
Assessment of Antifungal Pharmacodynamics


Abstract
:1. Introduction
2. Developing and Characterizing Disease Models
2.1. Host
2.2. Pathogen Species
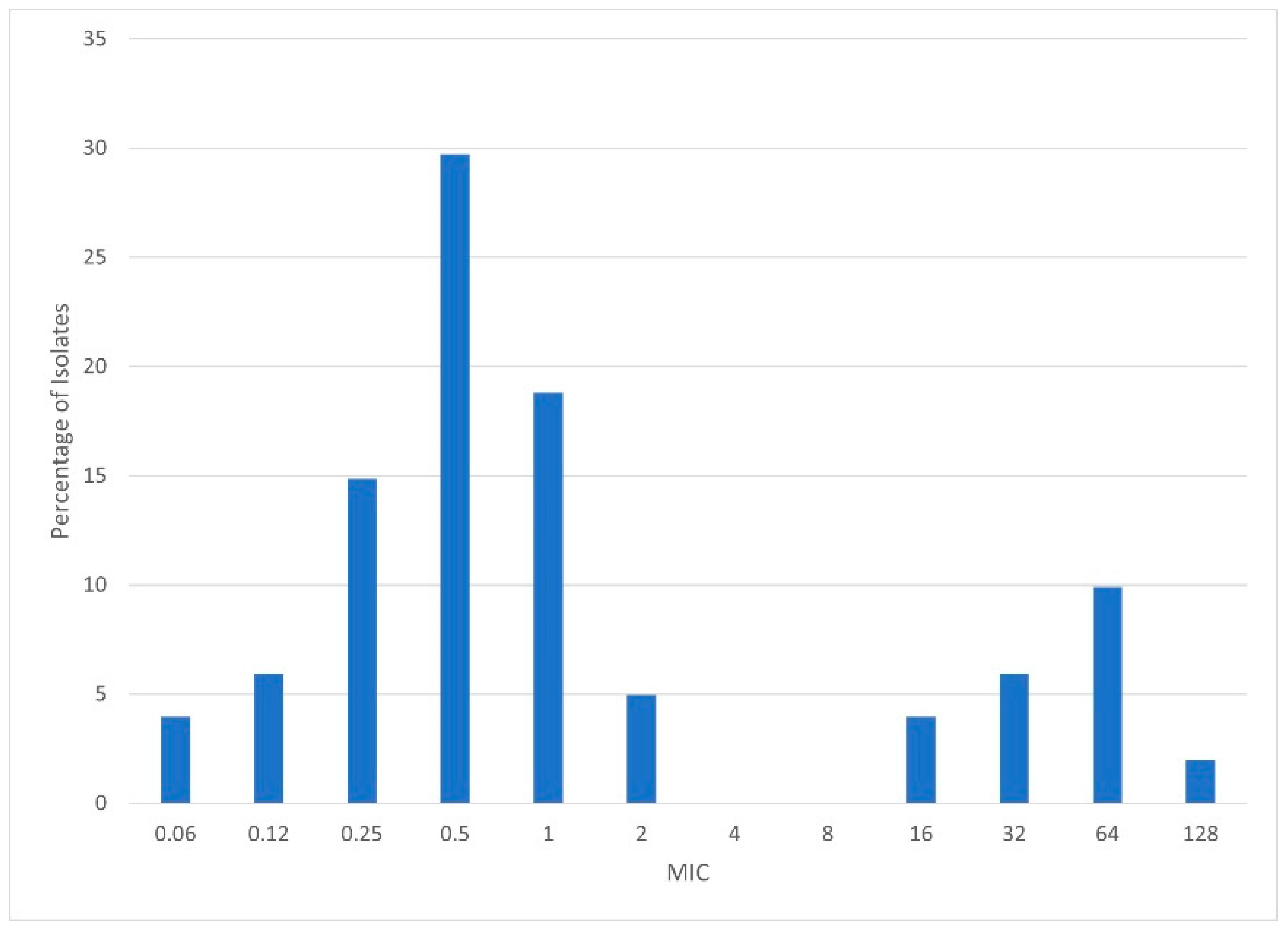
2.3. Minimum Inhibitory Concentration
2.4. Pathogenesis
3. Defining Outcome Measures
3.1. Quantifying the Antifungal Effect
3.2. Setting Endpoints
3.3. Establishing the Dose–Exposure–Response Relationship
4. Regimen Identification
4.1. Human Pharmacokinetic Data
4.2. Pharmacokinetic Modelling
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vallor, A.C.; Kirkpatrick, W.R.; Najvar, L.K.; Bocanegra, R.; Kinney, M.C.; Fothergill, A.W.; Herrera, M.L.; Wickes, B.L.; Graybill, J.R.; Patterson, T.F. Assessment of Aspergillus fumigatus burden in pulmonary tissue of guinea pigs by quantitative PCR, galactomannan enzyme immunoassay, and quantitative culture. Antimicrob. Agents Chemother. 2008, 52, 2593–2598. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.J.; de Marie, S.; Willemse, D.; Verbrugh, H.A.; Bakker-Woudenberg, I.A.J.M. Quantitative Galactomannan Detection Is Superior to PCR in Diagnosing and Monitoring Invasive Pulmonary Aspergillosis in an Experimental Rat Model. J. Clin. Microbiol. 2000, 38, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Lionakis, M.S. Drosophila and Galleria insect model hosts. Virulence 2011, 2, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Anastassopoulou, C.G.; Burgwyn Fuchs, B.; Mylonakis, E. Caenorhabditis elegans-based Model Systems for Antifungal Drug Discovery. Curr. Pharm. Des. 2011, 17, 1225–1233. [Google Scholar] [CrossRef]
- Lewis, R.E.; Lund, B.C.; Klepser, M.E.; Ernst, E.J.; Pfaller, M.A. Assessment of Antifungal Activities of Fluconazole and Amphotericin B Administered Alone and in Combination against Candida albicans by Using a Dynamic In Vitro Mycotic Infection Model. Antimicrob. Agents Chemother. 1998, 42, 1382–1386. [Google Scholar] [CrossRef]
- Beredaki, M.-I.; Arendrup, M.C.; Andes, D.; Mouton, J.W.; Meletiadis, J. The Role of New Posaconazole Formulations in the Treatment of Candida albicans Infections: Data from an In Vitro Pharmacokinetic-Pharmacodynamic Model. Antimicrob. Agents Chemother. 2021, 65, e01292-20. [Google Scholar] [CrossRef]
- Jeans, A.R.; Howard, S.J.; Al-Nakeeb, Z.; Goodwin, J.; Gregson, L.; Majithiya, J.B.; Lass-Flörl, C.; Cuenca-Estrella, M.; Arendrup, M.C.; Warn, P.A.; et al. Pharmacodynamics of voriconazole in a dynamic in vitro model of invasive pulmonary aspergillosis: Implications for in vitro susceptibility breakpoints. J. Infect. Dis. 2012, 206, 442–452. [Google Scholar] [CrossRef]
- Box, H.; Livermore, J.; Johnson, A.; McEntee, L.; Felton, T.W.; Whalley, S.; Goodwin, J.; Hope, W.W. Pharmacodynamics of isavuconazole in a dynamic In Vitro model of invasive pulmonary aspergillosis. Antimicrob. Agents Chemother. 2016, 60, 278–287. [Google Scholar] [CrossRef]
- Meletiadis, J.; Al-Saigh, R.; Velegraki, A.; Walsh, T.J.; Roilides, E.; Zerva, L. Pharmacodynamic effects of simulated standard doses of antifungal drugs against Aspergillus species in a new In Vitro pharmacokinetic/pharmacodynamic model. Antimicrob. Agents Chemother. 2012, 56, 403–410. [Google Scholar] [CrossRef]
- Jeans, A.R.; Howard, S.J.; Al-Nakeeb, Z.; Goodwin, J.; Gregson, L.; Warn, P.A.; Hope, W.W. Combination of voriconazole and anidulafungin for treatment of triazole-resistant Aspergillus fumigatus in an In Vitro model of invasive pulmonary aspergillosis. Antimicrob. Agents Chemother. 2012, 56, 5180–5185. [Google Scholar] [CrossRef] [Green Version]
- Hope, W.W.; Kruhlak, M.J.; Lyman, C.A.; Petraitiene, R.; Petraitis, V.; Francesconi, A.; Kasai, M.; Mickiene, D.; Sein, T.; Peter, J.; et al. Pathogenesis of Aspergillus fumigatus and the Kinetics of Galactomannan in an In Vitro Model of Early Invasive Pulmonary Aspergillosis: Implications for Antifungal Therapy. J. Infect. Dis. 2007, 195, 455–466. [Google Scholar] [CrossRef]
- Al-Nakeeb, Z.; Sudan, A.; Jeans, A.R.; Gregson, L.; Goodwin, J.; Warn, P.A.; Felton, T.W.; Howard, S.J.; Hope, W.W. Pharmacodynamics of itraconazole against Aspergillus fumigatus in an in vitro model of the human alveolus: Perspectives on the treatment of triazole-resistant infection and utility of airway administration. Antimicrob. Agents Chemother. 2012, 56, 4146–4153. [Google Scholar] [CrossRef]
- Negri, C.E.; Johnson, A.; McEntee, L.; Box, H.; Whalley, S.; Schwartz, J.A.; Ramos-Martín, V.; Livermore, J.; Kolamunnage-Dona, R.; Colombo, A.L.; et al. Pharmacodynamics of the Novel Antifungal Agent F901318 for Acute Sinopulmonary Aspergillosis Caused by Aspergillus flavus. J. Infect. Dis. 2018, 217, 1118–1127. [Google Scholar] [CrossRef]
- Larsen, R.A.; Bauer, M.; Weiner, J.M.; Diamond, D.M.; Leal, M.E.; Ding, J.C.; Rinaldi, M.G.; Graybill, A.J.R. Effect of Fluconazole on Fungicidal Activity of Flucytosine in Murine Cryptococcal Meningitis. Antimicrob. Agents Chemother. 1996, 40, 2178–2182. [Google Scholar] [CrossRef]
- Ding, J.C.; Bauer, M.; Diamond, D.M.; Leal, M.A.E.; Johnson, D.; Williams, B.K.; Thomas, A.M.; Najvar, L.; Graybill, J.R.; Larsen, R.A. Effect of Severity of Meningitis on Fungicidal Activity of Flucytosine Combined with Fluconazole in a Murine Model of Cryptococcal Meningitis. Antimicrob. Agents Chemother. 1997, 41, 1589–1593. [Google Scholar] [CrossRef]
- Gebremariam, T.; Gu, Y.; Singh, S.; Kitt, T.M.; Ibrahim, A.S. Combination treatment of liposomal amphotericin B and isavuconazole is synergistic in treating experimental mucormycosis. J. Antimicrob. Chemother. 2021, 76, 2636–2639. [Google Scholar] [CrossRef]
- Lewis, R.E.; Albert, N.D.; Liao, G.; Hou, J.; Prince, R.A.; Kontoyiannis, D.P. Comparative pharmacodynamics of amphotericin B lipid complex and liposomal amphotericin B in a murine model of pulmonary mucormycosis. Antimicrob. Agents Chemother. 2010, 54, 1298–1304. [Google Scholar] [CrossRef]
- Lewis, R.E.; Wiederhold, N.P.; Klepser, M.E. In vitro pharmacodynamics of amphotericin B, itraconazole, and voriconazole against Aspergillus, Fusarium, and Scedosporium spp. Antimicrob. Agents Chemother. 2005, 49, 945–951. [Google Scholar] [CrossRef]
- Andes, D.; Stamsted, T.; Conklin, R. Pharmacodynamics of amphotericin B in a neutropenic-mouse disseminated-candidiasis model. Antimicrob. Agents Chemother. 2001, 45, 922–926. [Google Scholar] [CrossRef]
- Andes, D.; Safdar, N.; Marchillo, K.; Conklin, R. Pharmacokinetic-pharmacodynamic comparison of amphotericin B (AMB) and two lipid-associated AMB preparations, liposomal AMB and AMB lipid complex, in murine candidiasis models. Antimicrob. Agents Chemother. 2006, 50, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Lepak, A.J.; Zhao, M.; Andesa, D.R. Pharmacodynamic evaluation of rezafungin (CD101) against Candida auris in the neutropenic mouse invasive candidiasis model. Antimicrob. Agents Chemother. 2018, 62, e01572-18. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.J.; Livermore, J.; Sharp, A.; Goodwin, J.; Gregson, L.; Alastruey-Izquierdo, A.; Perlin, D.S.; Warn, P.A.; Hope, W.W. Pharmacodynamics of echinocandins against Candida glabrata: Requirement for dosage escalation to achieve maximal antifungal activity in neutropenic hosts. Antimicrob. Agents Chemother. 2011, 55, 4880–4887. [Google Scholar] [CrossRef] [PubMed]
- Arendrup, M.C.; Perlin, D.S.; Jensen, R.H.; Howard, S.J.; Goodwin, J.; Hope, W. Differential in vivo activities of anidulafungin, caspofungin, and micafungin against Candida glabrata isolates with and without FKS resistance mutations. Antimicrob. Agents Chemother. 2012, 56, 2435–2442. [Google Scholar] [CrossRef] [PubMed]
- Andes, D.R.; Diekema, D.J.; Pfaller, M.A.; Marchillo, K.; Bohrmueller, J. In vivo pharmacodynamic target investigation for micafungin against Candida albicans and C. glabrata in a neutropenic murine candidiasis model. Antimicrob. Agents Chemother. 2008, 52, 3497–3503. [Google Scholar] [CrossRef]
- Mavridou, E.; Bruggemann, R.J.M.; Melchers, W.J.G.; Verweij, P.E.; Mouton, J.W. Impact of cyp51A mutations on the pharmacokinetic and pharmacodynamic properties of voriconazole in a murine model of disseminated aspergillosis. Antimicrob. Agents Chemother. 2010, 54, 4758–4764. [Google Scholar] [CrossRef]
- Pierce, C.G.; Srinivasan, A.; Uppuluri, P.; Ramasubramanian, A.K.; López-Ribot, J.L. Antifungal therapy with an emphasis on biofilms. Curr. Opin. Pharmacol. 2013, 13, 726–730. [Google Scholar] [CrossRef]
- Cannon, R.D.; Lamping, E.; Holmes, A.R.; Niimi, K.; Baret, P.V.; Keniya, M.V.; Tanabe, K.; Niimi, M.; Goffeau, A.; Monk, B.C. Efflux-Mediated Antifungal Drug Resistance. Clin. Microbiol. Rev. 2009, 22, 291–321. [Google Scholar] [CrossRef]
- Hope, W.W.; Mickiene, D.; Petraitis, V.; Petraitiene, R.; Kelaher, A.M.; Hughes, J.E.; Cotton, M.P.; Bacher, J.; Keirns, J.J.; Buell, D.; et al. The pharmacokinetics and pharmacodynamics of micafungin in experimental hematogenous Candida meningoencephalitis: Implications for echinocandin therapy in neonates. J. Infect. Dis. 2008, 197, 163–171. [Google Scholar] [CrossRef]
- Zhao, Y.; Prideaux, B.; Nagasaki, Y.; Hee Lee, M.; Chen, P.-Y.; Blanc, L.; Ho, H.; Clancy, C.J.; Hong Nguyen, M.; Dartois, V.; et al. Unraveling Drug Penetration of Echinocandin Antifungals at the Site of Infection in an Intra-abdominal Abscess Model. Antimicrob. Agents Chemother. 2017, 10, e01009-17. [Google Scholar] [CrossRef]
- Seyedmousavi, S.; Brüggemann, R.J.M.; Melchers, W.J.G.; Verweij, P.E.; Mouton, J.W. Pharmacodynamics of anidulafungin against clinical Aspergillus fumigatus isolates in a nonneutropenic murine model of disseminated aspergillosis. Antimicrob. Agents Chemother. 2013, 57, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Lepak, A.J.; Marchillo, K.; Van Hecker, J.; Andes, D.R. Posaconazole Pharmacodynamic Target Determination against Wild-Type and Cyp51 Mutant Isolates of Aspergillus fumigatus in an In Vivo Model of Invasive Pulmonary Aspergillosis. Antimicrob. Agents Chemother. 2013, 57, 579–585. [Google Scholar] [CrossRef]
- Sheppard, D.C.; Rieg, G.; Chiang, L.Y.; Filler, S.G.; Edwards, J.E.; Ibrahim, A.S. Novel Inhalational Murine Model of Invasive Pulmonary Aspergillosis. Antimicrob. Agents Chemother. 2004, 48, 1908–1911. [Google Scholar] [CrossRef]
- Sheppard, D.C.; Graybill, J.R.; Najvar, L.K.; Chiang, L.Y.; Doedt, T.; Kirkpatrick, W.R.; Bocanegra, R.; Vallor, A.C.; Patterson, T.F.; Filler, S.G. Standardization of an experimental murine model of invasive pulmonary aspergillosis. Antimicrob. Agents Chemother. 2006, 50, 3501–3503. [Google Scholar] [CrossRef]
- Chiller, T.M.; Luque, J.C.; Sobel, R.A.; Farrokhshad, K.; Clemons, K.V.; Stevens, D.A. Development of a Murine Model of Cerebral Aspergillosis. J. Infect. Dis. 2002, 186, 574–577. [Google Scholar] [CrossRef]
- Wang, X.; Fries, B.C. A murine model for catheter-associated candiduria. J. Med. Microbiol. 2011, 60, 1523–1529. [Google Scholar] [CrossRef]
- Nett, J.E.; Brooks, E.G.; Cabezas-Olcoz, J.; Sanchez, H.; Zarnowski, R.; Marchillo, K.; Andes, D.R. Rat Indwelling Urinary Catheter Model of Candida albicans Biofilm Infection. Infect. Immun. 2014, 82, 4931–4940. [Google Scholar] [CrossRef]
- Zhao, Y.; Lee, M.H.; Paderu, P.; Lee, A.; Jimenez-Ortigosa, C.; Park, S.; Mansbach, R.S.; Shaw, K.J.; Perlin, D.S. Significantly Improved Pharmacokinetics Enhances In Vivo Efficacy of APX001 against Echinocandin-and Multidrug-Resistant Candida Isolates in a Mouse Model of Invasive Candidiasis. Antimicrob. Agents Chemother. 2018, 62, e00425-18. [Google Scholar] [CrossRef]
- Andes, D.; van Ogtrop, M. In Vivo Characterization of the Pharmacodynamics of Flucytosine in a Neutropenic Murine Disseminated Candidiasis Model. Antimicrob. Agents Chemother. 2000, 44, 938–942. [Google Scholar] [CrossRef]
- Petraitiene, R.; Petraitis, V.; Hope, W.W.; Mickiene, D.; Kelaher, A.M.; Murray, H.A.; Mya-San, C.; Hughes, J.E.; Cotton, M.P.; Bacher, J.; et al. Cerebrospinal fluid and plasma (1→3)-β-D-glucan as surrogate markers for detection and monitoring of therapeutic response in experimental hematogenous Candida meningoencephalitis. Antimicrob. Agents Chemother. 2008, 52, 4121–4129. [Google Scholar] [CrossRef]
- Hope, W.; Drusano, G.L.; Rex, J.H. Pharmacodynamics for antifungal drug development: An approach for acceleration, risk minimization and demonstration of causality. J. Antimicrob. Chemother. 2016, 71, 3008–3019. [Google Scholar] [CrossRef] [Green Version]
- Petraitis, V.; Petraitiene, R.; Groll, A.H.; Bell, A.; Callender, D.P.; Sein, T.; Schaufele, R.L.; Mcmillian, C.L.; Bacher, J.; Walsh, T.J. Antifungal Efficacy, Safety, and Single-Dose Pharmacokinetics of LY303366, a Novel Echinocandin B, in Experimental Pulmonary Aspergillosis in Persistently Neutropenic Rabbits. Antimicrob. Agents Chemother. 1998, 42, 2898–2905. [Google Scholar] [CrossRef] [PubMed]
- Lepak, A.J.; Marchillo, K.; Andes, D.R. Pharmacodynamic target evaluation of a novel oral glucan synthase inhibitor, SCY-078 (MK-3118), using an in vivo murine invasive candidiasis model. Antimicrob. Agents Chemother. 2015, 59, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Venisse, N.; Grégoire, N.; Marliat, M.; Couet, W. Mechanism-Based Pharmacokinetic-Pharmacodynamic Models of In Vitro Fungistatic and Fungicidal Effects against Candida albicans. Antimicrob. Agents Chemother. 2008, 52, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zarychanski, R.; Pisipati, A.; Kumar, A.; Kethireddy, S.; Bow, E.J. Fungicidal versus fungistatic therapy of invasive Candida infection in non-neutropenic adults: A meta-analysis. Mycology 2018, 9, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Gangwar, M.; Cole, R.; Ramani, R.; Sheehan, D.J.; Chaturvedi, V. Application of fluorescent probes to study structural changes in Aspergillus fumigatus exposed to amphotericin B, itraconazole, and voriconazole. Mycopathologia 2006, 162, 103–109. [Google Scholar] [CrossRef]
- Wiederhold, N.P.; Najvar, L.K.; Vallor, A.C.; Kirkpatrick, W.R.; Bocanegra, R.; Molina, D.; Olivo, M.; Graybill, J.R.; Patterson, T.F. Assessment of serum (1→3)-β-D-glucan concentration as a measure of disease burden in a murine model of invasive pulmonary aspergillosis. Antimicrob. Agents Chemother. 2008, 52, 1176–1178. [Google Scholar] [CrossRef]
- Gastine, S.; Hope, W.; Hempel, G.; Petraitiene, R.; Petraitis, V.; Mickiene, D.; Bacher, J.; Walsh, T.J.; Groll, A.H. Pharmacodynamics of Posaconazole in Experimental Invasive Pulmonary Aspergillosis: Utility of Serum Galactomannan as a Dynamic Endpoint of Antifungal Efficacy. Antimicrob. Agents Chemother. 2021, 2, e01574-20. [Google Scholar] [CrossRef]
- Petraitiene, R.; Petraitis, V.; Groll, A.H.; Sein, T.; Schaufele, R.L.; Francesconi, A.; Bacher, J.; Avila, N.A.; Walsh, T.J. Antifungal efficacy of caspofungin (MK-0991) in experimental pulmonary aspergillosis in persistently neutropenic rabbits: Pharmacokinetics, drug disposition, and relationship to galactomannan antigenemia. Antimicrob. Agents Chemother. 2002, 46, 12–23. [Google Scholar] [CrossRef]
- Tortorano, A.M.; Esposto, M.C.; Prigitano, A.; Grancini, A.; Ossi, C.; Cavanna, C.; lo Cascio, G. Cross-reactivity of Fusarium spp. In the Aspergillus galactomannan enzyme-linked immunosorbent assay. J. Clin. Microbiol. 2012, 50, 1051–1053. [Google Scholar] [CrossRef]
- Bowman, J.C.; Abruzzo, G.K.; Anderson, J.W.; Flattery, A.M.; Gill, C.J.; Pikounis, V.B.; Schmatz, D.M.; Liberator, A.; Douglas, C.M. Quantitative PCR assay to measure Aspergillus fumigatus burden in a murine model of disseminated aspergillosis: Demonstration of efficacy of caspofungin acetate. Antimicrob. Agents Chemother. 2001, 45, 3474–3481. [Google Scholar] [CrossRef] [Green Version]
- Wong, B.; Leal, I.; Feau, N.; Dale, A.; Uzunovic, A.; Hamelin, R.C. Molecular assays to detect the presence and viability of Phytophthora ramorum and Grosmannia clavigera. PloS ONE 2020, 15, e0221742. [Google Scholar] [CrossRef]
- Henneberg, S.; Hasenberg, A.; Maurer, A.; Neumann, F.; Bornemann, L.; Gonzalez-Menendez, I.; Kraus, A.; Hasenberg, M.; Thornton, C.R.; Pichler, B.J.; et al. Antibody-guided in vivo imaging of Aspergillus fumigatus lung infections during antifungal azole treatment. Nat. Commun. 2021, 12, 1707. [Google Scholar] [CrossRef]
- Howard, S.J.; Lestner, J.M.; Sharp, A.; Gregson, L.; Goodwin, J.; Slater, J.; Majithiya, J.B.; Warn, P.A.; Hope, W.W. Pharmacokinetics and pharmacodynamics of posaconazole for invasive pulmonary aspergillosis: Clinical implications for antifungal therapy. J. Infect. Dis. 2011, 203, 1324–1332. [Google Scholar] [CrossRef]
- Louie, A.; Deziel, M.; Liu, W.; Drusano, M.F.; Gumbo, T.; Drusano, G.L. Pharmacodynamics of caspofungin in a murine model of systemic candidiasis: Importance of persistence of caspofungin in tissues to understanding drug activity. Antimicrob. Agents Chemother. 2005, 49, 5058–5068. [Google Scholar] [CrossRef]
- Groll, A.H.; Rijnders, B.J.A.; Walsh, T.J.; Adler-Moore, J.; Lewis, R.E.; Brüggemann, R.J.M. Clinical Pharmacokinetics, Pharmacodynamics, Safety and Efficacy of Liposomal Amphotericin B. Clin. Infect. Dis. 2019, 68, S260–S274. [Google Scholar] [CrossRef]
- Pound, M.W.; Townsend, M.L.; Drew, R.H. Echinocandin pharmacodynamics: Review and clinical implications. J. Antimicrob. Chemother. 2010, 65, 1108–1118. [Google Scholar] [CrossRef]
- Falagas, M.E.; Karageorgopoulos, D.E.; Tansarli, G.S. Continuous versus Conventional Infusion of Amphotericin B Deoxycholate: A Meta-Analysis. PLoS ONE 2013, 8, e77075. [Google Scholar] [CrossRef]
- Lepak, A.; Marchillo, K.; VanHecker, J.; Azie, N.; Andes, D. Efficacy of Extended-Interval Dosing of Micafungin Evaluated Using a Pharmacokinetic/Pharmacodynamic Study with Humanized Doses in Mice. Antimicrob. Agents Chemother. 2016, 60, 674–677. [Google Scholar] [CrossRef]
- Al-Nakeeb, Z.; Petraitis, V.; Goodwin, J.; Petraitiene, R.; Walsh, T.J.; Hope, W.W. Pharmacodynamics of amphotericin B deoxycholate, amphotericin B lipid complex, and liposomal amphotericin B against Aspergillus fumigatus. Antimicrob. Agents Chemother. 2015, 59, 2735–2745. [Google Scholar] [CrossRef]
- Murrell, D.; Bossaer, J.B.; Carico, R.; Harirforoosh, S.; Cluck, D. Isavuconazonium sulfate: A triazole prodrug for invasive fungal infections. Int. J. Pharm. Pract. 2017, 25, 18–30. [Google Scholar] [CrossRef]
- Heykants, J.; van Peer, A.; van de Velde, V.; van Rooy, P.; Meuldermans, W.; Lavrijsen, K.; Woestenborghs, R.; van Cutsem, J.; Cauwenbergh, G. The Clinical Pharmacokinetics of Itraconazole: An Overview. Mycoses 1989, 32, 67–87. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Khanal Lamichhane, A.; Kwon-Chung, K.J. Cryptococcus neoformans, Unlike Candida albicans, Forms Aneuploid Clones Directly from Uninucleated Cells under Fluconazole Stress. mBio 2018, 9, e01290-18. [Google Scholar] [CrossRef] [PubMed]
- Scholz, I.; Oberwittler, H.; Riedel, K.-D.; Burhenne, J.; Weiss, J.; Haefeli, W.E.; Mikus, G. Pharmacokinetics, metabolism and bioavailability of the triazole antifungal agent voriconazole in relation to CYP2C19 genotype. Br. J. Clin. Pharmacol. 2009, 68, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Thai, S.-F.; Lambert, G.R.; Wolf, D.C.; Tully, D.B.; Goetz, A.K.; George, M.H.; Grindstaff, R.D.; Dix, D.J.; Nesnow, S. Fluconazole-induced hepatic cytochrome P450 gene expression and enzymatic activities in rats and mice. Toxicol. Lett. 2006, 164, 44–53. [Google Scholar] [CrossRef]
- Kovanda, L.L.; Sullivan, S.M.; Smith, L.R.; Desai, A.V.; Bonate, P.L.; Hope, W.W. Population Pharmacokinetic Modeling of VL-2397, a Novel Systemic Antifungal Agent: Analysis of a Single- and Multiple-Ascending-Dose Study in Healthy Subjects. Antimicrob. Agents Chemother. 2019, 63, e00163-19. [Google Scholar] [CrossRef] [Green Version]
- Schmitt-Hoffmann, A.; Roos, B.; Heep, M.; Schleimer, M.; Weidekamm, E.; Brown, T.; Roehrle, M.; Beglinger, C. Single-Ascending-Dose Pharmacokinetics and Safety of the Novel Broad-Spectrum Antifungal Triazole BAL4815 after Intravenous Infusions (50, 100, and 200 Milligrams) and Oral Administrations (100, 200, and 400 Milligrams) of Its Prodrug, BAL8557, in Healthy Volunteers. Antimicrob. Agents Chemother. 2006, 50, 279–285. [Google Scholar]


{kind=link}
| Disease | Model(s) |
|---|---|
| Disseminated candidiasis | Murine tail vein injection, intravenous injection in rabbits |
| Haematogenous Candida meningoencephalitis | Intravenous injection in non-neutropenic rabbits |
| Candida intra-abdominal abscess | Murine intraperitoneal injection |
| Urinary catheter-associated candiduria | Urinary catheter lumen injection |
| Disseminated aspergillosis | Murine tail vein injection |
| Pulmonary aspergillosis | Murine nasal inoculation/aerosol chamber, rabbit endotracheal inoculation, in vitro models |
| Cerebral aspergillosis | Murine intracerebral injection |
| Sinopulmonary aspergillosis | In vitro and murine nasal inoculation |
| Cryptococcal meningitis | Murine tail vein injection and intracisternal inoculation in rabbits |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Howard, A.; Hope, W. Assessment of Antifungal Pharmacodynamics. J. Fungi 2023, 9, 192. https://doi.org/10.3390/jof9020192
Howard A, Hope W. Assessment of Antifungal Pharmacodynamics. Journal of Fungi. 2023; 9(2):192. https://doi.org/10.3390/jof9020192
Chicago/Turabian StyleHoward, Alex, and William Hope. 2023. "Assessment of Antifungal Pharmacodynamics" Journal of Fungi 9, no. 2: 192. https://doi.org/10.3390/jof9020192
APA StyleHoward, A., & Hope, W. (2023). Assessment of Antifungal Pharmacodynamics. Journal of Fungi, 9(2), 192. https://doi.org/10.3390/jof9020192