
Near Chromosome-Level Genome Assembly and Annotation of Rhodotorula babjevae Strains Reveals High Intraspecific Divergence

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Yeast Strains
2.2. DNA Purification
2.3. Library Preparation and Sequencing
2.4. Genome Assembly and Annotation
2.5. Genome Divergence Analysis
3. Results and Discussion
3.1. Genome Assembly, Ploidy Estimation, and Gene Annotation of R. babjevae Strains
3.2. Chromosome Organization
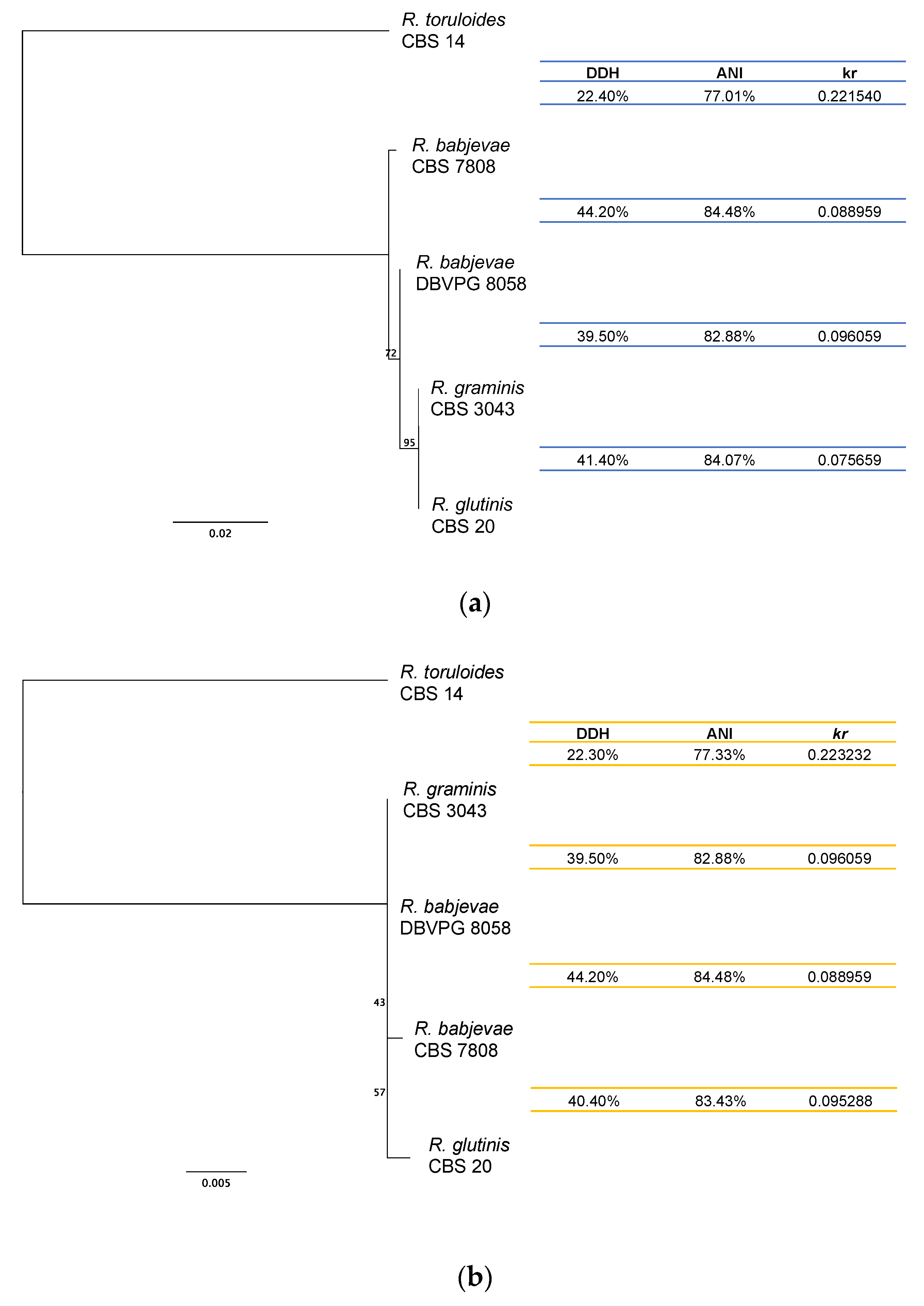
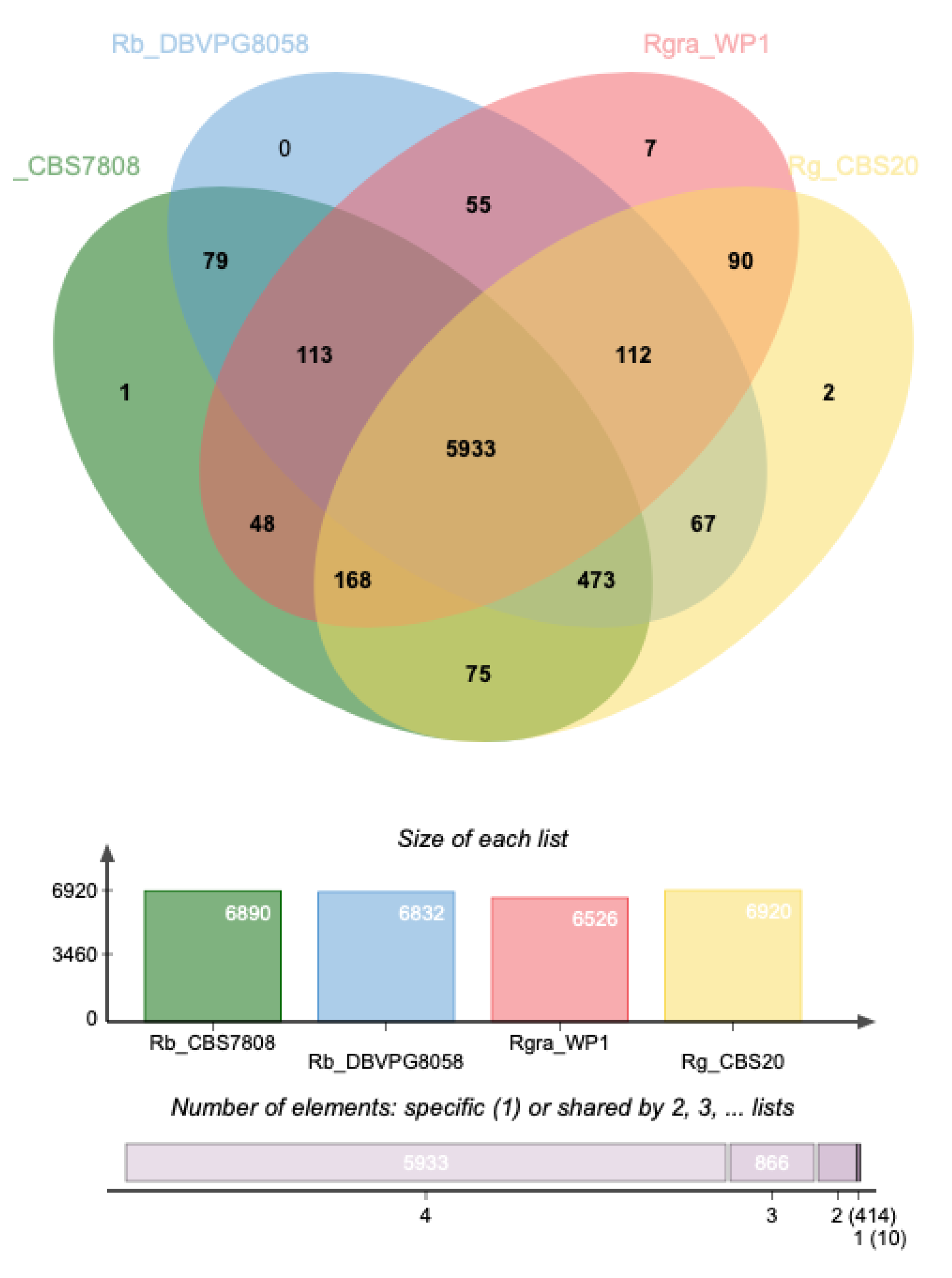
3.3. Genome Divergence Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poontawee, R.; Yongmanitchai, W.; Limtong, S. Efficient oleaginous yeasts for lipid production from lignocellulosic sugars and effects of lignocellulose degradation compounds on growth and lipid production. Process Biochem. 2017, 53, 44–60. [Google Scholar] [CrossRef]
- Brandenburg, J.; Poppele, I.; Blomqvist, J.; Puke, M.; Pickova, J.; Sandgren, M.; Rapoport, A.; Vedernikovs, N.; Passoth, V. Bioethanol and lipid production from the enzymatic hydrolysate of wheat straw after furfural extraction. Appl. Microbiol. Biotechnol. 2018, 102, 6269–6277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Hernández, G.C.; Müller, B.; Chmielarz, M.; Brandt, C.; Hölzer, M.; Viehweger, A.; Passoth, V. Chromosome-level genome assembly and transcriptome—Based annotation of the oleaginous yeast Rhodotorula toruloides CBS 14. Genomics 2021, 113, 4022–4027. [Google Scholar] [CrossRef]
- Chmielarz, M.; Blomqvist, J.; Sampels, S.; Sandgren, M.; Passoth, V. Microbial lipid production from crude glycerol and hemicellulosic hydrolysate with oleaginous yeasts. Biotechnol. Biofuels 2021, 14, 65. [Google Scholar] [CrossRef]
- Ayadi, I.; Belghith, H.; Gargouri, A.; Guerfali, M. Screening of new oleaginous yeasts for single cell oil production, hydrolytic potential exploitation and agro-industrial by-products valorization. Process Saf. Environ. Prot. 2018, 119, 104–114. [Google Scholar] [CrossRef]
- Blomqvist, J.; Pickova, J.; Tilami, S.K.; Sampels, S.; Mikkelsen, N.; Brandenburg, J.; Sandgren, M.; Passoth, V. Oleaginous yeast as a component in fish feed. Sci. Rep. 2018, 8, 15945. [Google Scholar] [CrossRef] [Green Version]
- Guerfali, M.; Ayadi, I.; Mohamed, N.; Ayadi, W.; Belghith, H.; Bronze, M.R.; Ribeiro, M.H.L.; Gargouri, A. Triacylglycerols accumulation and glycolipids secretion by the oleaginous yeast Rhodotorula babjevae Y-SL7: Structural identification and biotechnological applications. Bioresour. Technol. 2019, 273, 326–334. [Google Scholar] [CrossRef]
- Sen, S.; Borah, S.N.; Bora, A.; Deka, S. Production, characterization, and antifungal activity of a biosurfactant produced by Rhodotorula babjevae YS3. Microb. Cell Fact. 2017, 16, 95. [Google Scholar] [CrossRef] [Green Version]
- Seveiri, R. Characterization and prospective applications of the exopolysaccharides produced by Rhodosporidium babjevae. Adv. Pharm. Bull. 2020, 10, 254–263. [Google Scholar] [CrossRef]
- Sen, S.; Borah, S.N.; Kandimalla, R.; Bora, A.; Deka, S. Sophorolipid Biosurfactant Can Control Cutaneous Dermatophytosis Caused by Trichophyton mentagrophytes. Front. Microbiol. 2020, 11, 329. [Google Scholar] [CrossRef] [Green Version]
- Firrincieli, A.; Otillar, R.; Salamov, A.; Schmutz, J.; Khan, Z.; Redman, R.S.; Fleck, N.D.; Lindquist, E.; Grigoriev, I.V.; Doty, S.L. Genome sequence of the plant growth promoting endophytic yeast Rhodotorula graminis WP1. Front. Microbiol. 2015, 6, 978. [Google Scholar] [CrossRef] [Green Version]
- Sen, D.; Paul, K.; Saha, C.; Mukherjee, G.; Nag, M.; Ghosh, S.; Das, A.; Seal, A.; Tripathy, S. A unique life-strategy of an endophytic yeast Rhodotorula mucilaginosa JGTA-S1—A comparative genomics viewpoint. DNA Res. 2019, 26, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Li, C.J.; Zhao, D.; Cheng, P.; Zheng, L.; Yu, G.H. Genomics and lipidomics analysis of the biotechnologically important oleaginous red yeast Rhodotorula glutinis ZHK provides new insights into its lipid and carotenoid metabolism. BMC Genomics 2020, 21, 834. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.M.; Thomas, B.N.; Cavanaugh, N.T.; Morales, G.H.; Mayers, A.N.; Savka, M.A.; Hudson, A.O. Whole genome sequencing of Rhodotorula mucilaginosa isolated from the chewing stick (Distemonanthus benthamianus): Insights into Rhodotorula phylogeny, mitogenome dynamics and carotenoid biosynthesis. PeerJ 2017, 5, e4030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Zhang, S.; Liu, H.; Shen, H.; Lin, X.; Yang, F.; Zhou, Y.J.; Jin, G.; Ye, M.; Zou, H.; et al. A multi-omic map of the lipid-producing yeast Rhodosporidium toruloides. Nat. Commun. 2012, 3, 1111–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakankun, I.; Fristensky, B.; Levin, D.B. Genome sequence analysis of the oleaginous yeast, Rhodotorula diobovata, and comparison of the carotenogenic and oleaginous pathway genes and gene products with other oleaginous yeasts. J. Fungi 2021, 7, 320. [Google Scholar] [CrossRef] [PubMed]
- Tkavc, R.; Matrosova, V.Y.; Grichenko, O.E.; Gostincar, C.; Volpe, R.P.; Klimenkova, P.; Gaidamakova, E.K.; Zhou, C.E.; Stewart, B.J.; Lyman, M.G.; et al. Prospects for fungal bioremediation of acidic radioactive waste sites: Characterization and genome sequence of Rhodotorula taiwanensis MD1149. Front. Microbiol. 2018, 8, 2528. [Google Scholar] [CrossRef]
- Goordial, J.; Raymond-Bouchard, I.; Riley, R.; Ronholm, J.; Shapiro, N.; Woyke, T.; LaButti, K.M.; Tice, H.; Amirebrahimi, M.; Grigoriev, I.V.; et al. Improved high-quality draft genome sequence of the eurypsychrophile Rhodotorula sp. JG1b, isolated from permafrost in the hyperarid upper-elevation McMurdo Dry Valleys, Antarctica. Genome Announc. 2016, 4, 15–17. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.A.; Bunikis, I.; Tiukova, I.; Holmberg, K.; Lötstedt, B.; Pettersson, O.V.; Passoth, V.; Käller, M.; Vezzi, F. De novo assembly of Dekkera bruxellensis: A multi technology approach using short and long-read sequencing and optical mapping. Gigascience 2015, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Tiukova, I.A.; Pettersson, M.E.; Hoeppner, M.P.; Olsen, R.A.; Käller, M.; Nielsen, J.; Dainat, J.; Lantz, H.; Söderberg, J.; Passoth, V. Chromosomal genome assembly of the ethanol production strain CBS 11270 indicates a highly dynamic genome structure in the yeast species Brettanomyces bruxellensis. PLoS ONE 2019, 14, e0215077. [Google Scholar] [CrossRef]
- Chmielarz, M.; Sampels, S.; Blomqvist, J.; Brandenburg, J.; Wende, F.; Sandgren, M.; Passoth, V. FT-NIR: A tool for rapid intracellular lipid quantification in oleaginous yeasts. Biotechnol. Biofuels 2019, 12, 169. [Google Scholar] [CrossRef] [PubMed]
- Pi, H.W.; Anandharaj, M.; Kao, Y.Y.; Lin, Y.J.; Chang, J.J.; Li, W.H. Engineering the oleaginous red yeast Rhodotorula glutinis for simultaneous β-carotene and cellulase production. Sci. Rep. 2018, 8, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.; Bongcam-Rudloff, E.; Müller, B. Abundance tracking by long-read nanopore sequencing of complex microbial communities in samples from 20 different biogas/wastewater plants. Appl. Sci. 2020, 10, 7518. [Google Scholar] [CrossRef]
- Weiß, C.L.; Pais, M.; Cano, L.M.; Kamoun, S.; Burbano, H.A. nQuire: A statistical framework for ploidy estimation using next generation sequencing. BMC Bioinform. 2018, 19, 122. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef] [Green Version]
- Gremme, G.; Steinbiss, S.; Kurtz, S. Genome tools: A comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 645–656. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Konstantinidis, K.T. The enveomics collection: A toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016, 4, e1900v1. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S. Improved Pairwise Alignment of Genomic DNA. Ph.D. Thesis, The Pennsylvania State University, State College, PA, USA, 2007. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Dong, Z.; Fang, L.; Luo, Y.; Wei, Z.; Guo, H.; Zhang, G.; Gu, Y.Q.; Coleman-Derr, D.; Xia, Q.; et al. OrthoVenn2: A web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2019, 47, W52–W58. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsov, A.; Bollin, C.J. NCBI Genome Workbench: Desktop software for comparative genomics, visualization, and GenBank data submission. Methods Mol. Biol. 2021, 2231, 261–295. [Google Scholar] [CrossRef]
- Zou, C.; Chen, A.; Xiao, L.; Muller, H.M.; Ache, P.; Haberer, G.; Zhang, M.; Jia, W.; Deng, P.; Huang, R.; et al. A high-quality genome assembly of quinoa provides insights into the molecular basis of salt bladder-based salinity tolerance and the exceptional nutritional value. Cell Res. 2017, 27, 1327–1340. [Google Scholar] [CrossRef]
- Krahulec, J.; Lišková, V.; Boňková, H.; Lichvariková, A.; Šafranek, M.; Turňa, J. The ploidy determination of the biotechnologically important yeast Candida utilis. J. Appl. Genet. 2020, 61, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Fijarczyk, A.; Hénault, M.; Marsit, S.; Charron, G.; Fischborn, T.; Nicole-Labrie, L.; Landry, C.R. The genome sequence of the Jean-Talon strain, an archeological beer yeast from Québec, reveals traces of adaptation to specific brewing conditions. G3 Genes Genomes Genet. 2020, 10, 3087–3097. [Google Scholar] [CrossRef]
- Gallone, B.; Steensels, J.; Prahl, T.; Soriaga, L.; Saels, V.; Herrera-Malaver, B.; Merlevede, A.; Roncoroni, M.; Voordeckers, K.; Miraglia, L.; et al. Domestication and divergence of Saccharomyces cerevisiae beer yeasts. Cell 2016, 166, 1397–1410.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, J.; Chiara, M.D.; Friedrich, A.; Yue, J.; Pflieger, D.; Bergström, A.; Sigwalt, A.; Barre, B.; Freel, K.; Llored, A.; et al. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 2018, 556, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevers, A.; Doyen, A.; Malabat, C.; Néron, B.; Kergrohen, T.; Jacquier, A.; Badis, G. Antisense transcriptional interference mediates condition-specific gene repression in budding yeast. Nucleic Acids Res. 2018, 46, 6009–6025. [Google Scholar] [CrossRef] [Green Version]
- Wery, M.; Gautier, C.; Descrimes, M.; Yoda, M.; Vennin-Rendos, H.; Migeot, V.; Gautheret, D.; Hermand, D.; Morillon, A. Native elongating transcript sequencing reveals global anti-correlation between sense and antisense nascent transcription in fission yeast. RNA 2018, 24, 196–208. [Google Scholar] [CrossRef] [Green Version]
- Borovkova, A.N.; Michailova, Y.V.; Naumova, E.S. Molecular Genetic Features of Biological Species of the Genus Saccharomyces. Microbiology 2020, 89, 387–395. [Google Scholar] [CrossRef]
- Białkowska, A.M.; Szulczewska, K.M.; Krysiak, J.; Florczak, T.; Gromek, E.; Kassassir, H.; Kur, J.; Turkiewicz, M. Genetic and biochemical characterization of yeasts isolated from Antarctic soil samples. Polar Biol. 2017, 40, 1787–1803. [Google Scholar] [CrossRef]
- De Jonge, P.; De Jongh, F.C.M.; Meijers, R.; Steensma, H.Y.; Scheffers, W.A. Orthogonal-field-alternation gel electrophoresis banding patterns of DNA from yeasts. Yeast 1986, 2, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahan, P.; Kennell, J.C. Identification and distribution of sequences having similarity to mitochondrial plasmids in mitochondrial genomes of filamentous fungi. Mol. Genet. Genom. 2005, 273, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, F.; Hon, C.C.; Zhang, Y.; Leung, F.C.C. The mitochondrial genome of the Basidiomycete fungus Pleurotus ostreatus (oyster mushroom). FEMS Microbiol. Lett. 2008, 280, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Libkind, D.; Čadež, N.; Opulente, D.A.; Langdon, Q.K.; Rosa, C.A.; Sampaio, J.P.; Gonçalves, P.; Hittinger, C.T.; Lachance, M.A. Towards yeast taxogenomics: Lessons from novel species descriptions based on complete genome sequences. FEMS Yeast Res. 2020, 20, foaa042. [Google Scholar] [CrossRef] [PubMed]
- Lallemand, T.; Leduc, M.; Landès, C.; Rizzon, C.; Lerat, E. An overview of duplicated gene detection methods: Why the duplication mechanism has to be accounted for in their choice. Genes 2020, 11, 1046. [Google Scholar] [CrossRef]
- Dean, E.J.; Davis, J.C.; Davis, R.W.; Petrov, D.A. Pervasive and persistent redundancy among duplicated genes in yeast. PLOS Genet. 2008, 4, e1000113. [Google Scholar] [CrossRef] [Green Version]
- Grainger, R.J.; Barrass, J.D.; Jacquier, A.; Rain, J.-C.; Beggs, J.D. Physical and genetic interactions of yeast Cwc21p, an ortholog of human SRm300/SRRM2, suggest a role at the catalytic center of the spliceosome. RNA 2009, 15, 2161–2173. [Google Scholar] [CrossRef] [Green Version]
- Golubev, W. Rhodosporidium babjevae, a new heterothallic yeast species (Ustilaginales). Syst. Appl. Microbiol. 1993, 16, 445–449. [Google Scholar] [CrossRef]
- Chand Dakal, T.; Giudici, P.; Solieri, L. Contrasting patterns of rDNA homogenization within the Zygosaccharomyces rouxii species complex. PLoS ONE 2016, 11, e0160744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, A.; Corte, L.; Pierantoni, D.C.; Robert, V.; Cardinali, G. What is the best lens? Comparing the resolution power of genome-derived markers and standard barcodes. Microorganisms 2021, 9, 299. [Google Scholar] [CrossRef]
- Brandenburg, J.; Blomqvist, J.; Shapaval, V.; Kohler, A.; Sampels, S. Oleaginous yeasts respond differently to carbon sources present in lignocellulose hydrolysate. Biotechnol. Biofuels 2021, 14, 124. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, M.; Zhang, Y.; Song, Z.; Zhang, S.; Zhang, Q.; Xu, J.R.; Liu, H. Extensive chromosomal rearrangements and rapid evolution of novel effector superfamilies contribute to host adaptation and speciation in the basal ascomycetous fungi. Mol. Plant Pathol. 2020, 21, 330–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passoth, V.; Hansen, M.; Klinner, U.; Emeis, C.C. The electrophoretic banding pattern of the chromosomes of Pichia stipitis and Candida shehatae. Curr. Genet. 1992, 22, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Legrand, M.; Jaitly, P.; Feri, A.; D’Enfert, C.; Sanyal, K. Candida albicans: An emerging yeast model to study eukaryotic genome plasticity. Trends Genet. 2019, 35, 292–307. [Google Scholar] [CrossRef]
- Narayanan, A.; Vadnala, R.N.; Ganguly, P.; Selvakumar, P.; Rudramurthy, S.M.; Prasad, R.; Chakrabarti, A.; Siddharthan, R.; Sanyal, K. Functional and comparative analysis of centromeres reveals clade-specific genome rearrangements in Candida auris and a chromosome number change in related species. MBio 2021, 12, e00905-21. [Google Scholar] [CrossRef]
- Hagen, F.; Khayhan, K.; Theelen, B.; Kolecka, A.; Polacheck, I.; Sionov, E.; Falk, R.; Parnmen, S.; Lumbsch, H.T.; Boekhout, T. Recognition of seven species in the Cryptococcus gattii/Cryptococcus neoformans species complex. Fungal Genet. Biol. 2015, 78, 16–48. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.L.; Byrne, K.P.; Wolfe, K.H. Mechanisms of chromosome number evolution in yeast. PLoS Genet. 2011, 7, e1002190. [Google Scholar] [CrossRef] [Green Version]
- Hellborg, L.; Piškur, J. Complex nature of the genome in a wine spoilage yeast, Dekkera bruxellensis. Eukaryot. Cell 2009, 8, 1739–1749. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.L.; Lai, H.Y.; Tung, S.Y.; Leu, J.Y. Dynamic large-scale chromosomal rearrangements fuel rapid adaptation in yeast populations. PLoS Genet. 2013, 9, e1003232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassiliadis, D.; Wong, K.H.; Blinco, J.; Dumsday, G.; Andrianopoulos, A.; Monahan, B. Adaptation to industrial stressors through genomic and transcriptional plasticity in a bioethanol producing fission yeast isolate. G3 Genes Genomes Genet. 2020, 10, 1375–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, N.T.; Irber, L.; Reiter, T.; Brooks, P.; Brown, C.T. Large-scale sequence comparisons with sourmash. F1000 Res. 2019, 8, 1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Civiero, E.; Pintus, M.; Ruggeri, C.; Tamburini, E.; Sollai, F.; Sanjust, E.; Zucca, P. Physiological and phylogenetic characterization of Rhodotorula diobovata DSBCA06, a nitrophilous yeast. Biology 2018, 7, 39. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | This Study | This Study | [11] | [13] | [3] | [15] |
|---|---|---|---|---|---|---|
| Strain number | R. babjevae CBS 7808 | R. babjevae DBVPG 8058 | R. graminis WP1 | R. glutinis ZHK | R. toruloides CBS 14 | R. toruloides NP11 |
| Genome size (Mbp) | 21.9 | 21.5 | 21.0 | 21.8 | 20.5 | 20.2 |
| Coverage | 2058 | 2122 | 8.6 | 470 | 1514 | 96 |
| GC content (%) | 68.23 | 68.24 | 67.76 | 67.8 | 61.83 | 62.05 |
| Bases masked (%) | 5.93 | 6.73 | 6.5 | NA | 2.01 | 2.53 |
| No. Scaffolds | 3 | 1 | 26 | 30 | 3 | 34 |
| No. Contigs | 24 | 33 | 325 | NA | 23 | NA |
| Protein-coding genes | 7591 | 7481 | 7283 a | 6774 a | 9464 | 8171 |
| Avg. no. exons per gene | 4.0 | 3.9 | 6.2 | NA | 5.9 | NA |
| Sequencing platform | Nanopore and Illumina | Nanopore and Illumina | Sanger | PacBio and Illumina | Nanopore and Illumina | Illumina and Sanger |
| R. bajevae CBS 7808 | R. bajevae DBVPG 8058 | Genetic Structure | GC Content | Comments | Size (Mbp) |
|---|---|---|---|---|---|
| Contig_5 (2,415,752 bp) | Contig_69 (1,447,990 bp) | Putative chromosome 1 | 67–69% | Figure S8A | 2.4 |
| Scaffold_52 (977,625 bp) | |||||
| Contig_27 (320,063 bp) | Contig_38 (1,780,658 bp) | Putative chromosome 2 | 67–69% | Figure S8B | 1.8 |
| Contig_38 (881,966 bp) | |||||
| Contig_62 (644,441 bp) | |||||
| Contig_30 (1,569,459 bp) | Contig_20 (637,402 bp) | Putative chromosome 3 | 67–69% | Figure S8C | 1.6 |
| Contig_42 (1,446,680 bp) | Large translocation event between Chr. 3 and Chr.6 | ||||
| Contig_44 (357,974 bp) | |||||
| Contig_3 (1,574,520 bp) | Contig_46 (670,828 bp) | Putative chromosome 4 | 67–69% | Figure S8D | 1.6 |
| Contig_75 (900,917 bp) | |||||
| Contig_7 (1,460,653 bp) | Contig_48 (931,129 bp) | Putative chromosome 5 | 67–69% | Figure S8E | 1.5 |
| Contig_53 (571,073 bp) | |||||
| Contig_11 (1,300,441 bp) | Contig_42 (1,446,680 bp) | Putative chromosome 6 | 67–69% | Figure S8F | 1.3 |
| Contig_44 (357,974 bp) | Large translocation event between Chr. 3 and Chr.6 | ||||
| Contig_84 (425,340 bp) | |||||
| Scaffold_6 (1,337,997 bp) | Contig_33 (529,001 bp) | Putative chromosome 7 | 67–69% | Figure S8G | 1.3 |
| Contig_70 (789,767 bp) | |||||
| Scaffold_49 (1,089,446 bp) | Contig_40 (1,004,683 bp) | Putative chromosome 8 | 67–69% | Figure S8H | 1.1 |
| Contig_65 (41,334 bp) | |||||
| Contig_10 (1,067,634 bp) | Contig_47 (557,103 bp) | Putative chromosome 9 | 67–69% | Large translocation event between Chr. 9 and Chr.14 | 1.1 |
| Contig_54 (766,724 bp) | Figure S8I | ||||
| Contig_36 (1,056,323 bp) | Contig_57 (1,049,892 bp) | Putative chromosome 10 | 67–69% | Figure S8J | 1.1 |
| Contig_31 (979,228 bp) | Contig_73 (659,761 bp) | Putative chromosome 11 | 67–69% | Figure S8K | 1.0 |
| Contig_82 (299,180 bp) | |||||
| Scaffold_40 (948,604 bp) | Contig_51 (924,743 bp) | Putative chromosome 12 | 67–69% | Figure S8L | 0.9 |
| Contig 37 (362,520 bp) | Contig_68 (408,627 bp) | Putative chromosome 13 | 67–69% | Figure S8M | 0.9 |
| Contig_12 (511,897 bp) | Contig_71 (449,691 bp) | ||||
| Contig_45 (762,860 bp) | Contig_54 (766,724 bp) | Putative chromosome 14 | 67–69% | Figure S8N Large translocation event between Chr. 9 and Chr.14 | 0.8 |
| Contig_77 (446,828 bp) | |||||
| Contig_65 (630,535 bp) | Contig_74 (614,034 bp) | Putative chromosome 15 | 67–69% | Figure S8O | 0.6 |
| Contig_25 (627,118 bp) | Contig_85 (573,802 bp) | Putative chromosome 16 | 67–69% | Figure S8P | 0.6 |
| Contig_39 (564,129 bp) | Contig_1 (565,532 bp) | Putative chromosome 17 | 67–69% | Figure S8Q | 0.6 |
| Contig_9 (429,397 bp) | Contig_86 (443,617 bp) | Putative chromosome 18 | 67–69% | Figure S8R | 0.4 |
| Contig_28 (422,133 bp) | Contig_66 (419,035 bp) | Putative chromosome 19 | 67–69% | Figure S8S | 0.4 |
| Contig_4 (418,972 bp) | Contig_45 (394,205 bp) | Putative chromosome 20 | 67–69% | Figure S8T | 0.4 |
| Contig_66 (406,102 bp) | Contig_49 (396,114 bp) | Putative chromosome 21 | 67–69% | Figure S8U | 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Hernández, G.C.; Müller, B.; Brandt, C.; Hölzer, M.; Viehweger, A.; Passoth, V. Near Chromosome-Level Genome Assembly and Annotation of Rhodotorula babjevae Strains Reveals High Intraspecific Divergence. J. Fungi 2022, 8, 323. https://doi.org/10.3390/jof8040323
Martín-Hernández GC, Müller B, Brandt C, Hölzer M, Viehweger A, Passoth V. Near Chromosome-Level Genome Assembly and Annotation of Rhodotorula babjevae Strains Reveals High Intraspecific Divergence. Journal of Fungi. 2022; 8(4):323. https://doi.org/10.3390/jof8040323
Chicago/Turabian StyleMartín-Hernández, Giselle C., Bettina Müller, Christian Brandt, Martin Hölzer, Adrian Viehweger, and Volkmar Passoth. 2022. "Near Chromosome-Level Genome Assembly and Annotation of Rhodotorula babjevae Strains Reveals High Intraspecific Divergence" Journal of Fungi 8, no. 4: 323. https://doi.org/10.3390/jof8040323
APA StyleMartín-Hernández, G. C., Müller, B., Brandt, C., Hölzer, M., Viehweger, A., & Passoth, V. (2022). Near Chromosome-Level Genome Assembly and Annotation of Rhodotorula babjevae Strains Reveals High Intraspecific Divergence. Journal of Fungi, 8(4), 323. https://doi.org/10.3390/jof8040323