
Fungal Strains with Identical Genomes Were Found at a Distance of 2000 Kilometers after 40 Years

 ,
,  , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolates and Culture Methods of S. sclerotiorum
2.2. Sexual Reproduction of Strain PB4
2.3. Extraction of Genomic DNA
2.4. Library Construction and Data Filtering
2.5. Genome Assembly and Assessment
2.6. Gene Prediction and Annotation
2.7. Comparative Genomics
2.8. Phylogenetic Analyses
3. Results
3.1. De Novo Assembly and Functional Annotations of the Genome of the S. sclerotiorum Strain PB4
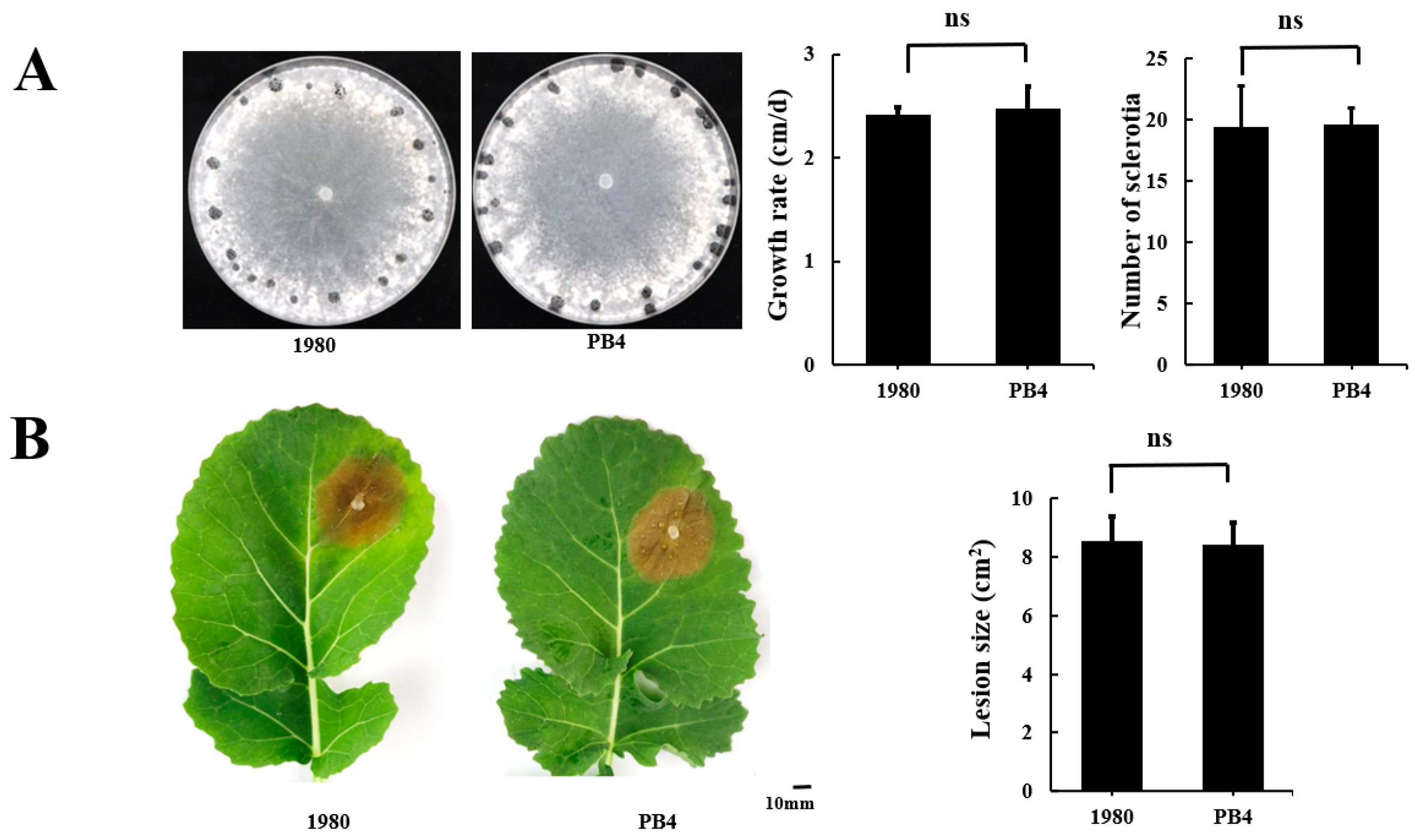
3.2. The Genome Sequences of Strains PB4 and 1980 Are Almost Identical
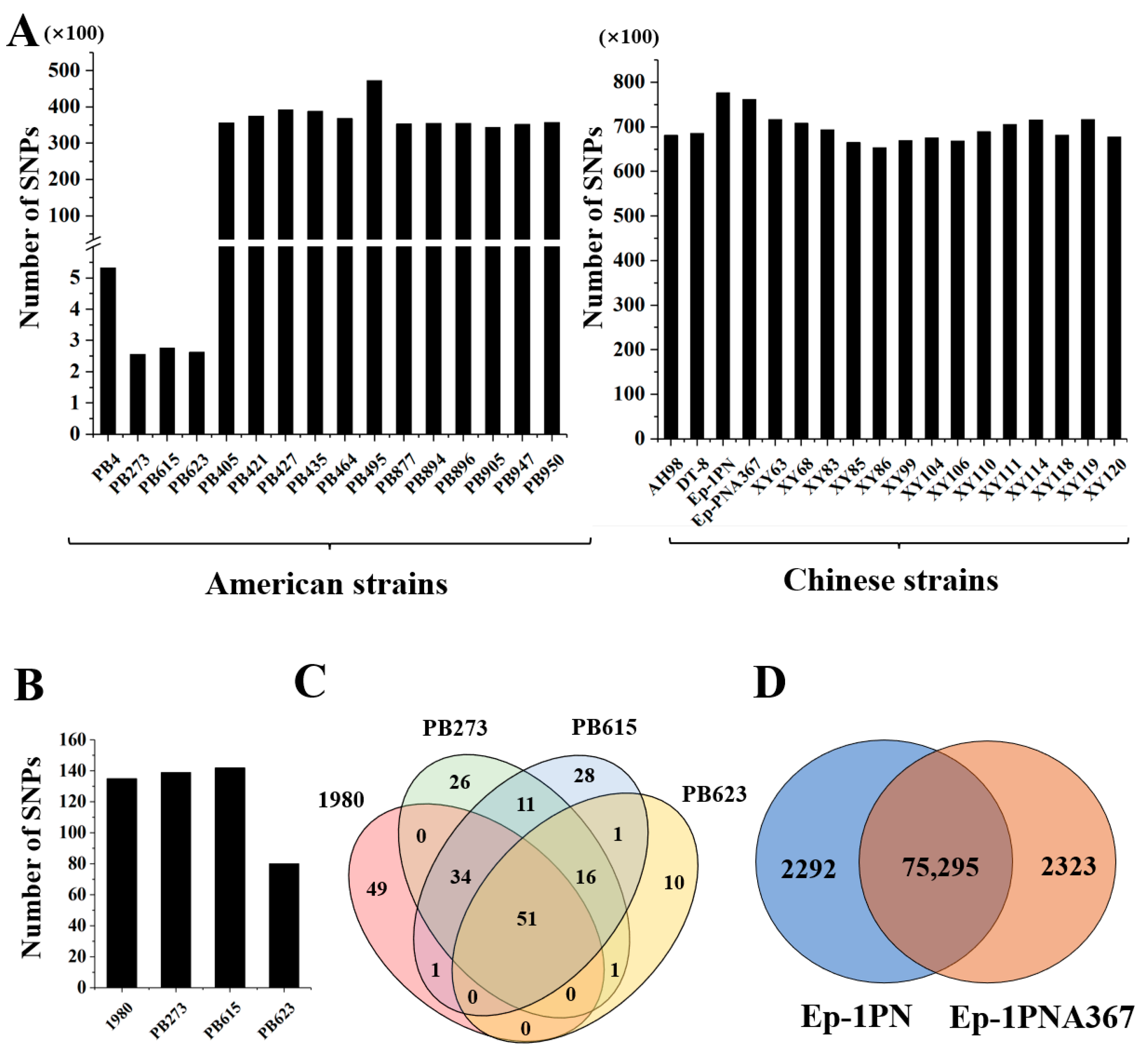
3.3. Distinct Population Evolution Patterns of Strains in the USA and China
3.4. The SNP of Offspring Compared with the Parent Strain PB4
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tamas, I.; Klasson, L.; Canbck, B.; Naslund, A.K.; Eriksson, A.-S.; Wernegreen, J.J.; Sandstrom, J.P.; Moran, N.A.; Andersson, S.G. 50 million years of genomic stasis in endosymbiotic bacteria. Science 2002, 296, 2376–2379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinsky, M.L.; Jensen, O.P.; Ricard, D.; Palumbia, S.R. Unexpected patterns of fisheries collapse in the world’s oceans. Proc. Natl. Acad. Sci. USA 2011, 108, 8317–8322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glen, C.D.; Dubrova, Y.E. Exposure to anticancer drugs can result in transgenerational genomic instability in mice. Proc. Natl. Acad. Sci. USA 2012, 209, 2984–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Therkildsen, N.O.; Wilder, A.P.; Conover, D.V.; Munch, S.B.; Baumann, H.; Palumbi, S. Contrasting genomic shifts underlie parallel phenotypic evolution in response to fishing. Science 2019, 365, 487–490. [Google Scholar] [CrossRef]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Raja, S.; Van Houten, B. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic Acids Res. 2020, 48, 11227–11243. [Google Scholar] [CrossRef]
- Huang, J.; Cook, D.E. The contribution of DNA repair pathways to genome editing and evolution in filamentous pathogens. FEMS Microbiol. Rev. 2022, fuac035. [Google Scholar] [CrossRef]
- Sharp, N.P.; Sandell, L.; James, C.G.; Otto, S.P. The genome-wide rate and spectrum of spontaneous mutations differ between haploid and diploid yeast. Proc. Natl. Acad. Sci. USA 2018, 115, e5046–e5055. [Google Scholar] [CrossRef] [Green Version]
- Trost, B.; Loureiro, L.O.; Scherer, S.W. Discovery of genomic variation across a generation. Hum. Mol. Genet. 2021, 30, R174–R186. [Google Scholar]
- Baranova, M.A.; Logacheva, M.D.; Penin, A.A.; Seplyarskiy, V.B.; Safonova, Y.Y.; Naumenko, S.A.; Klepikova, A.V.; Gerasimov, E.S.; Bazykin, G.A.; James, T.Y.; et al. Extraordinary genetic diversity in a wood decay mushroom. Mol. Biol. Evol. 2015, 32, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Wojcik, G.L.; Graff, M.; Nishimura, K.K.; Tao, R.; Haessler, J.; Gignoux, C.R.; Highland, H.M.; Patel, Y.M.; Sorokin, E.P.; Avery, C.L.; et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 2019, 570, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mauleon, R.; Hu, Z.; Chebotarov, D.; Tai, S.; Wu, Z.; Li, M.; Zheng, T.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Chen, M.; Lin, L.; Han, Y.; Bao, J.; Tang, W.; Lin, L.; Lin, Y.; Somai, R.; Lu, L.; et al. Population genomic analysis of the rice blast fungus reveals specific events associated with expansion of three main clades. ISME J. 2018, 12, 1867–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampaio, N.M.; Ajith, V.P.; Watson, R.A.; Heasley, L.R.; Chakraborty, P.; Rodrigues-Prause, A.; Malc, E.P.; Mieczkowski, P.A.; Nishant, K.T. Characterization of systemic genomic instability in budding yeast. Proc. Natl. Acad. Sci. USA 2020, 117, 28221–28231. [Google Scholar] [CrossRef] [PubMed]
- Million, K.M.; Bhattacharya, A. A DNA content variation and SNP diversity within a single population of asexual snails. J. Hered. 2021, 112, 58–66. [Google Scholar] [CrossRef]
- Badet, T.; Fouché, S.; Hartmann, F.E.; Zala, M.; Croll, D. Machine-learning predicts genomic determinants of meiosis-driven structural variation in a eukaryotic pathogen. Nat. Commun. 2021, 12, 3551. [Google Scholar] [CrossRef]
- Wachi, N.; Gau, J.J.; Fujie, S.; Fukano, K.; Maeto, K. Genomic population structure of sympatric sexual and asexual populations in a parasitic wasp, Meteorus pulchricornis (Hymenoptera: Braconidae), inferred from six hundred single-nucleotide polymorphism loci. Mol. Ecol. 2021, 30, 1612–1623. [Google Scholar] [CrossRef]
- Miska, E.A.; Ferguson-Smith, A.C. Transgenerational inheritance: Models and mechanisms of non-DNA sequence-based inheritance. Science 2016, 354, 59–63. [Google Scholar] [CrossRef]
- Habig, M.; Lorrain, C.; Feurtey, A.; Komluski, J.; Stukenbrock, E.H. Epigenetic modifications affect the rate of spontaneous mutations in a pathogenic fungus. Nat. Commun. 2021, 12, 5869. [Google Scholar] [CrossRef]
- McLeod, D.V.; Wild, G.; Ubeda, F. Epigenetic memories and the evolution of infectious diseases. Nat. Commun. 2021, 12, 4273. [Google Scholar] [CrossRef]
- Boyce, K.J.; Wang, Y.; Verma, S.; Shakya, V.P.S.; Xue, C.; Idnurm, A. Mismatch repair of DNA replication errors contributes to microevolution in the pathogenic fungus Cryptococcus neoformans. mBio 2017, 8, e00595-17. [Google Scholar] [CrossRef] [PubMed]
- Calo, S.; Shertz-Wall, C.; Lee, S.C.; Bastidas, R.J.; Nicolás, F.E.; Granek, J.A.; Mieczkowski, P.; Torres-Martínez, S.; Ruiz-Vázquez, M.; Cardenas, M.E. Antifungal drug resistance evoked via RNAi-dependent epimutations. Nature 2014, 513, 555–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Garcia, S.; Yaseen, I.; Shukla, M.; Audergon, P.N.; White, S.A.; Pidoux, A.L.; Allshire, R.C. Epigenetic gene silencing by heterochromatin primes fungal resistance. Nature 2020, 585, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Loeillet, S.; Herzog, M.; Puddu, F.; Legoix, P.; Baulande, S.; Jackson, S.P.; Nicolas, A.G. Trajectory and uniqueness of mutational signatures in yeast mutators. Proc. Natl. Acad. Sci. USA 2020, 117, 24947–24956. [Google Scholar] [CrossRef]
- Brookes, A.J. The essence of SNPs. Gene 1999, 234, 177–186. [Google Scholar] [CrossRef]
- López-Pérez, M.; Martin-Cuadrado, A.B.; Rodriguez-Valera, F. Homologous recombination is involved in the diversity of replacement flexible genomic islands in aquatic prokaryotes. Front. Genet. 2014, 5, 147. [Google Scholar]
- Wang, J.; Chu, S.; Zhang, H.; Yu, D. Development and application of a novel genome-wide SNP array reveals domestication history in soybean. Sci. Rep. 2016, 6, 20728. [Google Scholar] [CrossRef] [Green Version]
- Cusick, M.F.; Clark, L.; Tu, T.; Goforth, J.; Zhang, X.; LaRue, B.; Gutierrez, R.; Jindra, P.T. Performance characteristics of chimerism testing by next generation sequencing. Hum. Immunol. 2022, 83, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Goossens, S.N.; Heupink, T.H.; De Vos, E.; Dippenaar, A.; De Vos, M.; Warren, R.; Van Rie, A. Detection of minor variants in Mycobacterium tuberculosis whole genome sequencing data. Brief. Bioinform. 2022, 23, bbab541. [Google Scholar] [CrossRef]
- Wang, N.; Lysenkov, V.; Orte, K.; Kairisto, V.; Aakko, J.; Khan, S.; Elo, L.L. Tool evaluation for the detection of variably sized indels from next generation whole genome and targeted sequencing data. PLoS Comput. Biol. 2022, 18, e1009269. [Google Scholar] [CrossRef]
- Lam, I.; Keeney, S. Nonparadoxical evolutionary stability of the recombination initiation landscape in yeast. Science 2015, 350, 932–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallone, B.; Steensels, J.; Prahl, T.; Soriaga, L.; Saels, V.; Herrera-Malaver, B.; Merlevede, A.; Roncoroni, M.; Voordeckers, K.; Miraglia, L. Domestication and divergence of Saccharomyces cerevisiae beer yeasts. Cell 2016, 166, 1397–1410. [Google Scholar] [CrossRef]
- Peter, J.; De Chiara, M.; Friedrich, A.; Yue, J.X.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgins-Davis, A.; Duveau, F.; Walker, E.A.; Wittkopp, P.J. Empirical measures of mutational effects define neutral models of regulatory evolution in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2019, 116, 21085–21093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puddu, F.; Herzog, M.; Selivanova, A.; Wang, S.; Zhu, J.; Klein-Lavi, S.; Gordon, M.; Meirman, R.; Millan-Zambrano, G.; Ayestaran, I. Genome architecture and stability in the Saccharomyces cerevisiae knockout collection. Nature 2019, 573, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Bezmenova, A.V.; Zvyagina, E.A.; Fedotova, A.V.; Kasianov, A.S.; Neretina, T.V.; Penin, A.A.; Bazykin, G.A.; Kondrashov, A.S. Rapid accumulation of mutations in growing mycelia of a hypervariable fungus Schizophyllum commune. Mol. Biol. Evol. 2020, 37, 2279–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Q.; Niu, Y.; Gundry, M.; Zong, C. Single-cell damagenome profiling unveils vulnerable genes and functional pathways in human genome toward DNA damage. Sci. Adv. 2021, 7, eabf3329. [Google Scholar] [CrossRef]
- Hiltunen, M.; Grudzinska-Sterno, M.; Wallerman, O.; Ryberg, M.; Johannesson, H. Maintenance of high genome integrity over vegetative growth in the fairy-ring mushroom Marasmius oreades. Curr. Biol. 2019, 29, 2758–2765. [Google Scholar] [CrossRef]
- Anderson, J.B.; Bruhn, J.N.; Kasimer, D.; Wang, H.; Rodrigue, N.; Smith, M.L. Clonal evolution and genome stability in a 2500-year-old fungal individual. Proc. R. Soc. B Boil. Sci. 2018, 285, 2018–2233. [Google Scholar] [CrossRef] [Green Version]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Li, G.; Jiang, D.; Chen, W. Sclerotinia sclerotiorum: An evaluation of virulence theories. Annu. Rev. Phytopathol. 2018, 56, 311–338. [Google Scholar] [CrossRef] [PubMed]
- Bolton, M.D.; Thomma, B.P.; Nelson, B.D. Sclerotinia sclerotiorum (Lib.) de Bary: Biology and molecular traits of a cosmopolitan pathogen. Mol. Plant Pathol. 2006, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Derbyshire, M.C.; Denton-Giles, M. The control of sclerotinia stem rot on oilseed rape (Brassica napus): Current practices and future opportunities. Plant Pathol. 2016, 65, 859–877. [Google Scholar] [CrossRef] [Green Version]
- Roper, M.; Seminara, A.; Bandi, M.; Cobb, A.; Dillard, H.R.; Pringle, A. Dispersal of fungal spores on a cooperatively generated wind. Proc. Natl. Acad. Sci. USA 2010, 107, 17474–17479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, Y.; Kohn, L.M. Random association among alleles in clonal populations of Sclerotinia sclerotiorum. Fungal Genet. Biol. 1998, 23, 139–149. [Google Scholar] [CrossRef]
- Attanayake, R.N.; Carter, P.A.; Jiang, D.; del Río-Mendoza, L.; Chen, W. Sclerotinia sclerotiorum populations infecting canola from China and the United States are genetically and phenotypically distinct. Phytopathology 2013, 103, 750–761. [Google Scholar] [CrossRef] [Green Version]
- Dunn, A.R.; Kikkert, J.R.; Pethybridge, S.J. Genotypic characteristics in populations of Sclerotinia sclerotiorum from New York State, USA. Annu. Appl. Biol. 2017, 170, 219–228. [Google Scholar] [CrossRef]
- Shang, Y.; Xiao, G.; Zheng, P.; Cen, K.; Zhan, S.; Wang, C. Divergent and convergent evolution of fungal pathogenicity. Genome Biol. Evol. 2016, 8, 1374–1387. [Google Scholar] [CrossRef] [Green Version]
- Steadman, J.R.; Marcinkowska, J.; Rutledge, S.A. Semi-Selective Medium for Isolation of Sclerotinia sclerotiorum. Can. J. Plant Pathol. 1994, 16, 68–70. [Google Scholar] [CrossRef]
- Derbyshire, M.; Denton-Giles, M.; Hegedus, D.; Seifbarghy, S.; Rollins, J.; van Kan, J.; Seidl, M.F.; Faino, L.; Mbengue, M.; Navaud, O. The complete genome sequence of the phytopathogenic fungus Sclerotinia sclerotiorum reveals insights into the genome architecture of broad host range pathogens. Genome Biol. Evol. 2017, 9, 593–618. [Google Scholar] [CrossRef]
- Zhang, X.; Cheng, X.; Liu, L.; Liu, S. Genome sequence resource for the plant pathogen Sclerotinia sclerotiorum WH6 isolated in China. Plant Dis. 2021, 105, 3720–3722. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Li, G.; Fu, Y.; Yi, X.; Wang, D. Transmissible hypovirulent element in isolate Ep-1PN of Sclerotinia sclerotiorum. Chin. Sci. Bull. 1998, 43, 779–781. [Google Scholar] [CrossRef]
- Yu, X.; Li, B.; Fu, Y.; Jiang, D.; Ghabrial, S.A.; Li, G.; Peng, Y.; Xie, J.; Cheng, J.; Huang, J. A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc. Natl. Acad. Sci. USA 2010, 107, 8387–8392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Xie, J.; Cheng, J.; Fu, Y.; Li, G.; Yi, X.; Jiang, D. Fungal negative-stranded RNA virus that is related to bornaviruses and nyaviruses. Proc. Natl. Acad. Sci. USA 2014, 111, 12205–12210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, J.J. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 13–15. [Google Scholar]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Dujon, B. Yeast evolutionary genomics. Nat. Rev. Genet. 2010, 11, 512–524. [Google Scholar] [CrossRef]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast processing of NGS alignment formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarel, B.L.; Korf, I.; Robb, S.M.; Parra, G.; Ross, E.; Moore, B.; Holt, C.; Alvarado, A.S.; Yandell, M. MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008, 18, 188–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [PubMed]
- Lomsadze, A.; Burns, P.D.; Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 2014, 42, e119. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Cubeta, M.A.; Cody, B.R.; Kohli, Y.; Kohn, L.M. Clonality in Sclerotinia sclerotiorum on infected cabbage in eastern North Carolina. Phytopathology 1997, 87, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.A.; Ferro, C.G.; Lehner, M.S.; Paula, T.J.; Mizubuti, E.S. The population of Sclerotinia sclerotiorum in Brazil is structured by mycelial compatibility groups. Phytopathology 2021, 105, 3376–3384. [Google Scholar] [CrossRef]
- Clarkson, J.P.; Warmington, R.J.; Walley, P.G.; Denton-Gilesm, M.; Barbetti, M.J.; Brodal, G.; Nordskog, B. Population structure of Sclerotinia subarctica and Sclerotinia sclerotiorum in England, Scotland and Norway. Front. Microbiol. 2017, 8, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attanayake, R.N.; Xu, L.; Chen, W. Sclerotinia sclerotiorum populations: Clonal or recombining? Trop. Plant Pathol. 2019, 44, 23–31. [Google Scholar] [CrossRef]
- Pannullo, A.; Kamvar, Z.N.; Miorini, T.J.; Steadman, J.R.; Everhart, S.E. Genetic variation and structure of Sclerotinia sclerotiorum populations from soybean in Brazil. Trop. Plant Pathol. 2019, 44, 53–64. [Google Scholar] [CrossRef]
- Yu, Y.; Cai, J.; Ma, L.; Huang, Z.; Wang, Y.; Fang, A.; Yang, Y.; Qing, L.; Bi, C. Population structure and aggressiveness of Sclerotinia sclerotiorum from rapeseed (Brassica napus) in Chongqing city. Plant Dis. 2020, 104, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Borah, T.R.; Dutta, S.; Barman, A.R.; Helim, R.; Sen, K.J. Variability and host range of Sclerotinia sclerotiorum in Eastern and North Eastern India. J. Plant Pathol. 2021, 103, 809–822. [Google Scholar] [CrossRef]
- Abán, C.L.; Taboada, G.; Spedaletti, Y.; Maita, E.; Galván, M.Z. Population structure of the fungus Sclerotinia sclerotiorum in common bean fields of Argentina. Eur. J. Plant Pathol. 2021, 160, 841–853. [Google Scholar] [CrossRef]
- Malvárez, G.; Carbone, I.; Grünwald, N.J.; Subbarao, K.V.; Schafer, M.; Kohn, L.M. New populations of Sclerotinia sclerotiorum from lettuce in California and peas and lentils in Washington. Phytopathology 2007, 97, 470–483. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, T.; Guruge, B.; Somachandra, K.; Jayasekara, E.; Rajapakse, C.; Attanayake, R.J. Phenotypic variation of cabbage white mold pathogen, Sclerotinia sclerotiorum in the upcountry commercial cabbage fields in Sri Lanka. J. Nat. Sci. Found. Sri Lanka 2018, 46, 159–164. [Google Scholar] [CrossRef]
- Martincorena, I.; Seshasayee, A.S.; Luscombe, N.M. Evidence of non-random mutation rates suggests an evolutionary risk management strategy. Nature 2012, 485, 95–98. [Google Scholar] [CrossRef]
- Behringer, M.G.; Choi, B.I.; Miller, S.F.; Doak, T.G.; Karty, J.A.; Guo, W.; Lynch, M. Escherichia coli cultures maintain stable subpopulation structure during long-term evolution. Proc. Natl. Acad. Sci. USA 2018, 115, e4642–e4650. [Google Scholar] [CrossRef] [Green Version]
- Foster, P.L.; Lee, H.; Popodi, E.; Townes, J.P.; Tang, H. Determinants of spontaneous mutation in the bacterium Escherichia coli as revealed by whole-genome sequencing. Proc. Natl. Acad. Sci. USA 2015, 112, e5990–e5999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poetsch, A.R.; Boulton, S.J.; Luscombe, N.M. Genomic landscape of oxidative DNA damage and repair reveals regioselective protection from mutagenesis. Genome Biol. 2018, 19, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.; Ollodart, A.R.; Sudhesh, V.; Herr, A.J.; Dunham, M.J.; Harris, K. A modified fluctuation assay reveals a natural mutator phenotype that drives mutation spectrum variation within Saccharomyces cerevisiae. eLife 2021, 10, e68285. [Google Scholar] [CrossRef] [PubMed]
- Lujan, S.A.; Kunkel, T.A. Stability across the whole nuclear genome in the presence and absence of DNA mismatch repair. Cells 2021, 10, 1224. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.; Ackerman, M.S.; Miller, S.F.; Doak, T.G.; Lynch, M. Drift-barrier hypothesis and mutation-rate evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 18488–18492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M.; Ackerman, M.S.; Gout, J.F.; Long, H.; Sung, W.; Thomas, W.K. Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 2016, 17, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Rozhok, A.; Eldredge, N.; DeGregori, J. Stochastic modeling uncovers a novel mechanism underlying the evolution of mutation rates in sexually reproducing populations. Biorxiv 2021. [Google Scholar] [CrossRef]
- Sung, W.; Tucker, A.E.; Doak, T.G.; Choi, E.; Thomas, W.K.; Lynch, M.J. Extraordinary genome stability in the ciliate Paramecium tetraurelia. Proc. Natl. Acad. Sci. USA 2012, 109, 19339–19344. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Williams, E.; Sung, W.; Lynch, M.; Long, H. The insect-killing bacterium Photorhabdus luminescens has the lowest mutation rate among bacteria. Mar. Life Sci. Technol. 2021, 3, 20–27. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Collection Date | Host | Geographical Origin | Reference |
|---|---|---|---|---|
| 1980 | 1980 | Bean | Western, Nebraska, USA | [49] |
| PB4 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB273 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB405 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB421 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB427 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB435 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB464 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB495 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB615 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB623 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB877 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB894 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB896 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB905 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB947 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| PB950 | Oct 2018 | Pinto bean | Quincy, Washington, USA | This study |
| AH98 | 2009 | Rapeseed | Hefei, Anhui Province, China | [54] |
| DT-8 | 2007 | Rapeseed | Yiyang, Hunan Province, China | [53] |
| Ep-1PN | 1992 | Eggplant | Jiamusi, Heilongjiang Province, China | [52] |
| Ep-1PNA367 | 1997 | Eggplant | Jiamusi, Heilongjiang Province, China | [52] |
| XY63 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY68 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY83 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY85 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY86 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY99 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY104 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY106 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY110 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY111 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY114 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY118 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY119 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| XY120 | Oct 2020 | Rapeseed | Xinyang, Henan Province, China | This study |
| Size Range 50–500 (bp) | Size Range 500–10,000 (bp) | Total | ||
|---|---|---|---|---|
| Insertion | Count | 5 | 6 | 11 |
| Length | 1150 (bp) | 22,676 (bp) | 23,826 (bp) | |
| Deletion | Count | 2 | 5 | 7 |
| Length | 263 (bp) | 15,331 (bp) | 15,594 (bp) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Q.; Lin, Y.; Lyu, X.; Qu, Z.; Lu, Z.; Fu, Y.; Cheng, J.; Xie, J.; Chen, T.; Li, B.; et al. Fungal Strains with Identical Genomes Were Found at a Distance of 2000 Kilometers after 40 Years. J. Fungi 2022, 8, 1212. https://doi.org/10.3390/jof8111212
Zhu Q, Lin Y, Lyu X, Qu Z, Lu Z, Fu Y, Cheng J, Xie J, Chen T, Li B, et al. Fungal Strains with Identical Genomes Were Found at a Distance of 2000 Kilometers after 40 Years. Journal of Fungi. 2022; 8(11):1212. https://doi.org/10.3390/jof8111212
Chicago/Turabian StyleZhu, Qili, Yang Lin, Xueliang Lyu, Zheng Qu, Ziyang Lu, Yanping Fu, Jiasen Cheng, Jiatao Xie, Tao Chen, Bo Li, and et al. 2022. "Fungal Strains with Identical Genomes Were Found at a Distance of 2000 Kilometers after 40 Years" Journal of Fungi 8, no. 11: 1212. https://doi.org/10.3390/jof8111212
APA StyleZhu, Q., Lin, Y., Lyu, X., Qu, Z., Lu, Z., Fu, Y., Cheng, J., Xie, J., Chen, T., Li, B., Cheng, H., Chen, W., & Jiang, D. (2022). Fungal Strains with Identical Genomes Were Found at a Distance of 2000 Kilometers after 40 Years. Journal of Fungi, 8(11), 1212. https://doi.org/10.3390/jof8111212