Francino, O. Microbiota profiling with long amplicons using Nanopore sequencing: Full-length 16S rRNA gene and the 16S-ITS-23S of the rrn operon. F1000Research 2019, 7, 1755. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.; Gilmore, S.R.; Robertson, J.; Peakall, R. A grass molecular identification system for forensic botany: A critical evaluation of the strengths and limitations. J. Forensic Sci. 2009, 54, 1254–1260. [Google Scholar] [CrossRef]
- Miller, S.; Chiu, C. The role of metagenomics and next-generation sequencing in infectious disease diagnosis. Clin. Chem. 2022, 68, 115–124. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. Meta-IDBA: A de Novo assembler for metagenomic data. Bioinformatics 2011, 27, i94–i101. [Google Scholar] [CrossRef] [Green Version]
- Bakshi, A.; Moin, M.; Madhav, M.S. Metagenomics in Agriculture: State-of-the-Art. In Metagenomics: Techniques, Applications, Challenges and Opportunities; Springer: Singapore, 2020; pp. 167–187. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Bhat, A.H.; Prabhu, P.; Balakrishnan, K. A critical analysis of state-of-the-art metagenomics OTU clustering algorithms. J. Biosci. 2019, 44, 1–9. [Google Scholar] [CrossRef]
- Brady, A.; Salzberg, S.L. Phymm and PhymmBL: Metagenomic phylogenetic classification with interpolated Markov models. Nat. Methods 2009, 6, 673–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, M.J.; Callahan, B.J.; Fisher, D.S.; Holmes, S.P. Denoising PCR-amplified metagenome data. BMC Bioinform. 2012, 13, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Lee, K.H.; Yoon, S.W.; Kim, B.S.; Chun, J.; Yi, H. Analytical tools and databases for metagenomics in the next-generation sequencing era. Genom. Inform. 2013, 11, 102. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.-H.; Alexander, I.J.; Eberhardt, U.; Erland, S.; Høiland, K.; Kjøller, R.; Larsson, E.; Pennanen, T.; et al. The UNITE database for molecular identification of fungi recent updates and future perspectives. New Phytol. 2010, 186, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Huse, S.M.; Welch, D.M.; Morrison, H.G.; Sogin, M.L. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ. Microbiol. 2010, 12, 1889–1898. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Johnson, M.A.; Hansen, M.A.; Bush, E.; Li, S.; Vinatzer, B.A. Metagenomic sequencing for detection and identification of the boxwood blight pathogen Calonectria pseudonaviculata. Sci. Rep. 2022, 12, 1–14. [Google Scholar] [CrossRef]
- Barroso-Bergadà, D.; Massot, M.; Vignolles, N.; Faivre d’Arcier, J.; Chancerel, E.; Guichoux, E.; Walker, A.; Vacher, C.; Bohan, D.A.; Laval, V.; et al. Metagenomic next- generation sequencing (mNGS) data reveals the phyllosphere microbiome of wheat plants infected by the fungal pathogen Zymoseptoria tritici. Phytobiomes J. 2022. [Google Scholar] [CrossRef]
- Behrens, F.H.; Fischer, M. Evaluation of different phyllosphere sample types for parallel metabarcoding of Fungi and Oomycetes in Vitis vinifera. Phytobiomes J. 2022, 6, 207–213. [Google Scholar] [CrossRef]
- Cureau, N.; Threlfall, R.; Savin, M.; Marasini, D.; Lavefve, L.; Carbonero, F. Year, location, and variety impact on grape-, soil-, and leaf-associated fungal microbiota of Arkansas-grown table grapes. Microb. Ecol. 2021, 82, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Schiro, G.; Colangeli, P.; Müller, M.E. A metabarcoding analysis of the mycobiome of wheat ears across a topographically heterogeneous field. Front. Microbiol. 2019, 10, 2095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Frari, G.; Gobbi, A.; Aggerbeck, M.R.; Oliveira, H.; Hansen, L.H.; Ferreira, R.B. Characterization of the wood mycobiome of Vitis vinifera in a vineyard affected by esca. Spatial distribution of fungal communities and their putative relation with leaf symptoms. Front. Plant Sci. 2019, 10, 910. [Google Scholar] [CrossRef] [PubMed]
- Cobo-Díaz, J.F.; Baroncelli, R.; Le Floch, G.; Picot, A. Combined metabarcoding and co-occurrence network analysis to profile the bacterial, fungal and Fusarium communities and their interactions in maize stalks. Front. Microbiol. 2019, 10, 261. [Google Scholar] [CrossRef] [Green Version]
- Kerdraon, L.; Barret, M.; Laval, V.; Suffert, F. Differential dynamics of microbial community networks help identify microorganisms interacting with residue-borne pathogens: The case of Zymoseptoria tritici in wheat. Microbiome 2019, 7, 1–17. [Google Scholar] [CrossRef]
- Morales-Cruz, A.; Figueroa-Balderas, R.; García, J.F.; Tran, E.; Rolshausen, P.E.; Baumgartner, K.; Cantu, D. Profiling grapevine trunk pathogens in planta: A case for community-targeted DNA metabarcoding. BMC Microbiol. 2018, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Penton, C.R.; Lv, N.; Xue, C.; Yuan, X.; Ruan, Y.; Li, R.; Shen, Q. Banana Fusarium wilt disease incidence is influenced by shifts of soil microbial communities under different monoculture spans. Microb. Ecol. 2018, 75, 739–750. [Google Scholar] [CrossRef]
- Walder, F.; Schlaeppi, K.; Wittwer, R.; Held, A.Y.; Vogelgsang, S.; van der Heijden, M.G. Community profiling of Fusarium in combination with other plant-associated fungi in different crop species using SMRT sequencing. Front. Plant Sci. 2017, 8, 2019. [Google Scholar] [CrossRef]
- Xu, X.; Passey, T.; Wei, F.; Saville, R.; Harrison, R.J. Amplicon-based metagenomics identified candidate organisms in soils that caused yield decline in strawberry. Hortic. Res. 2015, 2, 15022. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Purpose | Algorithm Tools | References |
|---|---|---|
| OTU clustering | MOTHUR, SUMACLUST, SWARM, METACLUSTER, UCLUST, CD-HIT-OUT, TBC | [41,42] |
| Phylogenetic classifications | Phymm, BLAST, CARMA | [43] |
| Denoising | Pyronoise, Denoiser, DADA, Acacia | [44,45] |
| Chimera detection | UCHİME, ChimeraSlayer, Persus, DECIPHER | [45,46] |
| ITS database for fungal detection | UNITE | [47] |
| All in one | MOTHUR, QIIME, MEGAN | [45,48,49] |
| Plant | Aim of Study | Metagenomics Techniques | References |
|---|---|---|---|
| Grape | Determination of fungi and oomycetes in different phyllosphere samples | Metabarcoding | [52] |
| Grape | Determination of soil and leaf-associated fungal microbiota | mNGS-Ilumina | [53] |
| Wheat | Detection of fungal microorganisms in the wheat phyllosphere | Microbiome Metabarcoding using ITS barcodes | [54] |
| Grape | Identification of fungal diseases on the vine trunk | mNGS-Ilumina | [55] |
| Maize | Determination of fungal microbiota after harvest | Metabarcoding | [56] |
| Wheat | Determination of fungal communities in wheat residues | Metabarcoding | [57] |
| Grapevine | Determination of fungal disease agents associated with grapevine | Metabarcoding | [58] |
| Banana | Investigation of the effect of variable soil microbiota on fusarium disease | Metabarcoding | [59] |
| Wheat, maize | To determine Fusarium species in various plants | PaCBio SMRT Sequencing | [60] |
| Strawberry | Determination of microbial communities in strawberry growing soils with different yields | Amplicon Based Metagenomic | [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gökdemir, F.Ş.; İşeri, Ö.D.; Sharma, A.; Achar, P.N.; Eyidoğan, F. Metagenomics Next Generation Sequencing (mNGS): An Exciting Tool for Early and Accurate Diagnostic of Fungal Pathogens in Plants. J. Fungi 2022, 8, 1195. https://doi.org/10.3390/jof8111195
Gökdemir FŞ, İşeri ÖD, Sharma A, Achar PN, Eyidoğan F. Metagenomics Next Generation Sequencing (mNGS): An Exciting Tool for Early and Accurate Diagnostic of Fungal Pathogens in Plants. Journal of Fungi. 2022; 8(11):1195. https://doi.org/10.3390/jof8111195
Chicago/Turabian StyleGökdemir, Fatma Şeyma, Özlem Darcansoy İşeri, Abhishek Sharma, Premila N. Achar, and Füsun Eyidoğan. 2022. "Metagenomics Next Generation Sequencing (mNGS): An Exciting Tool for Early and Accurate Diagnostic of Fungal Pathogens in Plants" Journal of Fungi 8, no. 11: 1195. https://doi.org/10.3390/jof8111195
APA StyleGökdemir, F. Ş., İşeri, Ö. D., Sharma, A., Achar, P. N., & Eyidoğan, F. (2022). Metagenomics Next Generation Sequencing (mNGS): An Exciting Tool for Early and Accurate Diagnostic of Fungal Pathogens in Plants. Journal of Fungi, 8(11), 1195. https://doi.org/10.3390/jof8111195