
Comparative Genomic Analysis Reveals Gene Content Diversity, Phylogenomic Contour, Putative Virulence Determinants, and Potential Diagnostic Markers within Pythium insidiosum Traits

 ,
,  ,
, 
Abstract
:1. Introduction
2. Methods
2.1. Genome Sequences of P. insidiosum and Related Species
2.2. Homologous Gene Clusters for Gene-Content Comparison
2.3. Core Gene-Based Phylogenomic Analysis
2.4. Hierarchical Clustering of the Gene-Presence Profile Data
2.5. Annotation of P. insidiosum-Specific Genes
3. Results and Discussion
3.1. Genome Summary and Homologous Gene Cluster Data
3.2. Evolutionary Relationship between P. insidiosum Strains
3.3. Gene Content of P. insidiosum
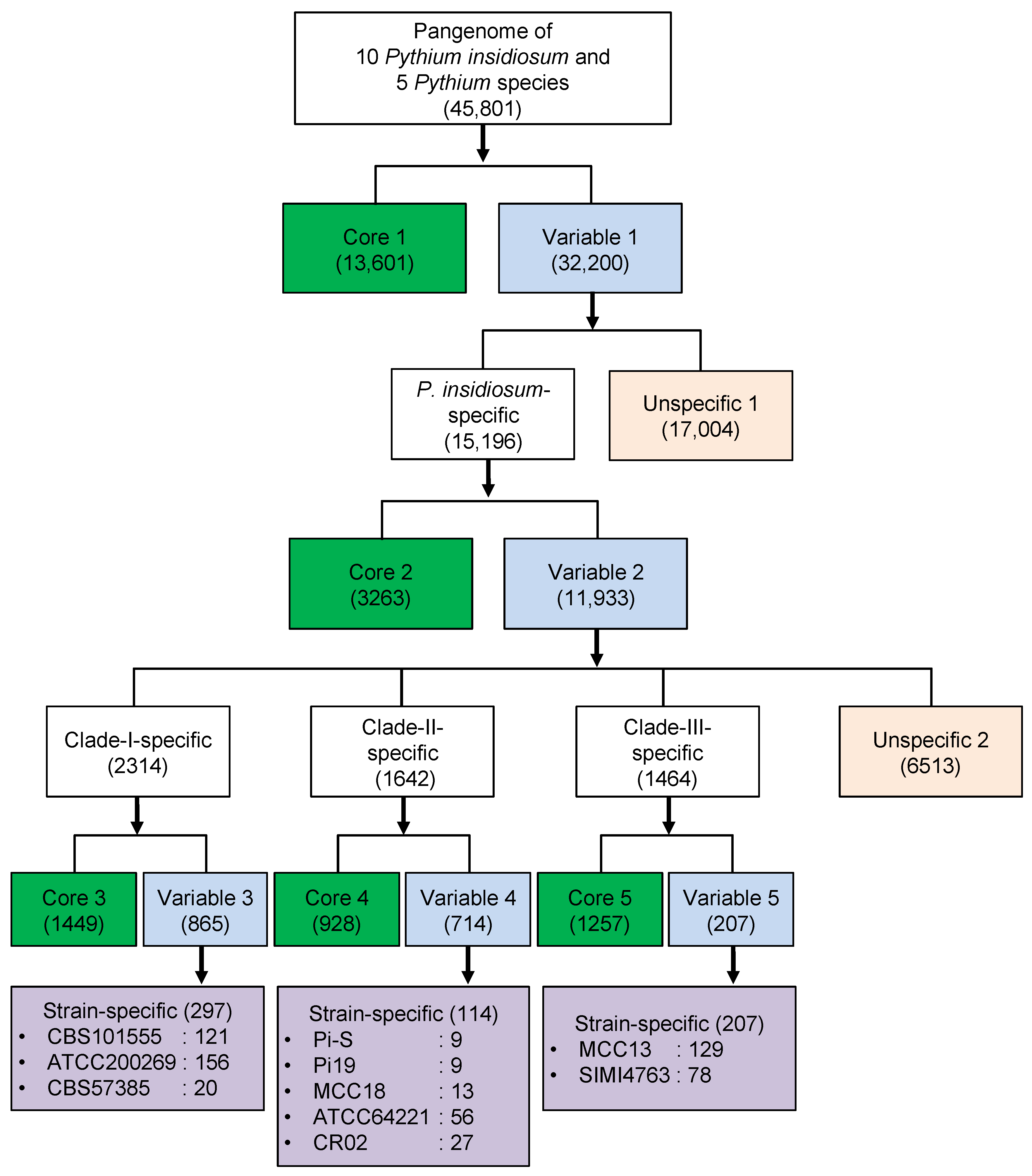
3.4. Core and Variable Genes of Pythium insidiosum
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yolanda, H.; Krajaejun, T. Global Distribution and Clinical Features of Pythiosis in Humans and Animals. J. Fungi 2022, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Krajaejun, T.; Sathapatayavongs, B.; Pracharktam, R.; Nitiyanant, P.; Leelachaikul, P.; Wanachiwanawin, W.; Chaiprasert, A.; Assanasen, P.; Saipetch, M.; Mootsikapun, P.; et al. Clinical and Epidemiological Analyses of Human Pythiosis in Thailand. Clin. Infect. Dis. 2006, 43, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, L.; Ajello, L.; McGinnis, M.R. Infection Caused by the Oomycetous Pathogen Pythium insidiosum. J Mycol Med 1996, 6, 151–164. [Google Scholar]
- Gaastra, W.; Lipman, L.J.; De Cock, A.W.; Exel, T.K.; Pegge, R.B.; Scheurwater, J.; Vilela, R.; Mendoza, L. Pythium insidiosum: An Overview. Vet. Microbiol. 2010, 146, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitasombat, M.N.; Jongkhajornpong, P.; Lekhanont, K.; Krajaejun, T. Recent Update in Diagnosis and Treatment of Human Pythiosis. PeerJ 2020, 8, e8555. [Google Scholar] [CrossRef] [Green Version]
- Rujirawat, T.; Sridapan, T.; Lohnoo, T.; Yingyong, W.; Kumsang, Y.; Sae-Chew, P.; Tonpitak, W.; Krajaejun, T. Single Nucleotide Polymorphism-Based Multiplex PCR for Identification and Genotyping of the Oomycete Pythium insidiosum from Humans, Animals and the Environment. Infect. Genet. Evol. 2017, 54, 429–436. [Google Scholar] [CrossRef]
- Keeratijarut, A.; Lohnoo, T.; Yingyong, W.; Sriwanichrak, K.; Krajaejun, T. A Peptide ELISA to Detect Antibodies against Pythium insidiosum Based on Predicted Antigenic Determinants of Exo-1,3-Beta-Glucanase. Southeast Asian J. Trop. Med. Public Health 2013, 44, 672–680. [Google Scholar]
- Keeratijarut, A.; Lohnoo, T.; Yingyong, W.; Nampoon, U.; Lerksuthirat, T.; Onpaew, P.; Chongtrakool, P.; Krajaejun, T. PCR Amplification of a Putative Gene for Exo-1, 3-Beta-Glucanase to Identify the Pathogenic Oomycete Pythium insidiosum. Asian Biomed. 2014, 8, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Inkomlue, R.; Larbcharoensub, N.; Karnsombut, P.; Lerksuthirat, T.; Aroonroch, R.; Lohnoo, T.; Yingyong, W.; Santanirand, P.; Sansopha, L.; Krajaejun, T. Development of an Anti-Elicitin Antibody-Based Immunohistochemical Assay for Diagnosis of Pythiosis. J. Clin. Microbiol. 2016, 54, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Krajaejun, T.; Kunakorn, M.; Niemhom, S.; Chongtrakool, P.; Pracharktam, R. Development and Evaluation of an In-House Enzyme-Linked Immunosorbent Assay for Early Diagnosis and Monitoring of Human Pythiosis. Clin. Diagn. Lab. Immunol. 2002, 9, 378–382. [Google Scholar] [CrossRef] [Green Version]
- Krajaejun, T.; Imkhieo, S.; Intaramat, A.; Ratanabanangkoon, K. Development of an Immunochromatographic Test for Rapid Serodiagnosis of Human Pythiosis. Clin. Vaccine Immunol. 2009, 16, 506–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jindayok, T.; Piromsontikorn, S.; Srimuang, S.; Khupulsup, K.; Krajaejun, T. Hemagglutination Test for Rapid Serodiagnosis of Human Pythiosis. Clin. Vaccine Immunol. 2009, 16, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Intaramat, A.; Sornprachum, T.; Chantrathonkul, B.; Chaisuriya, P.; Lohnoo, T.; Yingyong, W.; Jongruja, N.; Kumsang, Y.; Sandee, A.; Chaiprasert, A.; et al. Protein A/G-Based Immunochromatographic Test for Serodiagnosis of Pythiosis in Human and Animal Subjects from Asia and Americas. Med. Mycol. 2016, 54, 641–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeratijarut, A.; Lohnoo, T.; Yingyong, W.; Rujirawat, T.; Srichunrusami, C.; Onpeaw, P.; Chongtrakool, P.; Brandhorst, T.T.; Krajaejun, T. Detection of the Oomycete Pythium insidiosum by Real-Time PCR Targeting the Gene Coding for Exo-1,3-β-Glucanase. J. Med. Microbiol. 2015, 64, 971–977. [Google Scholar] [CrossRef]
- Keeratijarut, A.; Karnsombut, P.; Aroonroch, R.; Srimuang, S.; Sangruchi, T.; Sansopha, L.; Mootsikapun, P.; Larbcharoensub, N.; Krajaejun, T. Evaluation of an In-House Immunoperoxidase Staining Assay for Histodiagnosis of Human Pythiosis. Southeast Asian J. Trop. Med. Public Health 2009, 40, 1298–1305. [Google Scholar]
- Krajaejun, T.; Lohnoo, T.; Jittorntam, P.; Srimongkol, A.; Kumsang, Y.; Yingyong, W.; Rujirawat, T.; Reamtong, O.; Mangmee, S. Assessment of Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry for Identification and Biotyping of the Pathogenic Oomycete Pythium insidiosum. Int. J. Infect. Dis. 2018, 77, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerksuthirat, T.; Sangcakul, A.; Lohnoo, T.; Yingyong, W.; Rujirawat, T.; Krajaejun, T. Evolution of the Sterol Biosynthetic Pathway of Pythium insidiosum and Related Oomycetes Contributes to Antifungal Drug Resistance. Antimicrob. Agents Chemother. 2017, 61, e02352-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yolanda, H.; Krajaejun, T. Review of Methods and Antimicrobial Agents for Susceptibility Testing against Pythium insidiosum. Heliyon 2020, 6, e03737. [Google Scholar] [CrossRef] [PubMed]
- Rujirawat, T.; Patumcharoenpol, P.; Lohnoo, T.; Yingyong, W.; Kumsang, Y.; Payattikul, P.; Tangphatsornruang, S.; Suriyaphol, P.; Reamtong, O.; Garg, G.; et al. Probing the Phylogenomics and Putative Pathogenicity Genes of Pythium insidiosum by Oomycete Genome Analyses. Sci. Rep. 2018, 8, 4135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamoun, S. Molecular Genetics of Pathogenic Oomycetes. Eukaryot. Cell 2003, 2, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Kittichotirat, W.; Krajaejun, T. Application of Genome Sequencing to Study Infectious Diseases. J. Infect. Dis. Antimicrob. Agents 2019, 36, 47–58. [Google Scholar]
- Krajaejun, T.; Kittichotirat, W.; Patumcharoenpol, P.; Rujirawat, T.; Lohnoo, T.; Yingyong, W. Draft Genome Sequence of the Oomycete Pythium destruens Strain ATCC 64221 from a Horse with Pythiosis in Australia. BMC Res. Notes 2020, 13, 329. [Google Scholar] [CrossRef] [PubMed]
- Krajaejun, T.; Kittichotirat, W.; Patumcharoenpol, P.; Rujirawat, T.; Lohnoo, T.; Yingyong, W. Genome Data of Four Pythium insidiosum Strains from the Phylogenetically-Distinct Clades I, II, and III. BMC Res. Notes 2021, 14, 197. [Google Scholar] [CrossRef] [PubMed]
- Patumcharoenpol, P.; Rujirawat, T.; Lohnoo, T.; Yingyong, W.; Vanittanakom, N.; Kittichotirat, W.; Krajaejun, T. Draft Genome Sequences of the Oomycete Pythium insidiosum Strain CBS 573.85 from a Horse with Pythiosis and Strain CR02 from the Environment. Data Brief 2018, 16, 47–50. [Google Scholar] [CrossRef] [PubMed]
- Krajaejun, T.; Kittichotirat, W.; Patumcharoenpol, P.; Rujirawat, T.; Lohnoo, T.; Yingyong, W. Data on Whole Genome Sequencing of the Oomycete Pythium insidiosum Strain CBS 101555 from a Horse with Pythiosis in Brazil. BMC Res. Notes 2018, 11, 880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittichotirat, W.; Patumcharoenpol, P.; Rujirawat, T.; Lohnoo, T.; Yingyong, W.; Krajaejun, T. Draft Genome and Sequence Variant Data of the Oomycete Pythium insidiosum Strain Pi45 from the Phylogenetically-Distinct Clade-III. Data Brief 2017, 15, 896–900. [Google Scholar] [CrossRef]
- Ascunce, M.S.; Huguet-Tapia, J.C.; Braun, E.L.; Ortiz-Urquiza, A.; Keyhani, N.O.; Goss, E.M. Whole Genome Sequence of the Emerging Oomycete Pathogen Pythium insidiosum Strain CDC-B5653 Isolated from an Infected Human in the USA. Genomics Data 2016, 7, 60–61. [Google Scholar] [CrossRef] [Green Version]
- Rujirawat, T.; Patumcharoenpol, P.; Lohnoo, T.; Yingyong, W.; Lerksuthirat, T.; Tangphatsornruang, S.; Suriyaphol, P.; Grenville-Briggs, L.J.; Garg, G.; Kittichotirat, W.; et al. Draft Genome Sequence of the Pathogenic Oomycete Pythium insidiosum Strain Pi-S, Isolated from a Patient with Pythiosis. Genome Announc. 2015, 3, e00574-15. [Google Scholar] [CrossRef] [Green Version]
- Rujirawat, T.; Patumcharoenpol, P.; Kittichotirat, W.; Krajaejun, T. Oomycete Gene Table: An Online Database for Comparative Genomic Analyses of the Oomycete Microorganisms. Database 2019, 2019, baz082. [Google Scholar] [CrossRef]
- Lévesque, C.A.; Brouwer, H.; Cano, L.; Hamilton, J.P.; Holt, C.; Huitema, E.; Raffaele, S.; Robideau, G.P.; Thines, M.; Win, J.; et al. Genome Sequence of the Necrotrophic Plant Pathogen Pythium ultimum Reveals Original Pathogenicity Mechanisms and Effector Repertoire. Genome Biol. 2010, 11, R73. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, B.N.; Hamilton, J.P.; Zerillo, M.M.; Tisserat, N.; Lévesque, C.A.; Buell, C.R. Comparative Genomics Reveals Insight into Virulence Strategies of Plant Pathogenic Oomycetes. PLoS ONE 2013, 8, e75072. [Google Scholar] [CrossRef] [PubMed]
- Holt, C.; Yandell, M. MAKER2: An Annotation Pipeline and Genome-Database Management Tool for Second-Generation Genome Projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Nguyen, H.D.T.; Dodge, A.; Dadej, K.; Rintoul, T.L.; Ponomareva, E.; Martin, F.N.; de Cock, A.W.A.M.; Lévesque, C.A.; Redhead, S.A.; Spies, C.F.J. Whole Genome Sequencing and Phylogenomic Analysis Show Support for the Splitting of Genus Pythium. Mycologia 2022, 114, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Kittichotirat, W.; Bumgarner, R.E.; Asikainen, S.; Chen, C. Identification of the Pangenome and Its Components in 14 Distinct Aggregatibacter Actinomycetemcomitans Strains by Comparative Genomic Analysis. PLoS ONE 2011, 6, e22420. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinforma. Oxf. Engl. 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: A Free, Open-Source System for Microarray Data Management and Analysis. BioTechniques 2003, 34, 374–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Smith, J.; Lam, M.; Zemla, A.; Dyer, M.; Slezak, T. MvirDB—A Microbial Database of Protein Toxins, Virulence Factors and Antibiotic Resistance Genes for Bio-Defence Applications. Nucleic Acids Res. 2007, 35, D391–D394. [Google Scholar] [CrossRef] [PubMed]
- Chaiprasert, A.; Krajaejun, T.; Pannanusorn, S.; Prariyachatigul, C.; Wanachiwanawin, W.; Sathapatayavongs, B.; Juthayothin, T.; Smittipat, N.; Vanittanakom, N.; Chindamporn, A. Pythium insidiosum Thai Isolates: Molecular Phylogenetic Analysis. Asian Biomed. 2009, 3, 623–633. [Google Scholar]
- Schurko, A.M.; Mendoza, L.; Lévesque, C.A.; Désaulniers, N.L.; de Cock, A.W.A.M.; Klassen, G.R. A Molecular Phylogeny of Pythium insidiosum. Mycol. Res. 2003, 107, 537–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerksuthirat, T.; Lohnoo, T.; Inkomlue, R.; Rujirawat, T.; Yingyong, W.; Khositnithikul, R.; Phaonakrop, N.; Roytrakul, S.; Sullivan, T.D.; Krajaejun, T. The Elicitin-like Glycoprotein, ELI025, Is Secreted by the Pathogenic Oomycete Pythium insidiosum and Evades Host Antibody Responses. PLoS ONE 2015, 10, e0118547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajaejun, T.; Lerksuthirat, T.; Garg, G.; Lowhnoo, T.; Yingyong, W.; Khositnithikul, R.; Tangphatsornruang, S.; Suriyaphol, P.; Ranganathan, S.; Sullivan, T.D. Transcriptome Analysis Reveals Pathogenicity and Evolutionary History of the Pathogenic Oomycete Pythium insidiosum. Fungal Biol. 2014, 118, 640–653. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organisms and Strains (Genotype: clade I, II, or III) | Country of Origin | Source | Number of Contigs | Total Contig Length (bp) | G+C Content (%) | Numbers of Protein Coding Genes | Total Coding Sequence Length | Average CDS Size (bp) | Coding Density (%) | N50 Length (bp) | Accession Number [Reference] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P. insidiosum CBS 573.85 (I) | Costa Rica | Horse | 11,223 | 35,561,321 | 57.7 | 14,487 | 18,305,243 | 1264 | 51 | 12,261 | BCFO00000000.1 [24] |
| P. insidiosum CBS 101555 (I) | Brazil | Horse | 60,602 | 48,855,945 | 57.3 | 23,254 | 15,797,443 | 679 | 32 | 953 | BCFP00000000.1 [25] |
| P. insidiosum ATCC200269 (I) | USA | Human | 8992 | 45,609,708 | 57.3 | 20,359 | 31,572,237 | 1551 | 69 | 13,382 | BCFN00000000.1 [23] |
| P. insidiosum Pi-S (II) | Thailand | Human | 1192 | 53,239,050 | 57.9 | 14,962 | 22,867,201 | 1528 | 43 | 146,252 | BBXB00000000.1 [28] |
| P. insidiosum Pi19 (II) | Thailand | Human | 14,576 | 35,372,432 | 57.2 | 13,895 | 16,104,157 | 1159 | 46 | 6208 | BCFS00000000.1 [23] |
| P. insidiosum MCC18 (II) | Thailand | Human | 11,084 | 34,541,218 | 57.2 | 13,249 | 16,709,445 | 1261 | 48 | 8946 | BCFT00000000.1 [23] |
| P. insidiosum CR02 (II) | Thailand | Environment | 22,560 | 37,673,126 | 57.1 | 15,231 | 15,695,021 | 1030 | 42 | 3553 | BCFR00000000.1 [24] |
| P. insidiosum ATCC64221 (II) | Australia | Horse | 13,060 | 37,817,292 | 57.6 | 14,424 | 17,868,042 | 1239 | 47 | 11,370 | BCFQ01000000.1 [22] |
| P. insidiosum SIMI4763 (III) | Thailand | Human | 15,162 | 47,141,692 | 57.6 | 19,340 | 24,053,325 | 1244 | 51 | 11,187 | BCFU00000000.1 [23] |
| P. insidiosum Pi45 (III) | Thailand | Human | 17,277 | 65,230,783 | 57.8 | 26,058 | 33,835,683 | 1298 | 52 | 14,374 | BCFM00000000.1 [26] |
| P. arrhenomanes ATCC 12531 | - | - | 10,972 | 44,672,625 | 56.9 | 13,805 | 18,531,402 | 1342 | 41 | 9784 | AKXY00000000.2 [31] |
| P. irregulare DAOM BR486 | - | - | 5887 | 42,968,084 | 53.8 | 13,805 | 20,666,825 | 1497 | 48 | 23,217 | JAADWQ000000000.1 [34] |
| P. iwayamai DAOM BR242034 | - | - | 11,542 | 43,200,612 | 55.1 | 14,875 | 19,758,311 | 1328 | 46 | 11,008 | AKYA00000000.2 [31] |
| P. ultimum DAOM BR144 | - | - | 975 | 44,913,582 | 52.3 | 15,322 | 20,145,968 | 1315 | 45 | 837,833 | ADOS00000000.1 [30] |
| P. aphanidermatum DAOM BR444 | - | - | 1774 | 35,876,849 | 53.8 | 12,312 | 18,112,821 | 1471 | 50 | 37,384 | AKXX00000000.2 [31] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kittichotirat, W.; Rujirawat, T.; Patumcharoenpol, P.; Krajaejun, T. Comparative Genomic Analysis Reveals Gene Content Diversity, Phylogenomic Contour, Putative Virulence Determinants, and Potential Diagnostic Markers within Pythium insidiosum Traits. J. Fungi 2023, 9, 169. https://doi.org/10.3390/jof9020169
Kittichotirat W, Rujirawat T, Patumcharoenpol P, Krajaejun T. Comparative Genomic Analysis Reveals Gene Content Diversity, Phylogenomic Contour, Putative Virulence Determinants, and Potential Diagnostic Markers within Pythium insidiosum Traits. Journal of Fungi. 2023; 9(2):169. https://doi.org/10.3390/jof9020169
Chicago/Turabian StyleKittichotirat, Weerayuth, Thidarat Rujirawat, Preecha Patumcharoenpol, and Theerapong Krajaejun. 2023. "Comparative Genomic Analysis Reveals Gene Content Diversity, Phylogenomic Contour, Putative Virulence Determinants, and Potential Diagnostic Markers within Pythium insidiosum Traits" Journal of Fungi 9, no. 2: 169. https://doi.org/10.3390/jof9020169
APA StyleKittichotirat, W., Rujirawat, T., Patumcharoenpol, P., & Krajaejun, T. (2023). Comparative Genomic Analysis Reveals Gene Content Diversity, Phylogenomic Contour, Putative Virulence Determinants, and Potential Diagnostic Markers within Pythium insidiosum Traits. Journal of Fungi, 9(2), 169. https://doi.org/10.3390/jof9020169