

Integration of Metabolomes and Transcriptomes Provides Insights into Morphogenesis and Maturation in Morchella sextelata

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Metabolite Extraction
2.3. UHPLC-MS/MS Analysis
2.4. Metabolite Identification and Quantification
2.5. RNA Extraction and Sequencing
2.6. Transcriptomic Analysis
2.7. Correlation Analysis between the Metabolome and Transcriptome Data
2.8. Quantitative Real-Time PCR (qRT-PCR) Validation
3. Results
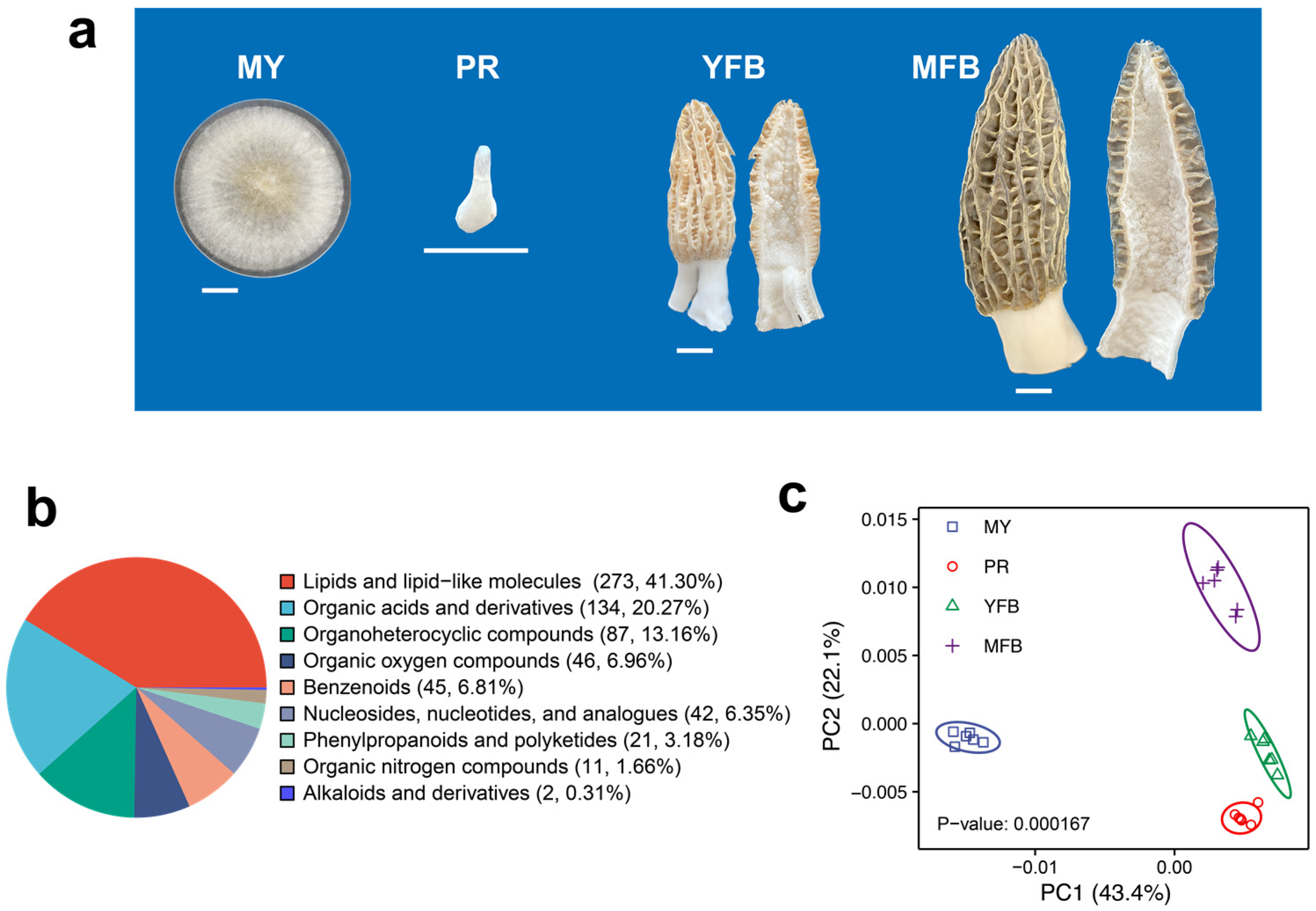
3.1. Metabolomics Analysis during Morphogenesis and Maturation in M. sextelata
3.1.1. Quality Control of the Metabolome Data
3.1.2. Identification and Screening of DEMs
3.1.3. K-Means Clustering and KEGG Enrichment Analysis of DEMs
3.2. Transcriptomics Analysis during Morphogenesis and Maturation in M. sextelata
3.2.1. Quality Assessment of Transcriptome Data
3.2.2. Identification and Enrichment of DEGs
3.2.3. Analysis of Transcription Factors and CAZymes
3.3. Integrated Metabolomics and Transcriptomics Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tietel, Z.; Masaphy, S. True morels (Morchella)-nutritional and phytochemical composition, health benefits and flavor: A review. Crit. Rev. Food Sci. 2018, 58, 1888–1901. [Google Scholar] [CrossRef] [PubMed]
- Pilz, D.; McLain, R.; Alexander, S.; Villarreal-Ruiz, L.; Berch, S.; Wurtz, T.L.; Parks, C.G.; McFarlane, E.; Baker, B.; Molina, R.; et al. Ecology and Management of Morels Harvested from the Forests of Western North America; United States Department of Agriculture, Forest Service, Pacific Northwest Research Station: Portland, OR, USA, 2007.
- Du, X.-H.; Zhao, Q.; Yang, Z.L. A review on research advances, issues, and perspectives of morels. Mycology 2015, 6, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Sunil, C.; Xu, B. Mycochemical profile and health-promoting effects of morel mushroom Morchella esculenta (L.)—A review. Food Res. Int. 2022, 159, 111571. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Chen, J.; Li, J.; Liu, Y.; Park, H.J.; Yang, L. Recent Advances on Bioactive Ingredients of Morchella esculenta. Appl. Biochem. Biotech. 2021, 193, 4197–4213. [Google Scholar] [CrossRef]
- Heleno, S.A.; Stojković, D.; Barros, L.; Glamočlija, J.; Soković, M.; Martins, A.; Queiroz, M.J.R.P.; Ferreira, I.C.F.R. A comparative study of chemical composition, antioxidant and antimicrobial properties of Morchella esculenta (L.) Pers. from Portugal and Serbia. Food Res. Int. 2013, 51, 236–243. [Google Scholar] [CrossRef]
- Dissanayake, A.A.; Mills, G.L.; Bonito, G.; Rennick, B.; Nair, M.G. Chemical Composition and Anti-Inflammatory and Antioxidant Activities of Extracts from Cultivated Morel Mushrooms, Species of Genus Morchella (Ascomycota). Int. J. Med. Mushrooms 2021, 23, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Y.; He, P. Morel Biology and Cultivation; Jilin Science and Technology Press: Changchun, China, 2017. [Google Scholar]
- Du, X.-H.; Yang, Z.L. Mating Systems in True Morels (Morchella). Microbiol. Mol. Biol. Rev. 2021, 85, e00220. [Google Scholar] [CrossRef]
- Ower, R. Notes on the Development of the Morel Ascocarp: Morchella Esculenta. Mycologia 1982, 74, 142–144. [Google Scholar] [CrossRef]
- Liu, Q.; Ma, H.; Zhang, Y.; Dong, C. Artificial cultivation of true morels: Current state, issues and perspectives. Crit. Rev. Biotechnol. 2018, 38, 259–271. [Google Scholar] [CrossRef]
- Shi, X.; Liu, D.; He, X.; Liu, W.; Yu, F. Epidemic Identification of Fungal Diseases in Morchella Cultivation across China. J. Fungi 2022, 8, 1107. [Google Scholar] [CrossRef]
- Tan, H.; Kohler, A.; Miao, R.; Liu, T.; Zhang, Q.; Zhang, B.; Jiang, L.; Wang, Y.; Xie, L.; Tang, J.; et al. Multi-omic analyses of exogenous nutrient bag decomposition by the black morel Morchella importuna reveal sustained carbon acquisition and transferring. Environ. Microbiol. 2019, 21, 3909–3926. [Google Scholar] [CrossRef]
- He, P.; Yu, M.; Cai, Y.; Liu, W.; Wang, W.; Wang, S.; Li, J. Effect of Aging on Culture and Cultivation of the Culinary-Medicinal Mushrooms Morchella importuna and M. sextelata (Ascomycetes). Int. J. Med. Mushrooms 2019, 21, 1089–1098. [Google Scholar] [CrossRef]
- He, P.; Li, C.; Cai, Y.; Zhang, Y.; Bian, Y.; Liu, W. First report of pileus rot disease on cultivated Morchella importuna caused by Diploospora longispora in China. J. Gen. Plant Pathol. 2018, 84, 65–69. [Google Scholar] [CrossRef]
- He, X.-L.; Peng, W.-H.; Miao, R.-Y.; Tang, J.; Chen, Y.; Liu, L.-X.; Wang, D.; Gan, B.-C. White mold on cultivated morels caused by Paecilomyces penicillatus. FEMS Microbiol. Lett. 2017, 364, fnx037. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Jiang, X.; Ma, L.; Xiao, D.; Liu, X.; Ying, Z.; Li, Y.; Lin, Y. Transcriptomic and Metabolomic Profiles Provide Insights into the Red-Stipe Symptom of Morel Fruiting Bodies. J. Fungi 2023, 9, 373. [Google Scholar] [CrossRef]
- Tan, H.; Liu, T.; Yu, Y.; Tang, J.; Jiang, L.; Martin, F.M.; Peng, W. Morel Production Related to Soil Microbial Diversity and Evenness. Microbiol. Spectr. 2021, 9, e00229-21. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Yu, Y.; Tang, J.; Liu, T.; Miao, R.; Huang, Z.; Martin, F.M.; Peng, W. Build Your Own Mushroom Soil: Microbiota Succession and Nutritional Accumulation in Semi-Synthetic Substratum Drive the Fructification of a Soil-Saprotrophic Morel. Front. Microbiol. 2021, 12, 656656. [Google Scholar] [CrossRef] [PubMed]
- Orlofsky, E.; Zabari, L.; Bonito, G.; Masaphy, S. Changes in soil bacteria functional ecology associated with Morchella rufobrunnea fruiting in a natural habitat. Environ. Microbiol. 2021, 23, 6651–6662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shi, X.; Zhang, J.; Zhang, Y.; Wang, W. Dynamics of soil microbiome throughout the cultivation life cycle of morel (Morchella sextelata). Front. Microbiol. 2023, 14, 979835. [Google Scholar] [CrossRef]
- Yu, F.-M.; Jayawardena, R.S.; Zhu, X.-T.; Zhao, Q. Morel Production Associated with Soil Nitrogen-Fixing and Nitrifying Microorganisms. J. Fungi 2022, 8, 299. [Google Scholar] [CrossRef]
- Du, X.-H.; Zhao, Q.; Xia, E.-H.; Gao, L.-Z.; Richard, F.; Yang, Z.L. Mixed-reproductive strategies, competitive mating-type distribution and life cycle of fourteen black morel species. Sci. Rep. 2017, 7, 1493. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Ma, X.; Zhang, Q.; Yu, F.; Zhao, Q.; Huang, W.; Song, J.; Liu, W. Physiological Characteristics and Comparative Secretome Analysis of Morchella importuna Grown on Glucose, Rice Straw, Sawdust, Wheat Grain, and MIX Substrates. Front. Microbiol. 2021, 12, 636344. [Google Scholar] [CrossRef]
- Want, E.J.; Masson, P.; Michopoulos, F.; Wilson, I.D.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Loftus, N.; Holmes, E.; Nicholson, J.K. Global metabolic profiling of animal and human tissues via UPLC-MS. Nat. Protoc. 2013, 8, 17–32. [Google Scholar] [CrossRef]
- Wen, B.; Mei, Z.; Zeng, C.; Liu, S. metaX: A flexible and comprehensive software for processing metabolomics data. BMC Bioinform. 2017, 18, 183. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Plaza, A.; Szklarczyk, D.; Botas, J.; Cantalapiedra, C.P.; Giner-Lamia, J.; Mende, D.R.; Kirsch, R.; Rattei, T.; Letunic, I.; Jensen, L.J.; et al. eggNOG 6.0: Enabling comparative genomics across 12,535 organisms. Nucleic Acids Res. 2022, 51, D389–D394. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, J.; Jang, S.; Kim, S.; Kong, S.; Choi, J.; Ahn, K.; Kim, J.; Lee, S.; Kim, S.; et al. FTFD: An informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 2008, 24, 1024–1025. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ge, Q.; Yan, Y.; Zhang, X.; Huang, L.; Yin, Y. dbCAN3: Automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 2023, 51, W115–W121. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; He, G.; Wei, J.; Dong, C. Comparative transcriptome analysis of cells from different areas reveals ROS responsive mechanism at sclerotial initiation stage in Morchella importuna. Sci. Rep. 2021, 11, 9418. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, W.; Cai, Y.; Lan, A.-F.; Bian, Y. Validation of Internal Control Genes for Quantitative Real-Time PCR Gene Expression Analysis in Morchella. Molecules 2018, 23, 2331. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rotzoll, N.; Dunkel, A.; Hofmann, T. Quantitative Studies, Taste Reconstitution, and Omission Experiments on the Key Taste Compounds in Morel Mushrooms (Morchella deliciosa Fr.). J. Agric. Food Chem. 2006, 54, 2705–2711. [Google Scholar] [CrossRef]
- Deng, K.; Lan, X.; Fang, Q.; Li, M.; Xie, G.; Xie, L. Untargeted Metabolomics Reveals Alterations in the Primary Metabolites and Potential Pathways in the Vegetative Growth of Morchella sextelata. Front. Mol. Biosci. 2021, 8, 632341. [Google Scholar] [CrossRef]
- Dong, H.; Zhao, X.; Cai, M.; Gu, H.; E, H.; Li, X.; Zhang, Y.; Lu, H.; Zhou, C. Metabolomics Analysis of Morchella sp. From Different Geographical Origins of China Using UPLC-Q-TOF-MS. Front. Nutr. 2022, 9, 865531. [Google Scholar] [CrossRef]
- Fan, T.; Ren, R.; Tang, S.; Zhou, Y.; Cai, M.; Zhao, W.; He, Y.; Xu, J. Transcriptomics combined with metabolomics unveiled the key genes and metabolites of mycelium growth in Morchella importuna. Front. Microbiol. 2023, 14, 1079353. [Google Scholar] [CrossRef] [PubMed]
- Brasaemle, D.L. The perilipin family of structural lipid droplet proteins: Stabilization of lipid droplets and control of lipolysis. J. Lipid Res. 2007, 48, 2547–2559. [Google Scholar] [CrossRef] [PubMed]
- Meer, G.V.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Bio. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Lipid signaling. Curr. Opin. Plant Biol. 2004, 7, 329–336. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Wang, K.; Cai, Y.; Hu, X.; Zheng, Y.; Zhang, J.; Liu, W. Involvement of autophagy and apoptosis and lipid accumulation in sclerotial morphogenesis of Morchella importuna. Micron 2018, 109, 34–40. [Google Scholar] [CrossRef]
- Liu, W.; Cai, Y.; He, P.; Chen, L.; Bian, Y. Comparative transcriptomics reveals potential genes involved in the vegetative growth of Morchella importuna. 3 Biotech 2019, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, C.W. Determination of organic acids by CE and CEC methods. Electrophoresis 2007, 28, 3362–3378. [Google Scholar] [CrossRef]
- Pelkmans, J.F.; Patil, M.B.; Gehrmann, T.; Reinders, M.J.T.; Wösten, H.A.B.; Lugones, L.G. Transcription factors of Schizophyllum commune involved in mushroom formation and modulation of vegetative growth. Sci. Rep. 2017, 7, 310. [Google Scholar] [CrossRef]
- Ohm, R.A.; Jong, J.F.d.; Bekker, C.d.; Wösten, H.A.B.; Lugones, L.G. Transcription factor genes of Schizophyllum commune involved in regulation of mushroom formation. Mol. Microbiol. 2011, 81, 1433–1445. [Google Scholar] [CrossRef]
- Pelkmans, J.F.; Vos, A.M.; Scholtmeijer, K.; Hendrix, E.; Baars, J.J.P.; Gehrmann, T.; Reinders, M.J.T.; Lugones, L.G.; Wösten, H.A.B. The transcriptional regulator c2h2 accelerates mushroom formation in Agaricus bisporus. Appl. Microbiol. Biotechnol. 2016, 100, 7151–7159. [Google Scholar] [CrossRef]
- Luan, Y.; Tang, Z.; He, Y.; Xie, Z. Intra-Domain Residue Coevolution in Transcription Factors Contributes to DNA Binding Specificity. Microbiol. Spectr. 2023, 11, e0365122. [Google Scholar] [CrossRef]
- Nagy, L.G.; Vonk, P.J.; Künzler, M.; Földi, C.; Virágh, M.; Ohm, R.A.; Hennicke, F.; Bálint, B.; Csernetics, Á.; Hegedüs, B.; et al. Lessons on fruiting body morphogenesis from genomes and transcriptomes of Agaricomycetes. Stud. Mycol. 2023, 104, 1–85. [Google Scholar] [CrossRef] [PubMed]
- Krizsán, K.; Almási, É.; Merényi, Z.; Sahu, N.; Virágh, M.; Kószó, T.; Mondo, S.; Kiss, B.; Bálint, B.; Kües, U.; et al. Transcriptomic atlas of mushroom development reveals conserved genes behind complex multicellularity in fungi. Proc. Natl. Acad. Sci. USA 2019, 116, 7409–7418. [Google Scholar] [CrossRef]
- Liu, Q.; Qu, S.; He, G.; Wei, J.; Dong, C. Mating-Type Genes Play an Important Role in Fruiting Body Development in Morchella sextelata. J. Fungi 2022, 8, 564. [Google Scholar] [CrossRef] [PubMed]
- Nowrousian, M. The Role of Chromatin and Transcriptional Control in the Formation of Sexual Fruiting Bodies in Fungi. Microbiol. Mol. Biol. Rev. 2022, 86, e0010422. [Google Scholar] [CrossRef]
- Ye, D.; Du, F.; Zou, Y.; Hu, Q. Transcriptomics Analysis of Primordium Formation in Pleurotus eryngii. Genes 2021, 12, 1863. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.; Dobrovin-Pennington, A.; Hobbs, P.J.; Pederby, J.; Rodger, A. Volatile C8 compounds and pseudomonads influence primordium formation of Agaricus bisporus. Mycologia 2009, 101, 583–591. [Google Scholar] [CrossRef]
- Sakamoto, Y. Influences of environmental factors on fruiting body induction, development and maturation in mushroom-forming fungi. Fungal Biol. Rev. 2018, 32, 236–248. [Google Scholar] [CrossRef]
- Camelini, C.M.; Maraschin, M.; Mendonça, M.M.D.; Zucco, C.; Ferreira, A.G.; Tavares, L.A. Structural characterization of β-glucans of Agaricus brasiliensis in different stages of fruiting body maturity and their use in nutraceutical products. Biotechnol. Lett. 2005, 27, 1295–1299. [Google Scholar] [CrossRef]
- Callac, P.; Moquet, F.; Imbernon, M.; Guedes-Lafargue, M.R.; Mamoun, M.; Olivier, J.-M. Evidence for PPC1, a determinant of the pilei-pellis color of Agaricus bisporus fruitbodies. Fungal Genet. Biol. 1998, 23, 181–188. [Google Scholar] [CrossRef]
- Fuller, K.K.; Loros, J.J.; Dunlap, J.C. Fungal photobiology: Visible light as a signal for stress, space and time. Curr. Genet. 2015, 61, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Corrochano, L.M. Light in the Fungal World: From Photoreception to Gene Transcription and Beyond. Annu. Rev. Genet. 2019, 53, 149–170. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Shi, X.; Zhang, J.; Zhang, Y.; Liu, W.; Wang, W. Integration of Metabolomes and Transcriptomes Provides Insights into Morphogenesis and Maturation in Morchella sextelata. J. Fungi 2023, 9, 1143. https://doi.org/10.3390/jof9121143
Zhang C, Shi X, Zhang J, Zhang Y, Liu W, Wang W. Integration of Metabolomes and Transcriptomes Provides Insights into Morphogenesis and Maturation in Morchella sextelata. Journal of Fungi. 2023; 9(12):1143. https://doi.org/10.3390/jof9121143
Chicago/Turabian StyleZhang, Chen, Xiaofei Shi, Jiexiong Zhang, Yesheng Zhang, Wei Liu, and Wen Wang. 2023. "Integration of Metabolomes and Transcriptomes Provides Insights into Morphogenesis and Maturation in Morchella sextelata" Journal of Fungi 9, no. 12: 1143. https://doi.org/10.3390/jof9121143
APA StyleZhang, C., Shi, X., Zhang, J., Zhang, Y., Liu, W., & Wang, W. (2023). Integration of Metabolomes and Transcriptomes Provides Insights into Morphogenesis and Maturation in Morchella sextelata. Journal of Fungi, 9(12), 1143. https://doi.org/10.3390/jof9121143