


Kernel Bioassay Evaluation of Maize Ear Rot and Genome-Wide Association Analysis for Identifying Genetic Loci Associated with Resistance to Fusarium graminearum Infection


Abstract
:1. Introduction
2. Materials and Methods
2.1. Maize Germplasm
2.2. Experimental Procedure for Kernel Bioassay
2.3. Phenotypic Analysis
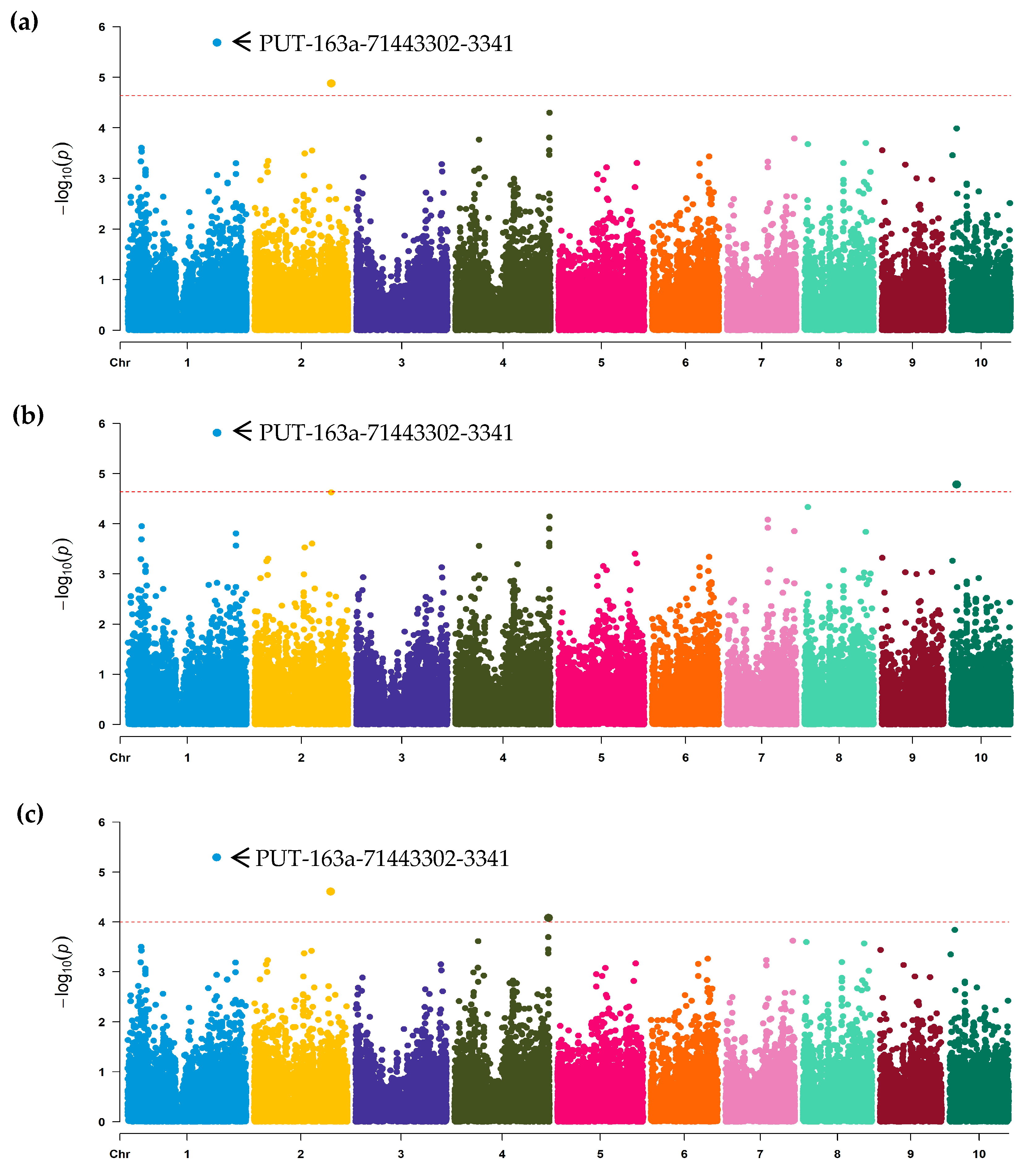
2.4. GWAS Analysis
3. Results
3.1. Phenotypic Evaluation of GER Severity
3.2. Association Analysis and SNPs Discovery
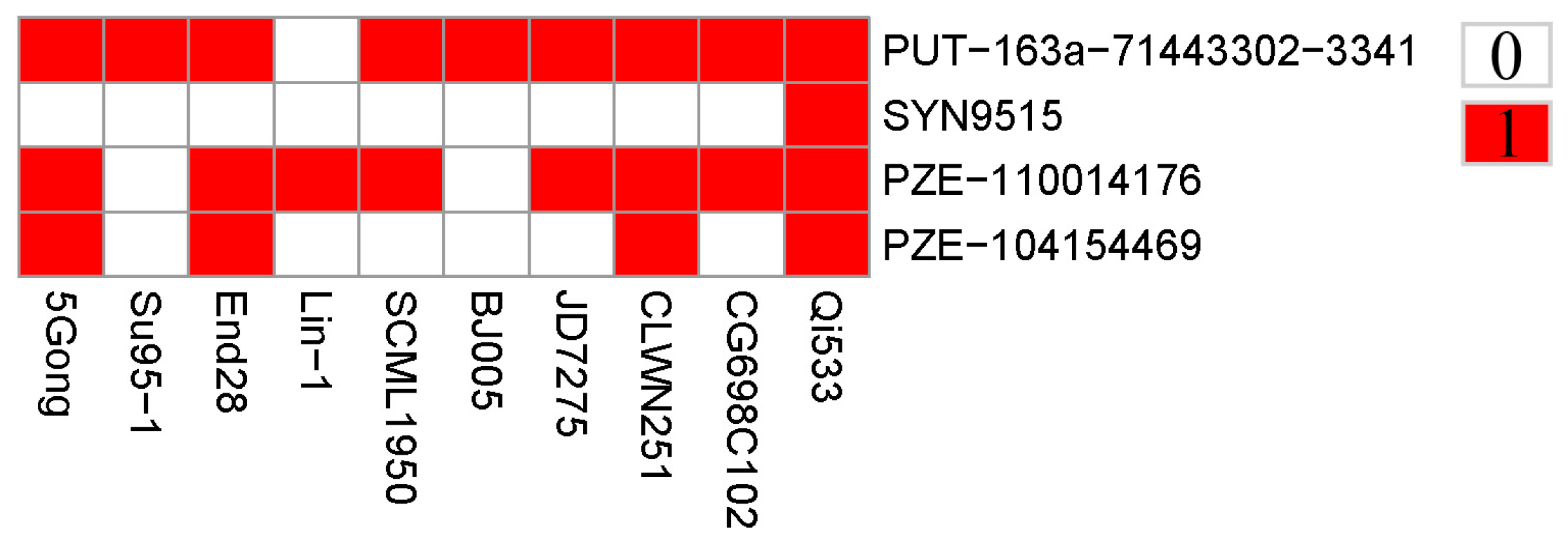
3.3. Genes Associated with GER Resistance
3.4. Distribution of Favorable Alleles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reid, L.M.; Nicol, R.W.; Ouellet, T.; Savard, M.; Miller, J.D.; Young, J.C.; Stewart, A.W.; Schaafsma, A.W. Interaction of Fusarium graminearum and F. moniliforme in maize ears: Disease progress, fungal biomass, and mycotoxin accumulation. Phytopathology 1999, 89, 1028–1037. [Google Scholar] [CrossRef]
- Yang, Q.; Balint-Kurtil, P.; Xu, M. Quantitative disease resistance: Dissection and adoption in maize. Mol. Plant 2017, 10, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Vigier, B.; Reid, L.M.; Dwyer, L.M.; Stewart, D.W.; Sinha, R.C.; Arnason, J.T.; Butler, G. Maize resistance to Gibberella ear rot: Symptoms, deoxynivalenol, and yield. Can. J. Plant Pathol. 2001, 23, 99–105. [Google Scholar] [CrossRef]
- Duan, C.X.; Qin, Z.H.; Yang, Z.H.; Li, W.X.; Sun, S.L.; Zhu, Z.D.; Wang, X.M. Identification of pathogenic Fusarium spp. causing maize ear rot and potential mycotoxin production in China. Toxins 2016, 8, 186. [Google Scholar] [CrossRef] [PubMed]
- Gaikpa, D.; Miedaner, T. Genomics-assisted breeding for ear rot resistances and reduced mycotoxin contamination in maize: Methods, advances and prospects. Theor. Appl. Genet. 2019, 132, 2721–2739. [Google Scholar] [CrossRef] [PubMed]
- Giomi, G.M.; Kreff, E.D.; Iglesias, J.; Fauguel, C.M.; Fernandez, M.; Oviedo, M.S.; Presello, D.A. Quantitative trait loci for Fusarium and Gibberella ear rot resistance in Argentinian maize germplasm. Euphytica 2016, 211, 287–294. [Google Scholar] [CrossRef]
- Han, S.; Miedaner, T.; Utz, H.F.; Schipprack, W.; Schrag, T.A.; Melchinger, A.E. Genomic prediction and GWAS of Gibberella ear rot resistance traits in dent and flint lines of a public maize breeding program. Euphytica 2018, 214, 6–11. [Google Scholar] [CrossRef]
- Kebede, A.Z.; Woldemariam, T.; Reid, L.M.; Harris, L.J. Quantitative trait loci mapping for Gibberella ear rot resistance and associated agronomic traits using genotyping-by-sequencing in maize. Theor. Appl. Genet. 2016, 129, 17–29. [Google Scholar] [CrossRef]
- Martin, M.; Miedaner, T.; Dhillon, B.S.; Ufermann, U.; Kessel, B.; Ouzunova, M.; Schipprack, W.; Melchinger, A.E. Colocalization of QTL for Gibberella ear rot resistance and low mycotoxin contamination in early European maize. Crop Sci. 2011, 51, 1935–1945. [Google Scholar] [CrossRef]
- Munkvold, G.P. Fusarium species and their associated mycotoxins. Methods Mol. Bio. 2017, 1542, 51–106. [Google Scholar]
- Ali, M.L.; Taylor, J.H.; Jie, L.; Sun, G.; William, M.; Kasha, K.J.; Reid, L.M.; Pauls, K.P. Molecular mapping of QTLs for resistance to Gibberella ear rot, in corn, caused by Fusarium graminearum. Genome 2005, 48, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Mesterházy, A.; Lemmens, M.; Reid, L.M. Breeding for resistance to ear rots caused by Fusarium spp. in maize—A review. Plant Breed. 2012, 131, 1–19. [Google Scholar] [CrossRef]
- Nerbass, F.R.; Casa, R.T.; Kuhnem, P.R.; Vieira, J.L., Jr.; Valente, J.B. Field evaluation of maize for Gibberella ear rot resistance using silk channel and kernel inoculation with Fusarium meridionale. Trop. Plant Pathol. 2015, 40, 388–393. [Google Scholar] [CrossRef]
- Nerbass, F.R.; Casa, R.T.; Kuhnem, P.R.; Bogo, A.; Sangoi, L.; Fingstag, M.D.; Vieira, J.J.; Stoltz, J.C. Evaluation of Fusarium graminearum inoculation methods in maize ears and hybrid reaction to Gibberella ear rot under southern Brazilian environmental conditions. Eur. J. Plant Pathol. 2016, 144, 45–53. [Google Scholar] [CrossRef]
- Reid, L.M.; Hamilton, R.L. Effects of inoculation position, timing, macroconidial concentration, and irrigation on resistance of maize to Fusarium graminearum infection through kernels. Can. J. Plant Pathol. 1996, 18, 279–285. [Google Scholar] [CrossRef]
- Reid, L.M.; Woldemariam, T.; Zhu, X.; Stewart, D.W.; Schaafsma, A.W. Effect of inoculation time and point of entry on disease severity in Fusarium graminearum, Fusarium verticillioides, or Fusarium subglutinans inoculated maize ears. Can. J. Plant Pathol. 2002, 24, 162–167. [Google Scholar] [CrossRef]
- Cobb, J.N.; DeClerck, G.; Greenberg, A.; Clark, R.; McCouch, S. Next-generation phenotyping: Requirements and strategies for enhancing our understanding of genotype-phenotype relationships and its relevance to crop improvement. Theor. Appl. Genet. 2013, 126, 867–887. [Google Scholar] [CrossRef]
- Butrón, A.; Reid, L.M.; Santiago, R.; Cao, A.; Malvar, R.A. Inheritance of maize resistance to Gibberella and Fusarium ear rots and kernel contamination with deoxynivalenol and fumonisins. Plant Pathol. 2015, 64, 1053–1060. [Google Scholar] [CrossRef]
- Kuska, M.T.; Mahlein, A.K. Aiming at decision making in plant disease protection and phenotyping by the use of optical sensors. Eur. J. Plant Pathol. 2018, 152, 987–992. [Google Scholar] [CrossRef]
- Mutka, A.M.; Bart, R.S. Image-based phenotyping of plant disease symptoms. Front. Plant Sci. 2015, 5, 734. [Google Scholar] [CrossRef]
- Christensen, S.; Borrego, E.; Shim, W.B.; Isakeit, T.; Kolomiets, M. Quantification of fungal colonization, sporogenesis, and production of mycotoxins using kernel bioassays. J. Vis. Exp. 2012, 62, e3727. [Google Scholar]
- Gao, X.; Shim, W.B.; Göbel, C.; Kunze, S.; Feussner, I.; Meeley, R.; Balint-Kurti, P.; Kolomiets, M. Disruption of a maize 9-lipoxygenase results in increased resistance to fungal pathogens and reduced levels of contamination with mycotoxin fumonisin. Mol. Plant-Microbe Interact. 2007, 20, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Li, S.; Ma, L.; Wang, F.; Jiang, F.; Sun, Y.; Ruan, X.; Cao, Y.; Wang, Q.; Zhang, Y.; et al. Mapping and validation of a stable quantitative trait locus conferring maize resistance to Gibberella ear rot. Plant Dis. 2021, 7, 1984–1991. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Li, H.; Liu, E.; He, K.; Song, Y.; Dong, C.; Wang, Z.; Zhang, X.; Zhou, Z.; Xu, Y.; et al. Evaluation and identification of resistance lines and QTLs of maize to seedborne Fusarium verticillioides. Plant Dis. 2022, 106, 2066–2073. [Google Scholar] [CrossRef] [PubMed]
- Ju, M.; Zhou, Z.; Mu, C.; Zhang, X.; Gao, J.; Liang, Y.; Chen, J.; Wu, Y.; Li, X.; Wang, S.; et al. Dissecting the genetic architecture of Fusarium verticillioides seed rot resistance in maize by combining QTL mapping and genome-wide association analysis. Sci. Rep. 2017, 7, 46446. [Google Scholar] [CrossRef]
- Miedaner, T.; Boeven, A.L.G.; Gaikpa, D.S.; Kistner, M.B.; Grote, C.P. Genomics-assisted breeding for quantitative disease resistances in small-grain cereals and maize. Int. J. Mol. Sci. 2020, 21, 9717. [Google Scholar] [CrossRef]
- Yuan, G.; He, X.; Li, H.; Xiang, K.; Liu, L.; Zou, C.; Lin, H.; Zhang, Z.; Pan, G. Transcriptomic responses in resistant and susceptible maize infected with Fusarium graminearum. Crop J. 2020, 8, 153–163. [Google Scholar] [CrossRef]
- Yuan, G.; Chen, B.; Peng, H.; Zheng, Q.; Li, Y.; Xiang, K.; Liu, L.; Zou, C.; Lin, H.; Ding, H.; et al. QTL mapping for resistance to ear rot caused by Fusarium graminearum using an IBM Syn10 DH population in maize. Mol. Breed. 2020, 40, 91. [Google Scholar] [CrossRef]
- Yuan, G.; Li, Y.; He, D.; Shi, J.; Yang, Y.; Du, J.; Zou, C.; Ma, L.; Pan, G.; Shen, Y. A combination of QTL mapping and GradedPool-Seq to dissect genetic complexity for Gibberella ear rot resistance in maize using an IBM Syn10 DH population. Plant Dis. 2023, 107, 1115–1121. [Google Scholar] [CrossRef]
- Yuan, G.; He, D.; Shi, J.; Li, Y.; Yang, Y.; Du, J.; Zou, C.; Ma, L.; Gao, S.; Pan, G.; et al. Genome-wide association study discovers novel germplasm resources and genetic loci with resistance to Gibberella ear rot caused by Fusarium graminearum. Phytopathology 2023, 113, 1317–1324. [Google Scholar] [CrossRef]
- Yu, J.; Buckler, E.S. Genetic association mapping and genome organization of maize. Curr. Opin. Biotechnol. 2006, 17, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Shikha, K.; Shahi, J.P.; Vinayan, M.T.; Zaidi, P.H.; Singh, A.K.; Sinha, B. Genome-wide association mapping in maize: Status and prospects. 3 Biotech. 2021, 11, 244. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zou, C.; Liu, X.; Wang, S.; Li, W.; Jeffers, D.; Fan, X.; Xu, M.; Xu, Y. Complex genetic system involved in Fusarium ear rot resistance in maize as revealed by GWAS, bulked sample analysis, and genomic prediction. Plant Dis. 2020, 104, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shrestha, R.; Ding, J.; Zheng, H.; Mu, C.; Wu, J.; Mahuku, G. Genome-wide association study and QTL mapping reveal genomic loci associated with Fusarium ear rot resistance in tropical maize germplasm. G3-Genes·Genomes·Genet. 2016, 6, 3803–3815. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, G.; Zhang, A.; Loladze, A.; Hu, Y.; Wang, H.; Qu, J.; Zhang, X.; Olsen, M.; Vicent, F.; et al. Genome-wide association study and genomic prediction of Fusarium ear rot resistance in tropical maize germplasm. Crop J. 2021, 2, 325–341. [Google Scholar] [CrossRef]
- Zila, C.; Samayoa, L.F.; Santiago, R.; Butrón, A.; Holland, J.B. A genome-wide association study reveals genes associated with Fusarium ear rot resistance in a maize core diversity panel. G3-Genes·Genomes·Genet. 2013, 3, 2095–2104. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Li, L.; Lan, H.; Ren, Z.; Liu, D.; Wu, L.; Jaqueth, J.; Li, B.; Pan, G.; et al. Characterizing the population structure and genetic diversity of maize breeding germplasm in southwest China using genome-wide SNP markers. BMC Genom. 2016, 17, 697. [Google Scholar] [CrossRef]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X. rMVP: A memory-efficient, visualization-enhanced, and parallel-accelerated tool for genome-wide association study. Genom. Proteom. Bioinf. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Bates, D.; Maechler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Yang, J.; Manolio, T.A.; Pasquale, L.R.; Boerwinkle, E.; Caporaso, N.; Cunningham, J.M.; de Andrade, M.; Feenstra, B.; Feingold, E.; Hayes, M.G.; et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat. Genet. 2011, 43, 519–525. [Google Scholar] [CrossRef]
- Gao, X.; Brodhagen, M.; Isakeit, T.; Brown, S.H.; Göbel, C.; Betran, J.; Feussner, I.; Keller, N.P.; Kolomiets, M.V. Inactivation of thelipoxygenase ZmLOX3 increases susceptibility of maize to Aspergillus spp. Mol. Plant-Microbe Interact. 2009, 22, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Tamborski, J.; Krasileva, K.V. Evolution of plant NLR: From natural history to precise modification. Annu. Rev. Plant Biol. 2020, 71, 355–378. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Zhang, F.; Zhang, C.; Wang, D.; Shen, S.; He, F.; Tao, H.; Wang, R.; Wang, M.; Wang, D.; et al. Function of hydroxycinnamoyl transferases for the biosynthesis of phenolamides in rice resistance to Magnaporthe oryzae. J. Genet. Genom. 2022, 49, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Roseenblad, A.M.; Zwieb, C.; Samuelsson, T. Identification and comparative analysis of components from the signal recognition particle in protozoa and fungi. BMC Genom. 2002, 5, 5. [Google Scholar] [CrossRef]
- Niu, Y.; Chen, M.; Xu, Z.; Li, L.; Chen, X.; Ma, Y. Characterization of ethylene receptors and their interactions with GmTPRA novel tetratricopeptide repeat protein (TPR) in Soybean (Glycine max L.). J. Integr. Agric. 2013, 12, 571–581. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Days Post-Inoculation (dpi) | Range (×106 mL−1) a | Mean ± SD (×106 mL−1) b | CV (%) c |
|---|---|---|---|
| 7 | 0.00–14.40 | 0.71 ± 1.47 | 207 |
| 14 | 0.00–64.00 | 2.77 ± 6.18 | 223 |
| 21 | 0.00–108.96 | 4.67 ± 9.38 | 200.8 |
| 28 | 0.00–84.70 | 7.25 ± 12.27 | 169 |
| Model a | SNP | Chr. b | Position | Allele | MAF c | p Value | PVE (%) d |
|---|---|---|---|---|---|---|---|
| FarmCPU | PUT-163a-71443302-3341 | 1 | 226,136,399 | G/A | 0.29 | 2.06 × 10−6 | 6.42 |
| FarmCPU | SYN9515 | 2 | 194,393,324 | C/A | 0.36 | 1.32 × 10−5 | 5.54 |
| GLM | PUT-163a-71443302-3341 | 1 | 226,136,399 | G/A | 0.29 | 1.53 × 10−6 | 6.42 |
| GLM | PZE-110014176 | 10 | 13,338,854 | A/C | 0.43 | 1.65 × 10−5 | 3.51 |
| MLM | PUT-163a-71443302-3341 | 1 | 226,136,399 | G/A | 0.29 | 5.12 × 10−6 | 6.42 |
| MLM | SYN9515 | 2 | 194,393,324 | C/A | 0.36 | 2.46 × 10−5 | 5.54 |
| MLM | PZE-104154469 | 4 | 238,758,660 | A/G | 0.49 | 8.28 × 10−5 | 5.35 |
| Physical Position | Candidate Genes | Annotation |
|---|---|---|
| 229515984-229517919 | Zm00001d032527 | hydroxycinnamoyltransferase13 |
| 229718687-229725690 | Zm00001d032530 | F-box/LRR-repeat protein |
| 229758635-229765316 | Zm00001d032531 | Membrane steroid-binding protein 1 |
| 229766139-229766442 | ENSRNA049476973 | Plant signal recognition particle RNA |
| 229816856-229817035 | ENSRNA049476978 | Plant signal recognition particle RNA |
| 229829797-229830137 | Zm00001d032533 | -- |
| 229830451-229831175 | Zm00001d022929 | -- |
| 229831273-229832169 | Zm00001d032534 | -- |
| 229847655-229849510 | Zm00001d022930 | -- |
| 229847655-229849510 | Zm00001d032535 | Tetratricopeptide repeat (TPR)-like superfamily protein |
| 229900429-229901097 | Zm00001d032538 | -- |
| 229992052-229992483 | Zm00001d032542 | plant/MXO21-9 protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Shi, H.; Yang, Y.; Zeng, C.; Jia, Z.; Ma, T.; Wu, M.; Du, J.; Huang, N.; Pan, G.; et al. Kernel Bioassay Evaluation of Maize Ear Rot and Genome-Wide Association Analysis for Identifying Genetic Loci Associated with Resistance to Fusarium graminearum Infection. J. Fungi 2023, 9, 1157. https://doi.org/10.3390/jof9121157
Zhang J, Shi H, Yang Y, Zeng C, Jia Z, Ma T, Wu M, Du J, Huang N, Pan G, et al. Kernel Bioassay Evaluation of Maize Ear Rot and Genome-Wide Association Analysis for Identifying Genetic Loci Associated with Resistance to Fusarium graminearum Infection. Journal of Fungi. 2023; 9(12):1157. https://doi.org/10.3390/jof9121157
Chicago/Turabian StyleZhang, Jihai, Haoya Shi, Yong Yang, Cheng Zeng, Zheyi Jia, Tieli Ma, Mengyang Wu, Juan Du, Ning Huang, Guangtang Pan, and et al. 2023. "Kernel Bioassay Evaluation of Maize Ear Rot and Genome-Wide Association Analysis for Identifying Genetic Loci Associated with Resistance to Fusarium graminearum Infection" Journal of Fungi 9, no. 12: 1157. https://doi.org/10.3390/jof9121157
APA StyleZhang, J., Shi, H., Yang, Y., Zeng, C., Jia, Z., Ma, T., Wu, M., Du, J., Huang, N., Pan, G., Li, Z., & Yuan, G. (2023). Kernel Bioassay Evaluation of Maize Ear Rot and Genome-Wide Association Analysis for Identifying Genetic Loci Associated with Resistance to Fusarium graminearum Infection. Journal of Fungi, 9(12), 1157. https://doi.org/10.3390/jof9121157