
Genomic and Metabolomic Analyses of the Medicinal Fungus Inonotus hispidus for Its Metabolite’s Biosynthesis and Medicinal Application


 ,
, 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Fungal Strain and Strain Culture
2.2. Genome Sequencing, De Novo Assembly, and Annotation
2.2.1. Extraction of Genome DNA
2.2.2. De Novo Sequencing
2.2.3. Gene Prediction and Annotation
2.3. Comparative Genomics Analysis
2.4. Phylogenomic Analysis
2.5. CAZy Family Analysis and Structural Prediction
2.6. Predictive Analysis of Candidate Genes for Secondary Metabolites
2.7. Prediction and Analysis of P450s
2.8. Metabolites Analysis and Structural Evaluation
3. Results
3.1. Fungal Species Identity and Artificial Cultivation
3.2. Genome Sequence, Assembly, and Annotation
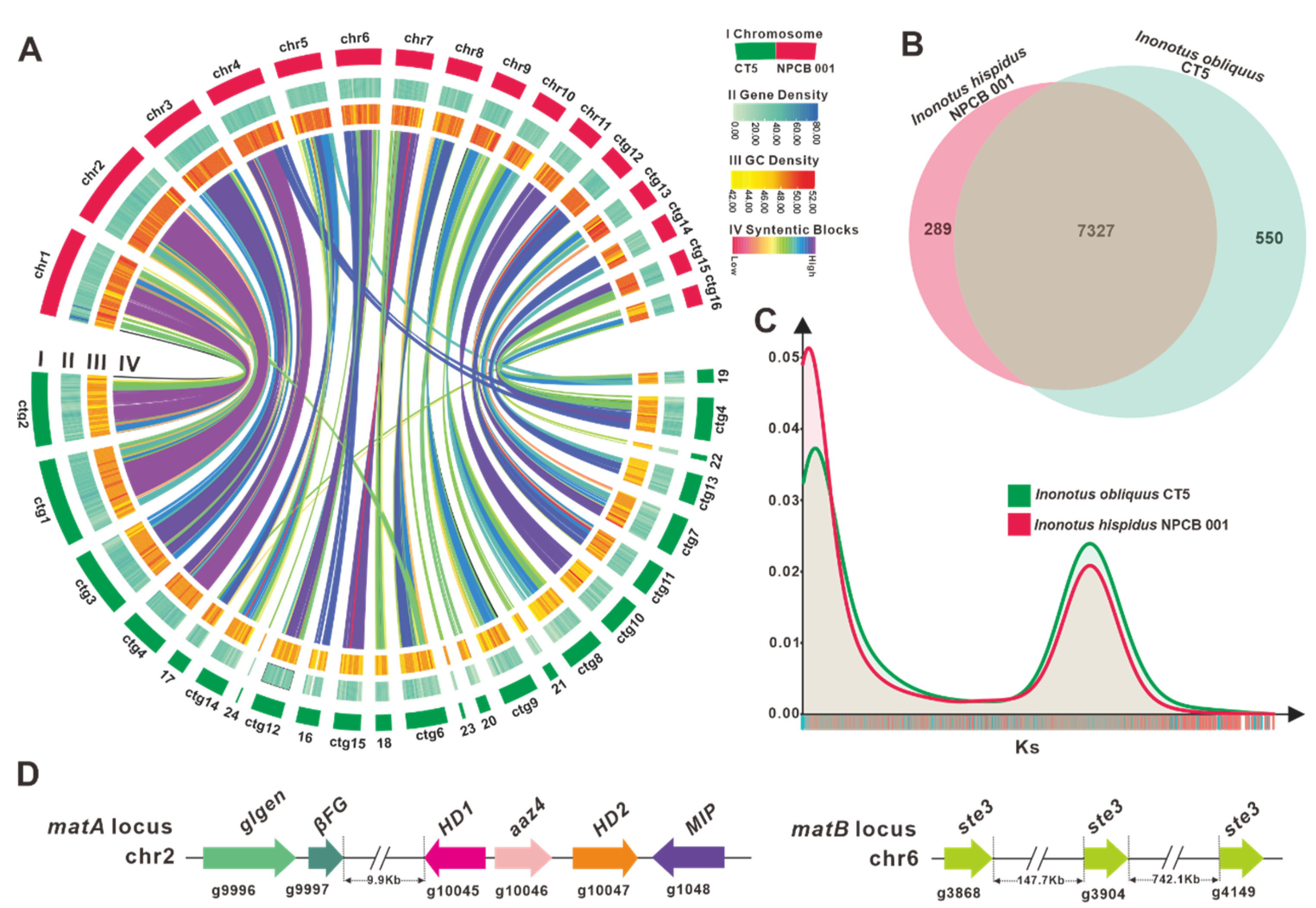
3.3. Comparative Genomic Analysis within Inonotus Species
3.4. Identification of the Mating Genes
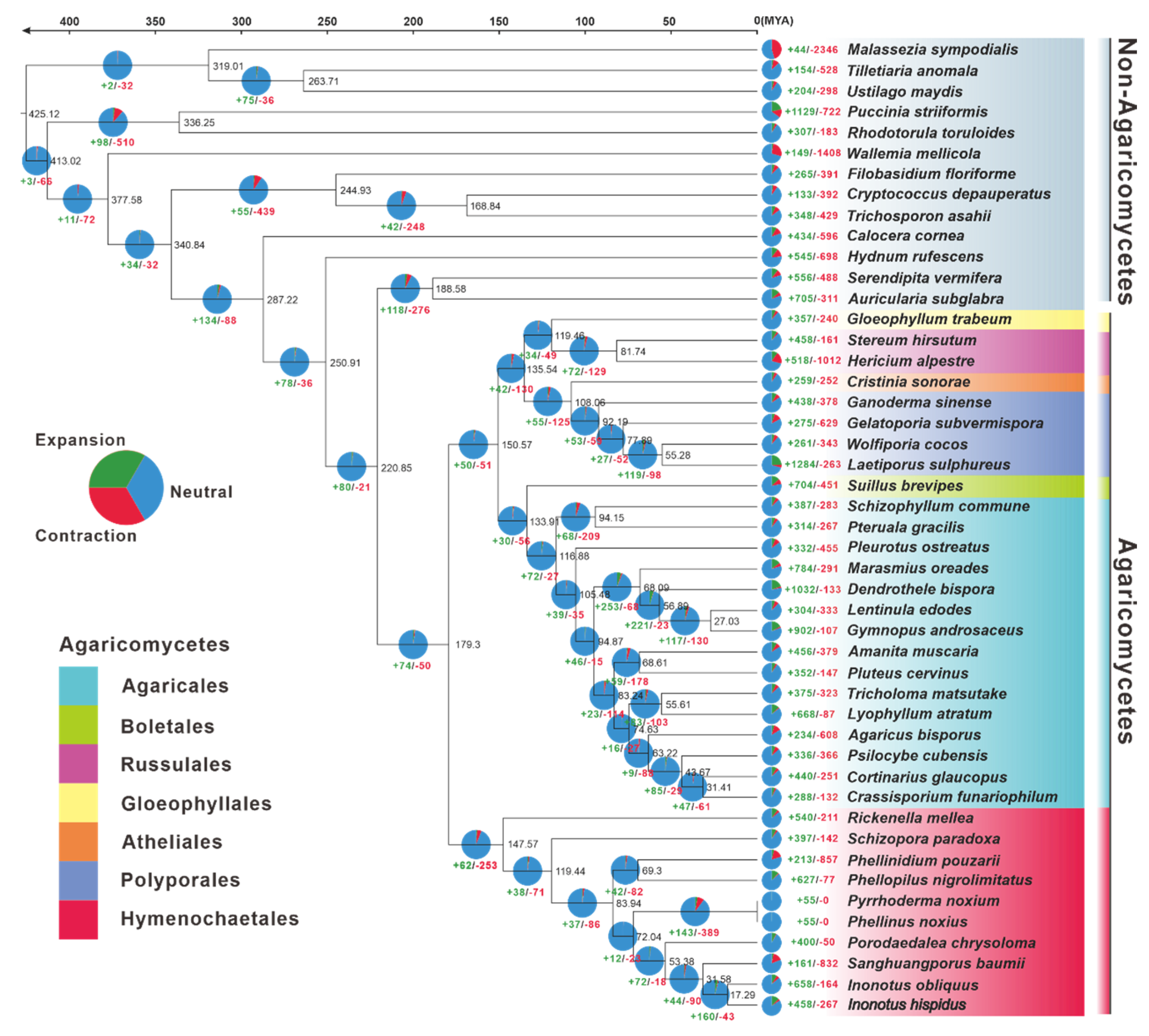
3.5. Phylogenomic and Evolutionary Analysis
3.6. CAZyme Analysis and Synthesis of Polysaccharides
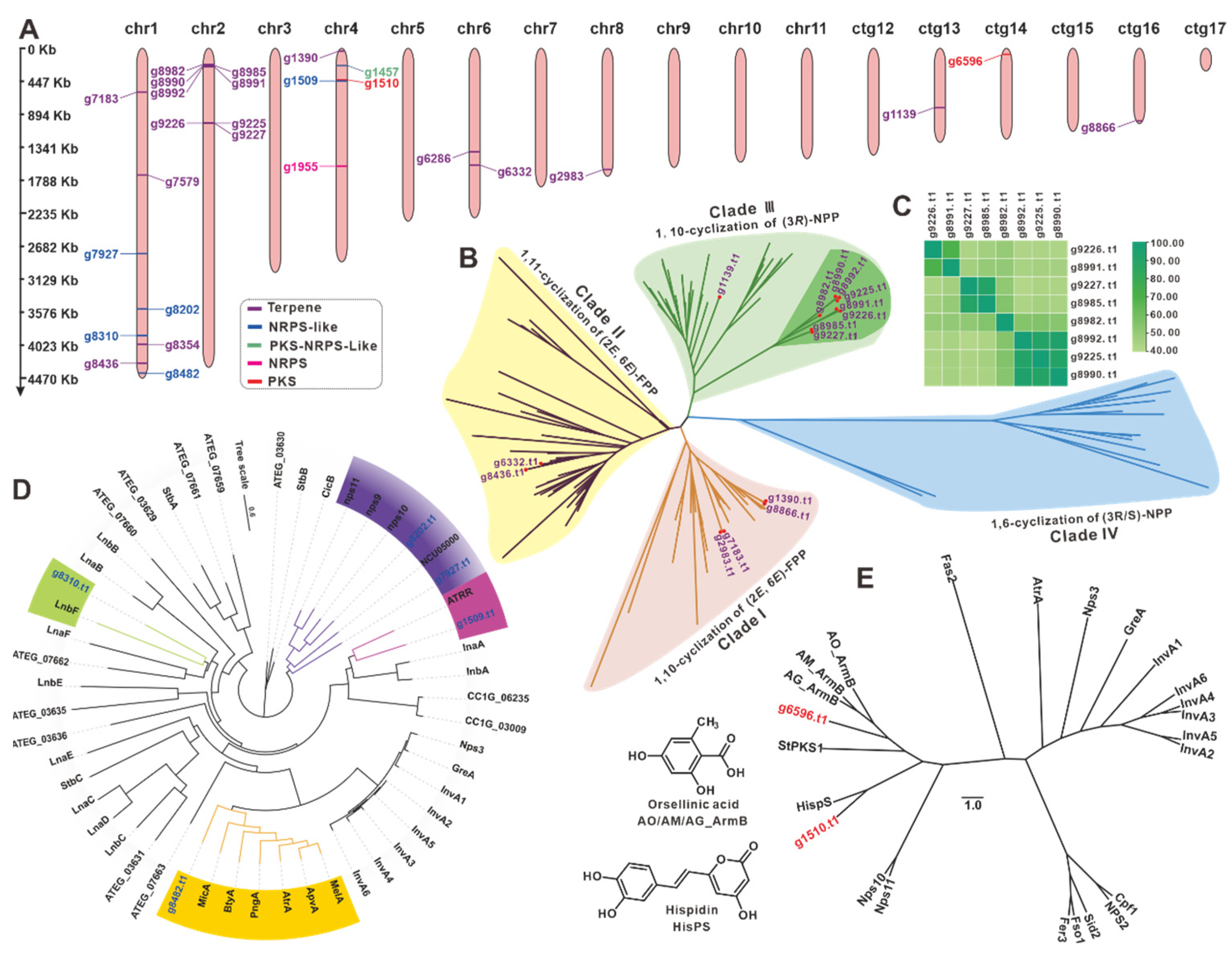
3.7. The BGCs for Secondary Metabolite Analysis
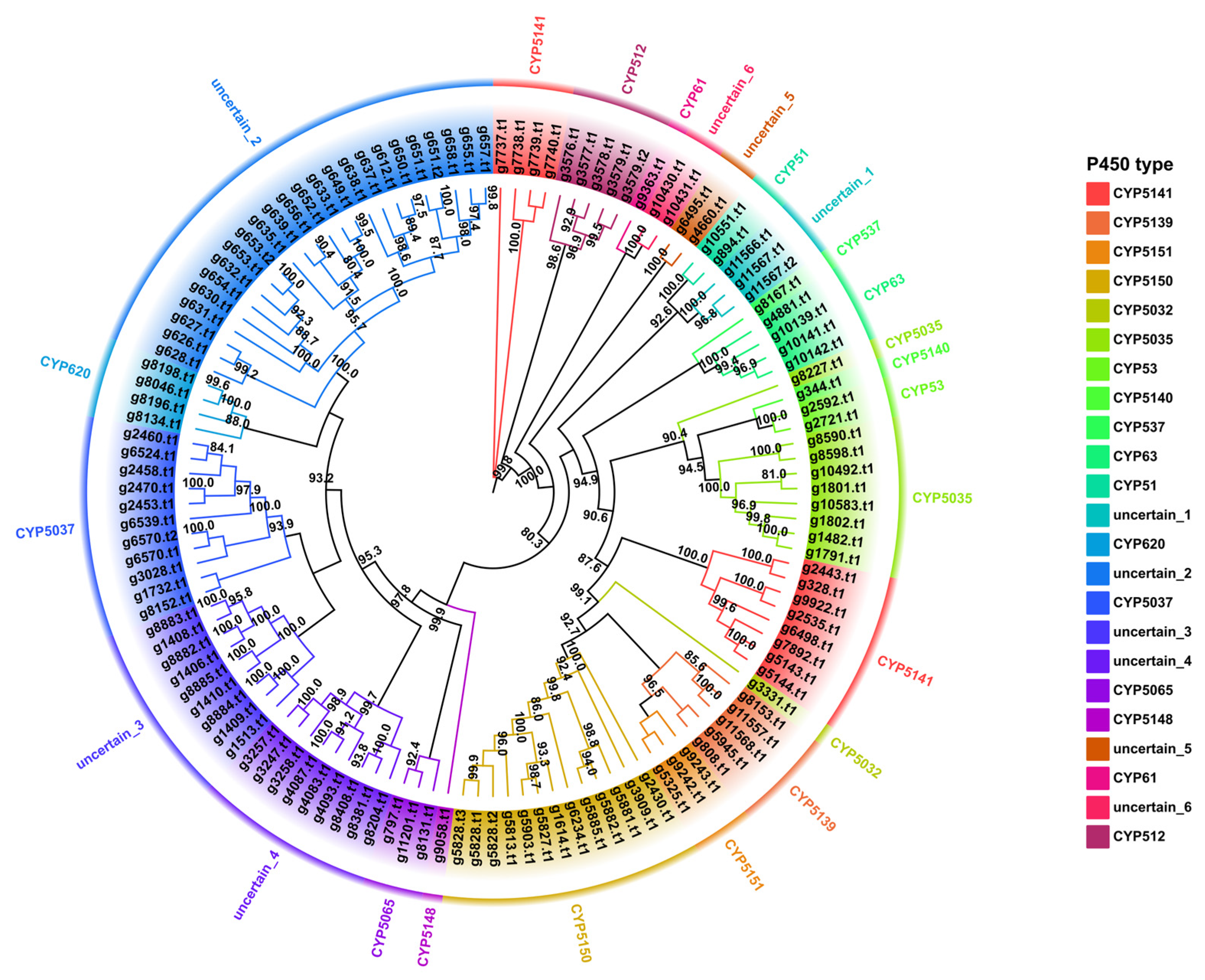
3.8. Cytochrome P450 Family Analysis and Identification
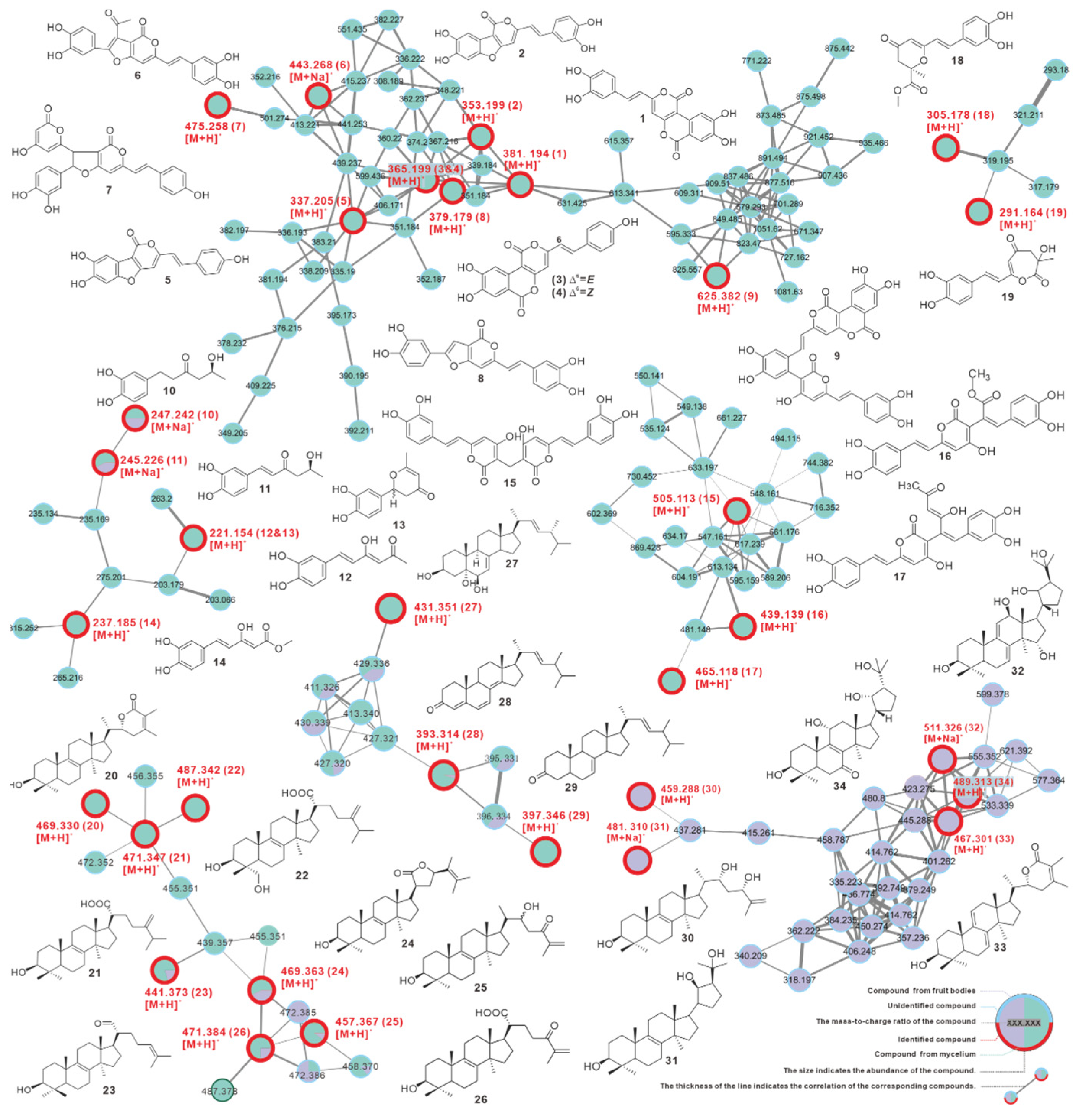
3.9. Identification and Difference of Metabolites from Fruiting Bodies and Mycelium
4. Discussion
4.1. Inonotus Hispidus and Sanghuang-like Fungi
4.2. The Metabolites and Medicinal Properties of Inonotus Hispidus
4.3. Genome Sequencing Helps Decipher Biosynthesis of Bioactive Ingredients in Medicinal Macrofungi
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Piątek, M. Inonotus hispidus (Bull.: Fr.) Karst. In Atlas of the Geographical Distribution of Fungi in Poland; Wojewoda, W., Ed.; W. Szafer Institute of Botany, Polish Academy of Sciences: Warsaw, Poland, 2000; pp. 35–40. [Google Scholar]
- Song, F.; Su, D.; Keyhani, N.O.; Wang, C.; Shen, L.; Qiu, J. Influence of selenium on the mycelia of the shaggy bracket fungus, Inonotus hispidus. J. Sci. Food Agric. 2022, 102, 3762–3770. [Google Scholar] [CrossRef] [PubMed]
- Zan, L.-F.; Bao, H.-Y. Progress in Inonotus hispidus research. Acta Edulis Fungi 2011, 18, 78–82. [Google Scholar] [CrossRef]
- Ali, N.A.A.; Jansen, R.; Pilgrim, H.; Liberra, K.; Lindequist, U. Hispolon, a yellow pigment from Inonotus hispidus. Phytochemistry 1996, 41, 927–929. [Google Scholar] [CrossRef]
- Yousfi, M.; Djeridane, A.; Bombarda, I.; Chahrazed, H.; Duhem, B.; Gaydou, E.M. Isolation and Characterization of a New Hispolone Derivative from Antioxidant Extracts of Pistacia atlantica. Phytother. Res. 2009, 23, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, H. Deciphering key regulators of Inonotus hispidus petroleum ether extract involved in anti-tumor through whole transcriptome and proteome analysis in H22 tumor-bearing mice model. J. Ethnopharmacol. 2022, 296, 115468. [Google Scholar] [CrossRef] [PubMed]
- Kou, R.W.; Du, S.T.; Xia, B.; Zhang, Q.; Yin, X.; Gao, J.M. Phenolic and Steroidal Metabolites from the Cultivated Edible Inonotus hispidus Mushroom and Their Bioactivities. J. Agric. Food Chem. 2021, 69, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Lu, X.-Y.; Han, J.-X.; Aisa, H.A.; Yuan, T. Triterpenoids and phenolics from the fruiting bodies of Inonotus hispidus and their activations of melanogenesis and tyrosinase. Chin. Chem. Lett. 2017, 28, 1052–1056. [Google Scholar] [CrossRef]
- Perrin, P.W.; Towers, G.H.N. Hispidin biosynthesis in cultures of Polyporus hispidus. Phytochemistry 1973, 12, 589–592. [Google Scholar] [CrossRef]
- Wang, Z.; Feng, X.; Liu, C.; Gao, J.; Qi, J. Diverse Metabolites and Pharmacological Effects from the Basidiomycetes Inonotus hispidus. Antibiotics 2022, 11, 1097. [Google Scholar] [CrossRef] [PubMed]
- Kahlos, K.; Hiltunen, R.; v Schantz, M. 3β-Hydroxy-lanosta-8,24-dien-21-al, a New Triterpene from Inontus obliquus. Planta Med. 1984, 50, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Liu, X.; Xiang, K.; Chen, L.; Wu, X.; Lin, W.; Zheng, M.; Fu, J. Characterization of the physicochemical properties and extraction optimization of natural melanin from Inonotus hispidus mushroom. Food Chem. 2019, 277, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bao, H. Anti-tumor effect of Inonotus hispidus petroleum ether extract in H22 tumor-bearing mice and analysis its mechanism by untargeted metabonomic. J. Ethnopharmacol. 2022, 285, 114898. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Bao, H.; Wang, H.; Li, Q. Anti-tumour Effect and Pharmacokinetics of an Active Ingredient Isolated from Inonotus hispidus. Biol. Pharm. Bull. 2019, 42, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zan, L.-F.; Qin, J.-C.; Zhang, Y.-M.; Yao, Y.-H.; Bao, H.-Y.; Li, X. Antioxidant Hispidin Derivatives from Medicinal Mushroom Inonotus hispidus. Chem. Pharm. Bull. 2011, 59, 770–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, P.; Girometta, C.; Tirillini, B.; Moretti, S.; Covino, S.; Cipriani, M.; D’Ellena, E.; Angeles, G.; Federici, E.; Savino, E.; et al. A comparative study of the antimicrobial and antioxidant activities of Inonotus hispidus fruit and their mycelia extracts. Int. J. Food Prop. 2019, 22, 768–783. [Google Scholar] [CrossRef] [Green Version]
- Min, T.; Ye, D.; Yang, Y.; Xie, M.-L.; Wang, S.-M.; Chen, C.-B.; Wang, H.; Li, Y. α-glucosidase Inhibition and Antioxidant Activities of Different Polar Extracts of Inonotus hispidus. Edible Fungi China 2021, 40, 37–41+46. [Google Scholar] [CrossRef]
- Shomali, N.; Onar, O.; Alkan, T.; Demirtaş, N.; Akata, I.; Yildirim, Ö. Investigation of the Polyphenol Composition, Biological Activities, and Detoxification Properties of Some Medicinal Mushrooms from Turkey. Turk J. Pharm. Sci. 2019, 16, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hou, Y. Determination of trace elements in anti-influenza virus mushrooms. Biol. Trace Elem. Res. 2011, 143, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.A.A.; Mothana, R.A.A.; Lesnau, A.; Pilgrim, H.; Lindequist, U. Antiviral activity of Inonotus hispidus. Fitoterapia 2003, 74, 483–485. [Google Scholar] [CrossRef]
- Gruendemann, C.; Arnhold, M.; Meier, S.; Baecker, C.; Garcia-Kaeufer, M.; Grunewald, F.; Steinborn, C.; Klemd, A.M.; Wille, R.; Huber, R.; et al. Effects of Inonotus hispidus Extracts and Compounds on Human Immunocompetent Cells. Planta Med. 2016, 82, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hou, R.; Yan, J.; Xu, K.; Wu, X.; Lin, W.; Zheng, M.; Fu, J. Purification and characterization of Inonotus hispidus exopolysaccharide and its protective effect on acute alcoholic liver injury in mice. Int. J. Biol. Macromol. 2019, 129, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Benarous, K.; Djeridane, A.; Kameli, A.; Yousfi, M. Inhibition of Candida rugosa Lipase by Secondary Metabolites Extracts of Three Algerian Plants and their Antioxydant Activities. Curr. Enzym. Inhib. 2013, 9, 75–82. [Google Scholar] [CrossRef]
- Benarous, K.; Bombarda, I.; Iriepa, I.; Moraleda, I.; Gaetan, H.; Linani, A.; Tahri, D.; Sebaa, M.; Yousfi, M. Harmaline and hispidin from Peganum harmala and Inonotus hispidus with binding affinity to Candida rugosa lipase: In silico and in vitro studies. Bioorg. Chem. 2015, 62, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ying, Y.-M.; Zhang, L.-Y.; Zhang, X.; Bai, H.-B.; Liang, D.-E.; Ma, L.-F.; Shan, W.-G.; Zhan, Z.-J. Terpenoids with alpha-glucosidase inhibitory activity from the submerged culture of Inonotus obliquus. Phytochemistry 2014, 108, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Hu, S.; Peng, B.; Li, Z.; Yuan, X.; Xiao, S.; Fu, Y. Genome of Ganoderma Species Provides Insights Into the Evolution, Conifers Substrate Utilization, and Terpene Synthesis for Ganoderma tsugae. Front. Microbiol. 2021, 12, 724451. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xu, J.; Liu, C.; Zhu, Y.; Nelson, D.R.; Zhou, S.; Li, C.; Wang, L.; Guo, X.; Sun, Y.; et al. Genome sequence of the model medicinal mushroom Ganoderma lucidum. Nat. Commun. 2012, 3, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.Y.; Fan, W.L.; Wang, W.F.; Chen, T.; Tang, Y.C.; Chu, F.H.; Chang, T.T.; Wang, S.Y.; Li, M.Y.; Chen, Y.H.; et al. Genomic and transcriptomic analyses of the medicinal fungus Antrodia cinnamomea for its metabolite biosynthesis and sexual development. Proc. Natl. Acad. Sci. USA 2014, 111, E4743–E4752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, W.; Wang, Y.; Xie, C.; Zhou, Y.; Zhu, Z.; Peng, Y. Whole genome sequence of an edible and medicinal mushroom, Hericium erinaceus (Basidiomycota, Fungi). Genomics 2020, 112, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Han, H.; Qi, J.; Gao, J.-M.; Xu, Z.; Wang, P.; Zhang, J.; Liu, C. Genome sequencing of Inonotus obliquus reveals insights into candidate genes involved in secondary metabolite biosynthesis. BMC Genom. 2022, 23, 314. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.-G.; Wang, Z.-X.; Feng, X.-L.; Zhang, R.-Q.; Shen, D.-Y.; Du, S.; Gao, J.-M.; Qi, J. Chromosome-Level Genome Sequences, Comparative Genomic Analyses, and Secondary-Metabolite Biosynthesis Evaluation of the Medicinal Edible Mushroom Laetiporus sulphureus. Microbiol. Spectr. 2022, 10, e02439-22. [Google Scholar] [CrossRef]
- Wu, S.H.; Chang, C.C.; Wei, C.L.; Jiang, G.Z.; Cui, B.K. Sanghuangporus toxicodendri sp. nov. (Hymenochaetales, Basidiomycota) from China. MycoKeys 2019, 57, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kües, U.; Casselton, L.A. Homeodomains and regulation of sexual development in basidiomycetes. Trends Genet. 1992, 8, 154–155. [Google Scholar] [CrossRef] [PubMed]
- Casselton, L.A.; Olesnicky, N.S. Molecular Genetics of Mating Recognition in Basidiomycete Fungi. Microbiol. Mol. Biol. Rev. 1998, 62, 55–70. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-L.; Li, W.-C.; Chuang, Y.-C.; Liu, H.-C.; Huang, C.-H.; Lo, K.-Y.; Chen, C.-Y.; Chang, F.-M.; Chang, G.-A.; Lin, Y.-L.; et al. Sexual Crossing, Chromosome-Level Genome Sequences, and Comparative Genomic Analyses for the Medicinal Mushroom Taiwanofungus Camphoratus (Syn. Antrodia Cinnamomea, Antrodia Camphorata). Microbiol. Spectr. 2022, 10, e02032-21. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Y.; Chen, L.; Wang, H.; Guo, L.; Zhou, X.; Dou, M.; Wang, B.; Lin, J.; Liu, L.; et al. Genetic structure and evolutionary diversity of mating-type (MAT) loci in Hypsizygus marmoreus. IMA Fungus 2021, 12, 35. [Google Scholar] [CrossRef]
- Ohm, R.A.; de Jong, J.F.; Lugones, L.G.; Aerts, A.; Kothe, E.; Stajich, J.E.; de Vries, R.P.; Record, E.; Levasseur, A.; Baker, S.E.; et al. Genome sequence of the model mushroom Schizophyllum commune. Nat. Biotech. 2010, 28, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, K.-E.L.; Blanchette, R.A.; Ander, P. Morphological Aspects of Wood Degradation by Fungi and Bacteria. In Microbial and Enzymatic Degradation of Wood and Wood Components; Springer: Berlin/Heidelberg, Germany, 1990; pp. 1–87. [Google Scholar]
- Sista Kameshwar, A.K.; Qin, W. Comparative study of genome-wide plant biomass-degrading CAZymes in white rot, brown rot and soft rot fungi. Mycology 2018, 9, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rytioja, J.; Hildén, K.; Yuzon, J.; Hatakka, A.; Vries, R.P.D.; Mäkelä, M.R. Plant-Polysaccharide-Degrading Enzymes from Basidiomycetes. Microbiol. Mol. Biol. Rev. 2014, 78, 614–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Qiao, S.; Xu, Z.; Guan, F.; Ding, Z.; Gu, Z.; Zhang, L.; Shi, G. Effects of culture conditions on monosaccharide composition of Ganoderma lucidum exopolysaccharide and on activities of related enzymes. Carbohyd. Polym. 2015, 133, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Tang, Z.; Zhang, J.; Li, X.; Yang, Z.; Yang, C.; Zhang, Z.; Huang, Z. Comparative transcriptome analysis reveals the genetic basis underlying the biosynthesis of polysaccharides in Hericium erinaceus. Bot. Stud. 2019, 60, 15. [Google Scholar] [CrossRef]
- Wu, J.; Yang, X.; Duan, Y.; Wang, P.; Qi, J.; Gao, J.-M.; Liu, C. Biosynthesis of Sesquiterpenes in Basidiomycetes: A Review. J. Fungi 2022, 8, 913. [Google Scholar] [CrossRef] [PubMed]
- Hai, Y.; Huang, A.M.; Tang, Y. Structure-guided function discovery of an NRPS-like glycine betaine reductase for choline biosynthesis in fungi. Proc. Natl. Acad. Sci. USA 2019, 116, 10348–10353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forseth, R.R.; Amaike, S.; Schwenk, D.; Affeldt, K.J.; Hoffmeister, D.; Schroeder, F.C.; Keller, N.P. Homologous NRPS-like Gene Clusters Mediate Redundant Small-Molecule Biosynthesis in Aspergillus flavus. Angew. Chem. Int. Edit. 2013, 52, 1590–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, H.-H.; Chiang, Y.-M.; Entwistle, R.; Ahuja, M.; Lee, K.-H.; Bruno, K.S.; Wu, T.-K.; Oakley, B.R.; Wang, C.C.C. Molecular genetic analysis reveals that a nonribosomal peptide synthetase-like (NRPS-like) gene in Aspergillus nidulans is responsible for microperfuranone biosynthesis. Appl. Microbiol. Biotechnol. 2012, 96, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotlobay, A.A.; Sarkisyan, K.S.; Mokrushina, Y.A.; Marcet-Houben, M.; Serebrovskaya, E.O.; Markina, N.M.; Gonzalez Somermeyer, L.; Gorokhovatsky, A.Y.; Vvedensky, A.; Purtov, K.V.; et al. Genetically encodable bioluminescent system from fungi. Proc. Natl. Acad. Sci. USA 2018, 115, 12728–12732. [Google Scholar] [CrossRef] [Green Version]
- Engels, B.; Heinig, U.; Grothe, T.; Stadler, M.; Jennewein, S. Cloning and Characterization of an Armillaria gallica cDNA Encoding Protoilludene Synthase, Which Catalyzes the First Committed Step in the Synthesis of Antimicrobial Melleolides *. J. Biol. Chem. 2011, 286, 6871–6878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunematsu, Y.; Takanishi, J.; Asai, S.; Masuya, T.; Nakazawa, T.; Watanabe, K. Genomic Mushroom Hunting Decrypts Coprinoferrin, A Siderophore Secondary Metabolite Vital to Fungal Cell Development. Org. Lett. 2019, 21, 7582–7586. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Sinha, K.; Sil, C.P. Cytochrome P450s: Mechanisms and Biological Implications in Drug Metabolism and its Interaction with Oxidative Stress. Curr. Drug Metab. 2014, 15, 719–742. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, J.; Cheng, F.; Li, S. Cytochrome P450 enzymes in fungal natural product biosynthesis. Nat. Prod. Rep. 2021, 38, 1072–1099. [Google Scholar] [CrossRef]
- Durairaj, P.; Hur, J.-S.; Yun, H. Versatile biocatalysis of fungal cytochrome P450 monooxygenases. Micro. Cell Fact. 2016, 15, 125. [Google Scholar] [CrossRef] [PubMed]
- Fessner, N.D.; Nelson, D.R.; Glieder, A. Evolution and enrichment of CYP5035 in Polyporales: Functionality of an understudied P450 family. Appl. Microbiol. Biotechnol. 2021, 105, 6779–6792. [Google Scholar] [CrossRef]
- Lee, I.-K.; Yun, B.-S. Styrylpyrone-class compounds from medicinal fungi Phellinus and Inonotus spp., and their medicinal importance. J. Antibiot. 2011, 64, 349–359. [Google Scholar] [CrossRef]
- Qing-Jie, L. Study on the active substances and quality standards of Sanghuang fungus. Ph.D. Thesis, Jilin Agricultural University, Jilin, China, 2017. [Google Scholar]
- Lifeng, Z. Studies on the Chemical Constituents and Pharmacological Activities of Inonotus Hispidus and Fomitiporia Ellipsoidea. Ph.D. Thesis, Jilin Agricultural University, Jilin, China, 2012. [Google Scholar]
- Lee, I.K.; Kim, Y.S.; Seok, S.J.; Yun, B.S. Inoscavin E, a Free Radical Scavenger from the Fruiting Bodies of Inonotus xeranticus. J. Antibiot. 2007, 60, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.K.; Kim, Y.S.; Jang, Y.W.; Jung, J.Y.; Yun, B.S. New antioxidant polyphenols from the medicinal mushroom Inonotus obliquus. Bioorg. Med. Chem. Lett. 2007, 17, 6678–6681. [Google Scholar] [CrossRef]
- Yang, S.-D.; Bao, H.-Y.; Wang, H. Chemical components and anti-tumour compounds from Inonotus hispidus. Mycosystema 2019, 38, 127–133. [Google Scholar] [CrossRef]
- Lee, I.K.; Yun, B.S. Highly oxygenated and unsaturated metabolites providing a diversity of hispidin class antioxidants in the medicinal mushrooms Inonotus and Phellinus. Bioorg. Med. Chem. 2007, 15, 3309–3314. [Google Scholar] [CrossRef]
- Xiu-Zhen, Y. Studies on the Chemical Constituents of Xanthochrous Hispidus. Master’s Thesis, Shandong University of Traditional Chinese Medicine, Shandong, China, 2008. [Google Scholar]
- Liu, C.; Zhao, C.; Pan, H.H.; Kang, J.; Yu, X.T.; Wang, H.Q.; Li, B.M.; Xie, Y.Z.; Chen, R.Y. Chemical constituents from Inonotus obliquus and their biological activities. J. Nat. Prod. 2014, 77, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Xia, G.; Chen, L.; Zhao, J.; Xie, Z.; Qiu, F.; Han, G. Chemical constituents from Inonotus obliquus and their antitumor activities. J. Nat. Med. 2016, 70, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Toyoshima, M.; Yamada, T. New lanostane-type triterpenoids, inonotsutriols D, and E, from Inonotus obliquus. Phytochem. Lett. 2011, 4, 328–332. [Google Scholar] [CrossRef]
- Taji, S.; Yamada, T.; Tanaka, R. Three New Lanostane Triterpenoids, Inonotsutriols A, B, and C, from Inonotus obliquus. Helv. Chim. Acta 2008, 91, 1513–1524. [Google Scholar] [CrossRef]
- Chen, W.; Tan, H.; Liu, Q.; Zheng, X.; Zhang, H.; Liu, Y.; Xu, L. A Review: The Bioactivities and Pharmacological Applications of Phellinus linteus. Molecules 2019, 24, 1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, P.; Zhang, Y.; Li, N. The phytochemistry and pharmacology of medicinal fungi of the genus Phellinus: A review. Food Funct. 2021, 12, 1856–1881. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tian, T.; Miao, H.; Zhao, Y.-Y. Traditional uses, fermentation, phytochemistry and pharmacology of Phellinus linteus: A review. Fitoterapia 2016, 113, 6–26. [Google Scholar] [CrossRef]
- Zhu, L.; Song, J.; Zhou, J.-L.; Si, J.; Cui, B.-K. Species Diversity, Phylogeny, Divergence Time, and Biogeography of the Genus Sanghuangporus (Basidiomycota). Front. Microbiol. 2019, 10, 00812. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Dai, Y.; Hattori, T.; Yu, T.; Wang, D.; Parmasto, É.K.; Zhang, H.; Shi, S. Species clarification for the medicinally valuable ‘sanghuang’ mushroom. Bot. Stud. 2012, 53, 135–149. [Google Scholar]
- Huo, J.; Zhong, S.; Du, X.; Cao, Y.; Wang, W.; Sun, Y.; Tian, Y.; Zhu, J.; Chen, J.; Xuan, L.; et al. Whole-genome sequence of Phellinus gilvus (mulberry Sanghuang) reveals its unique medicinal values. J. Adv. Res. 2020, 24, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Suabjakyong, P.; Saiki, R.; Van Griensven, L.J.L.D.; Higashi, K.; Nishimura, K.; Igarashi, K.; Toida, T. Polyphenol Extract from Phellinus igniarius Protects against Acrolein Toxicity In Vitro and Provides Protection in a Mouse Stroke Model. PLoS ONE 2015, 10, e0122733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-S.; Lin, Z.-M.; Wang, L.-N.; Guo, D.-X.; Wang, S.-Q.; Liu, Y.-Q.; Yuan, H.-Q.; Lou, H.-X. Phenolic compounds with NF-κB inhibitory effects from the fungus Phellinus baumii. Bio. Med. Chem. Lett. 2011, 21, 3261–3267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-J.; Chen, B.-S.; Dai, H.-Q.; Ren, J.-W.; Zhou, L.-W.; Wu, S.-H.; Liu, H.-W. Sesquiterpenes and polyphenols with glucose-uptake stimulatory and antioxidant activities from the medicinal mushroom Sanghuangporus sanghuang. Chin. J. Nat. Med. 2021, 19, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, R.; Zhang, J.; Bu, Q.; Wang, W.; Liu, Y.; Li, Q.; Guo, Y.; Zhang, L.; Yang, Y. The integration of metabolome and proteome reveals bioactive polyphenols and hispidin in ARTP mutagenized Phellinus baumii. Sci. Rep. 2019, 9, 16172. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Sui, D.; Wang, L.; Zhang, X.; Wang, C.; Liu, C. Research Progress of Small Molecule Chemical Components and Pharmacological Values of Inonotus obliquus. J. Fungal Res. 2021, 20, 214–227. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamada, K.D.; Tomii, K.; Katoh, K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018, 34, 2490–2492. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | I. hispidus NPCB_001 | I. obliquus CT5 |
|---|---|---|
| Sequencing technology | Illumina NovaSeq 6000 | Illumina HiSeq 6000 |
| Sequencing depth | 230.0× | 200.0× |
| Number of scaffolds | 17 | 31 |
| Total assembly length | 34,017,109 | 38,061,412 |
| largest length | 4,469,123 | 4,380,421 |
| Scaffold N50(bp) | 2,340,722 | 1,971,511 |
| Scaffold L50 | 5 | 7 |
| GC content (%) | 48.39 | 47.60 |
| No. of proteins | 12,304 | 12,525 |
| Genome accession | GCA_024712875.1 | GCA_023101745.1 |
| Isolate information | Mycelium | Mycelium |
| Cluster | Location | Start(bp) | Stop(bp) | Core Gene IDs | Core Gene Type |
|---|---|---|---|---|---|
| 1 | chr1 | 586,000 | 604,625 | g7183.t1 | terpene terpene |
| 2 | chr1 | 1,703,424 | 1,723,315 | g7579.t1 | |
| 3 | chr1 | 2,757,672 | 2,797,202 | g7927.t1 | NRPS-like |
| 4 | chr1 | 3,514,470 | 3,555,756 | g8202.t1 | NRPS-like |
| 5 | chr1 | 3,885,519 | 3,920,464 | g8310.t1 | NRPS-like |
| 6 | chr1 | 4,003,561 | 4,021,717 | g8354.t1 | terpene |
| 7 | chr1 | 4,255,843 | 4,277,160 | g8436.t1 | terpene |
| 8 | chr1 | 4,388,087 | 4,431,254 | g8482.t1 | NRPS-like |
| 9 | chr2 | 205,256 | 258,843 | g8982.t1 | terpene |
| g8985.t1 | |||||
| g8990.t1 | |||||
| g8991.t1 | |||||
| g8992.t1 | |||||
| 10 | chr2 | 999,219 | 1,027,559 | g9225.t1 | terpene |
| g9226.t1 | |||||
| g9227.t1 | |||||
| 11 | chr4 | 29,271 | 50,737 | g1390.t1 | terpene |
| 12 | chr4 | 213,291 | 257,392 | g1457.t1 | T1PKS- NRPS-like |
| 13 | chr4 | 407,063 | 453,131 | g1509.t1 | NRPS-like |
| g1510.t1 | T1PKS | ||||
| 14 | chr4 | 1,576,159 | 1,624,146 | g1955.t1 | NRPS |
| 15 | chr6 | 1,395,705 | 1,411,230 | g6286.t1 | terpene |
| 16 | chr6 | 1,570,068 | 1,591,381 | g6332.t1 | terpene |
| 17 | chr8 | 1,624,840 | 1,646,078 | g2983.t1 | terpene |
| 18 | ctg13 | 785,200 | 806,403 | g1139.t1 | terpene |
| 19 | ctg14 | 61,058 | 108,404 | g6596.t1 | T1PKS |
| 20 | ctg16 | 977,869 | 999,335 | g8866.t1 | terpene |
| No | Source | Putative Metabolite | Molecular Formula | Adduct | m/z | Reference |
|---|---|---|---|---|---|---|
| 1 | fruiting body | phelligridin D | C20H12O8 | [M + H]+ | 381.194 | Li, et al. [55] |
| 2 | fruiting body | phellibaumin A | C19H12O7 | [M + H]+ | 353.199 | Li, et al. [55] |
| 3 | fruiting body | phelligridin C | C20H12O7 | [M + H]+ | 365.199 | Li, et al. [55] |
| 4 | fruiting body | phelligridin C′ | C20H12O7 | [M + H]+ | 365.199 | Li, et al. [55] |
| 5 | fruiting body | 3′4′-dihydroxy-5-[11- hydroxyphenyl]-6,7-vinyl]-3,5-dioxafluoren-5-one | C19H12O6 | [M + H]+ | 337.205 | Li, et al. [55] |
| 6 | fruiting body | inoscavin C | C23H16O8 | [M + Na]+ | 443.268 | Zan, et al. [56] |
| 7 * | fruiting body | hypholomine A | C26H18O9 | [M + H]+ | 475.258 | Lee, et al. [54] |
| 8 * | fruiting body | inoscavin E | C21H14O7 | [M + H]+ | 379.179 | Lee, et al. [57] |
| 9 * | fruiting body | inonoblin A | C33H20O13 | [M + H]+ | 625.382 | Lee, et al. [58] |
| 10 | both | inonophenol A | C12H16O4 | [M + Na]+ | 247.242 | Kou, et al. [7] |
| 11 | both | inonophenol B | C12H14O4 | [M + Na]+ | 245.226 | Kou, et al. [7] |
| 12 | fruiting body | hispolon | C12H12O4 | [M + H]+ | 221.154 | Ali, N.A.A., et al. [4] |
| 13 | fruiting body | hispinine | C12H12O4 | [M + H]+ | 221.154 | Ren, et al. [8] |
| 14 | fruiting body | methyl 5-(3,4-dihydroxyphenyl)-3-hydroxypenta-2,4-dienoate | C12H12O5 | [M + H]+ | 237.185 | Yousfi, et al. [5] |
| 15 | fruiting body | MBP | C27H20O10 | [M + H]+ | 505.113 | Yang, et al. [59] |
| 16 * | fruiting body | interfungin C | C23H18O9 | [M + H]+ | 439.139 | Lee, et al. [60] |
| 17 * | fruiting body | interfungin A | C25H20O9 | [M + H]+ | 465.118 | Lee, et al. [60] |
| 18 | fruiting body | inonophenol C | C16H16O6 | [M + H]+ | 305.178 | Kou, et al. [7] |
| 19 | fruiting body | inonotusin A | C15H14O6 | [M + H]+ | 291.164 | Zan, et al. [56] |
| 20 | fruiting body | inotolactone B | C31H48O3 | [M + H]+ | 469.330 | Ying, et al. [25] |
| 21 | fruiting body | eburicoic acid | C31H50O3 | [M + H]+ | 471.347 | Yang, et al. [61] |
| 22 | fruiting body | hispindic acid B | C31H50O4 | [M + H]+ | 487.342 | Ren, et al. [8] |
| 23 | both | 3β-hydroxy-lanosta-8,24-dien-21-al | C30H48O2 | [M + H]+ | 441.373 | Kou, et al. [7] |
| 24 * | both | inonotusol F | C31H48O3 | [M + H]+ | 469.368 | Liu, et al. [62] |
| 25 * | both | inonotusol G | C30H48O3 | [M + H]+ | 457.367 | Liu, et al. [62] |
| 26 * | both | inonotusane F | C30H46O4 | [M + H]+ | 471.384 | Zhao, et al. [63] |
| 27 | fruiting body | cerevisterol | C28H46O3 | [M + H]+ | 431.351 | Kou, et al. [7] |
| 28 | both | 4,6,8(14),22(23)-tetraen-3-one-ergostane | C28H40O | [M + H]+ | 393.314 | Zan, et al. [56] |
| 29 | fruiting body | 7(8),22(23)-dien-3-one-ergostane | C28H40O | [M + H]+ | 397.346 | Zan, et al. [56] |
| 30 * | mycelium | inonotsutriol E | C30H50O3 | [M + H]+ | 459.288 | Reiko Tanaka, et al. [64] |
| 31 * | mycelium | inonotsutriol A | C30H50O3 | [M + Na]+ | 481.310 | Sayaka Taji, et al. [65] |
| 32 * | mycelium | inonotusane E | C30H50O3 | [M + Na]+ | 511.326 | Zhao, et al. [63] |
| 33 * | mycelium | inotolactone A | C31H46O3 | [M + H]+ | 467.301 | Ying, et al. [25] |
| 34 * | mycelium | inonotusol E | C30H48O5 | [M + H]+ | 489.313 | Liu, et al. [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.-q.; Feng, X.-l.; Wang, Z.-x.; Xie, T.-c.; Duan, Y.; Liu, C.; Gao, J.-m.; Qi, J. Genomic and Metabolomic Analyses of the Medicinal Fungus Inonotus hispidus for Its Metabolite’s Biosynthesis and Medicinal Application. J. Fungi 2022, 8, 1245. https://doi.org/10.3390/jof8121245
Zhang R-q, Feng X-l, Wang Z-x, Xie T-c, Duan Y, Liu C, Gao J-m, Qi J. Genomic and Metabolomic Analyses of the Medicinal Fungus Inonotus hispidus for Its Metabolite’s Biosynthesis and Medicinal Application. Journal of Fungi. 2022; 8(12):1245. https://doi.org/10.3390/jof8121245
Chicago/Turabian StyleZhang, Rui-qi, Xi-long Feng, Zhen-xin Wang, Tian-chen Xie, Yingce Duan, Chengwei Liu, Jin-ming Gao, and Jianzhao Qi. 2022. "Genomic and Metabolomic Analyses of the Medicinal Fungus Inonotus hispidus for Its Metabolite’s Biosynthesis and Medicinal Application" Journal of Fungi 8, no. 12: 1245. https://doi.org/10.3390/jof8121245
APA StyleZhang, R. -q., Feng, X. -l., Wang, Z. -x., Xie, T. -c., Duan, Y., Liu, C., Gao, J. -m., & Qi, J. (2022). Genomic and Metabolomic Analyses of the Medicinal Fungus Inonotus hispidus for Its Metabolite’s Biosynthesis and Medicinal Application. Journal of Fungi, 8(12), 1245. https://doi.org/10.3390/jof8121245