
Conservation and Expansion of Transcriptional Factor Repertoire in the Fusarium oxysporum Species Complex

 , , ,
, , , 

Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Fungal TFomes
2.2. RNA-Seq Analysis
2.3. Genome Partition
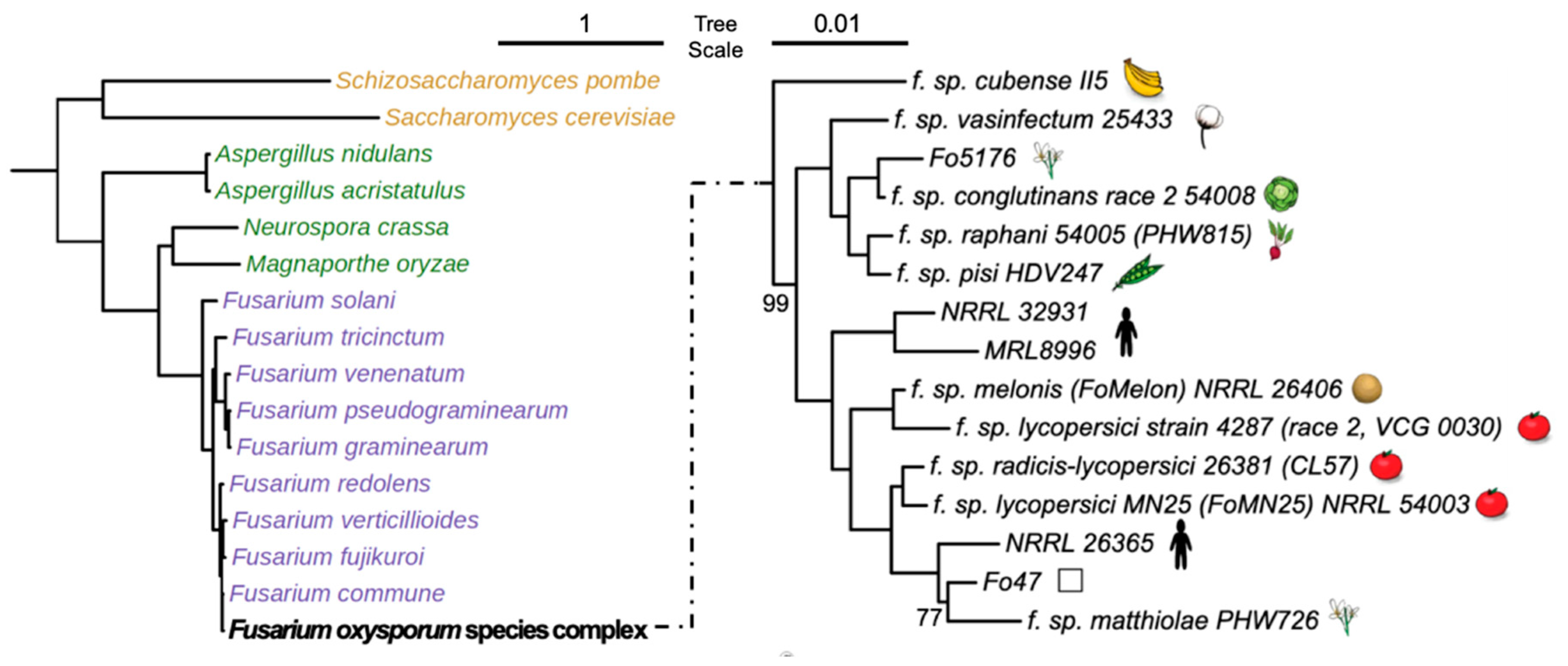
2.4. Phylogenetics Analysis
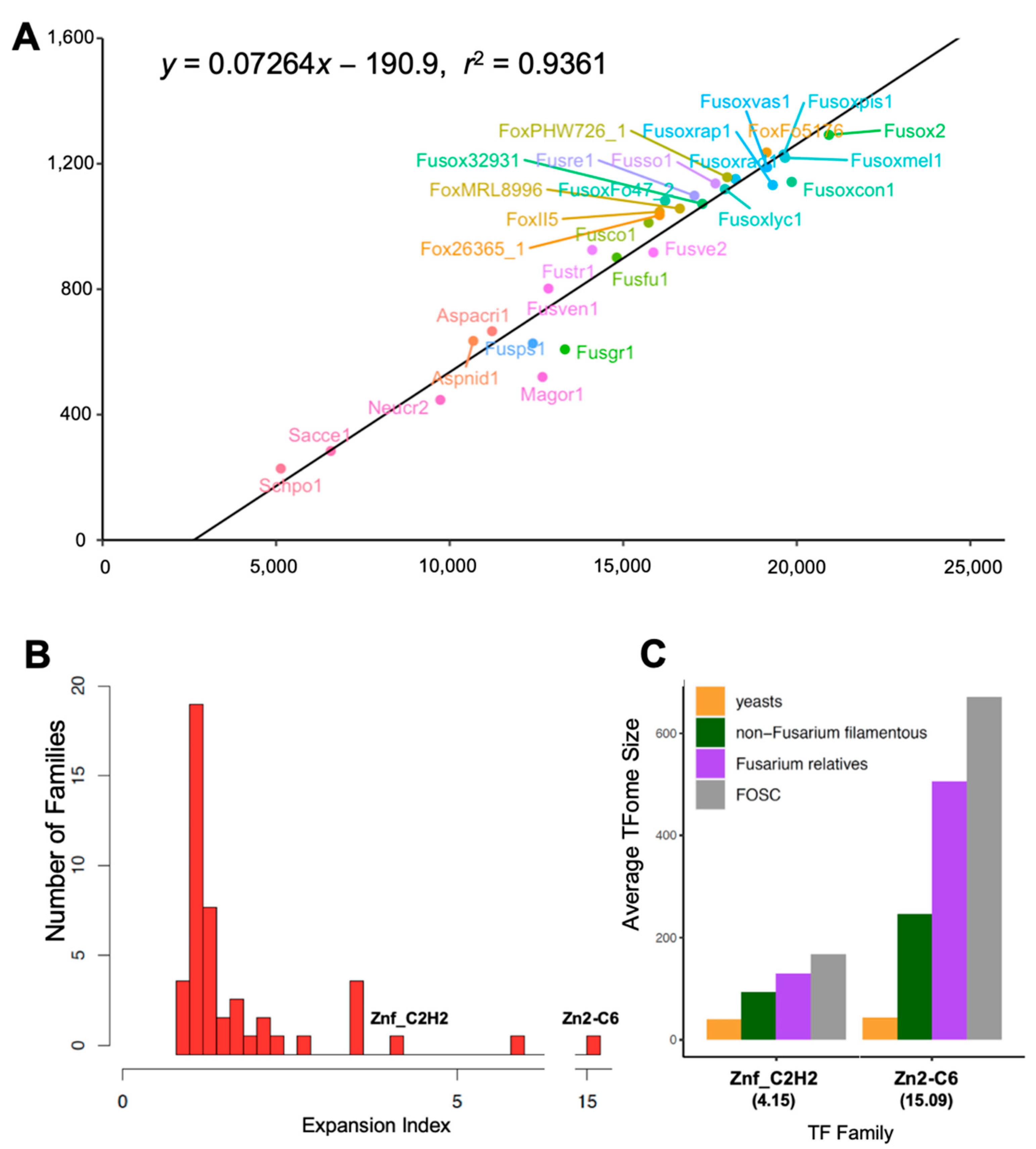
2.5. Expansion Index Calculation
3. Results
3.1. FOSC TFome Expansion Resulted from the Acquisition of ACs
3.2. Conserved TF Families That Are Primarily Associated with General/Global Transcription Factors
3.2.1. Transcription/Translation Regulation
3.2.2. Cell Cycle Control
3.3. Gene Family Contractions in FOSC Partially Caused by Whole Genome Duplication in Yeast
3.4. Significant FOSC TFome Expansion Driven by a Few Exceedingly Expanded TF Families
3.4.1. Gain-of-Function among Filamentous Ascomycete Fungi
3.4.2. Seven Exceedingly Expanded TF Families
3.4.3. Other Families
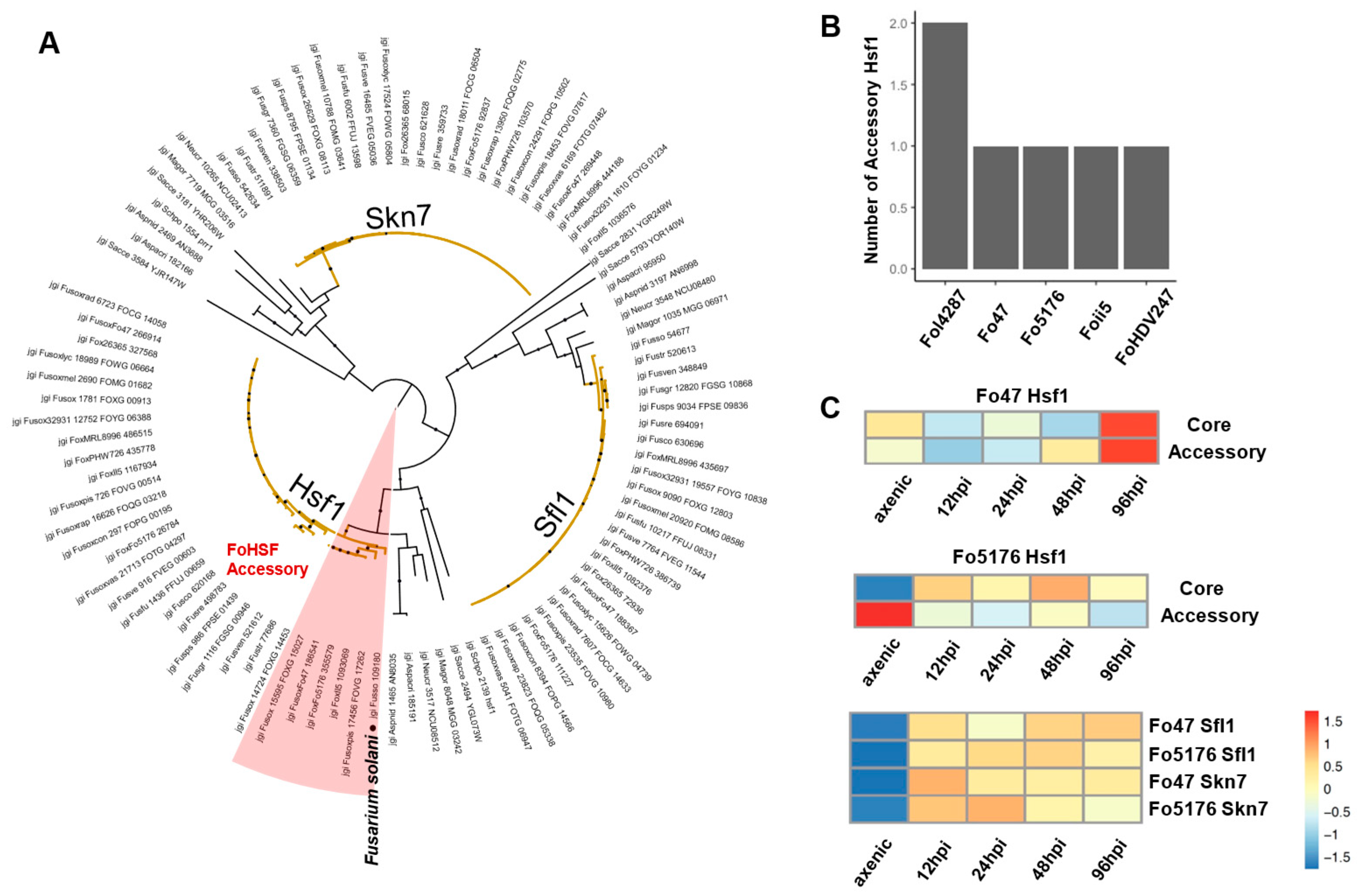
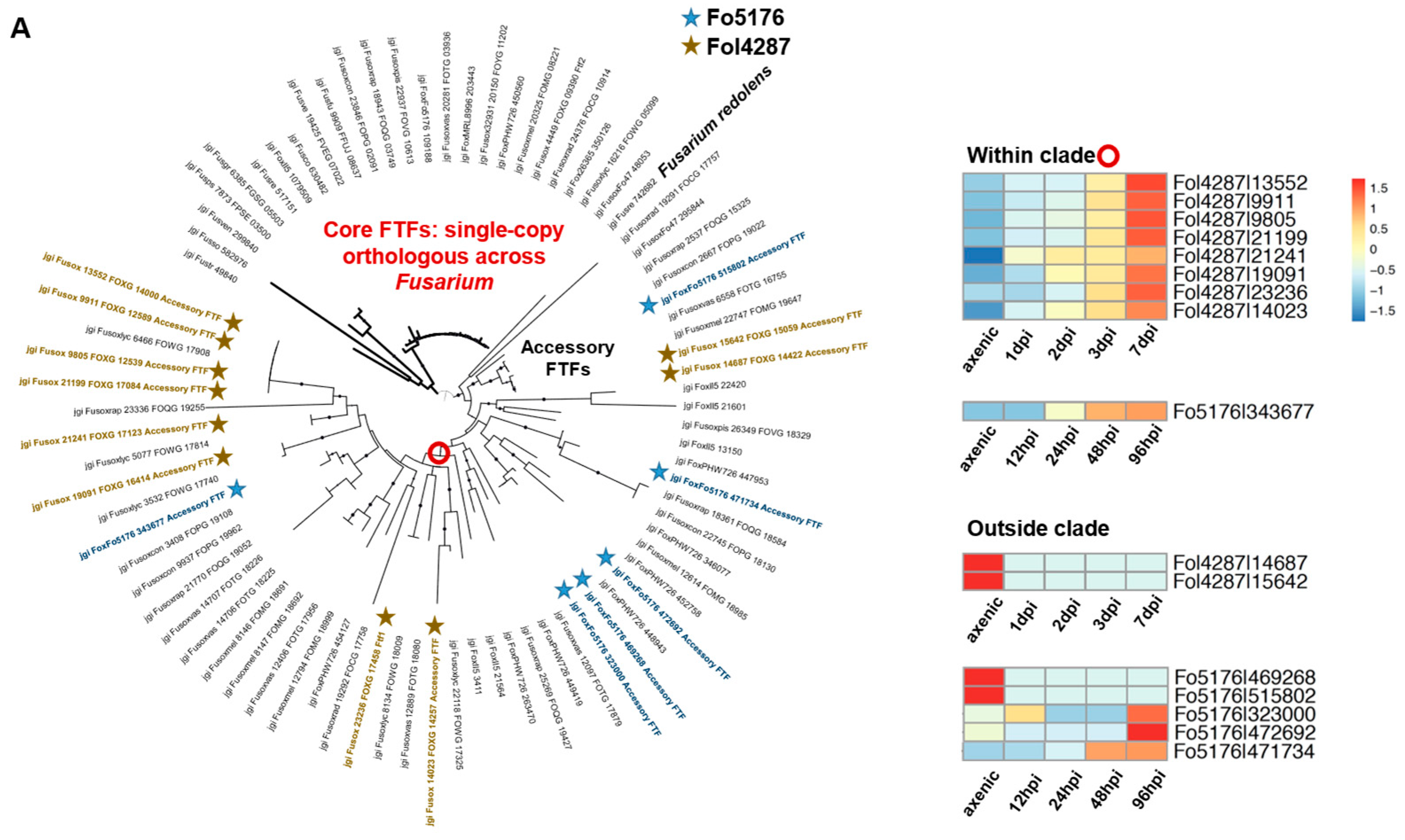
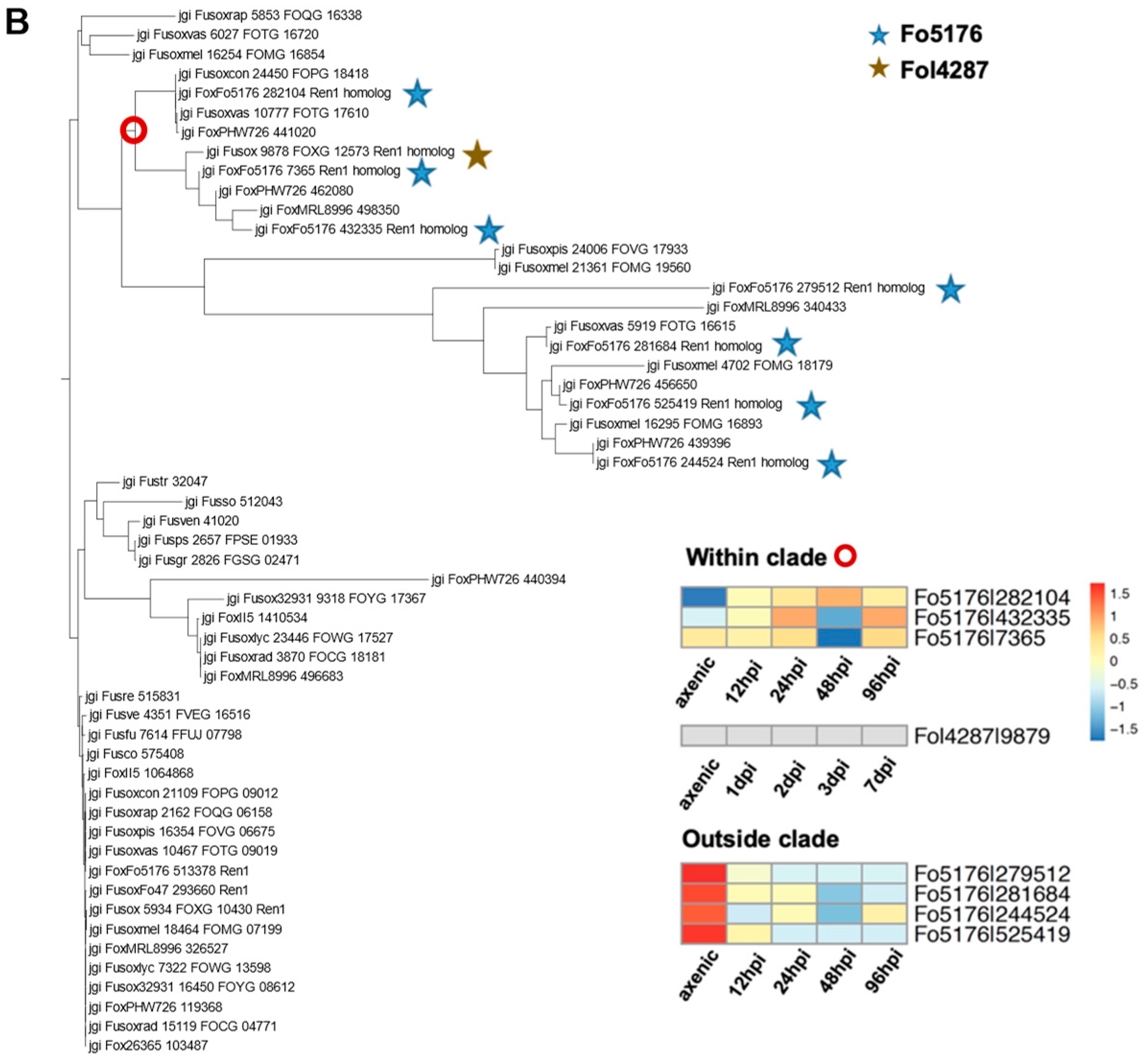
3.5. Orthologous Survey of TF Families That Were Manually Curated
3.6. Transcriptome Analysis to Probe the Essential TFs during Host Colonization
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, L.-J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium Pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.-J.; Shea, T.; Young, S.; Zeng, Q.; Kistler, H.C. Genome Sequence of Fusarium oxysporum f. sp. melonis Strain NRRL 26406, a Fungus Causing Wilt Disease on Melon. Genome Announc. 2014, 2, e00730-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michielse, C.B.; Rep, M. Pathogen profile update: Fusarium oxysporum. Mol. Plant Pathol. 2009, 10, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Ploetz, R.C. Fusarium Wilt of Banana. Phytopathology 2015, 105, 1512–1521. [Google Scholar] [CrossRef] [Green Version]
- Edel-Hermann, V.; Lecomte, C. Current Status of Fusarium oxysporum Formae Speciales and Races. Phytopathology 2019, 109, 512–530. [Google Scholar] [CrossRef] [Green Version]
- Pegg, K.G.; Coates, L.M.; O’Neill, W.T.; Turner, D.W. The Epidemiology of Fusarium Wilt of Banana. Front. Plant Sci. 2019, 10, 1395. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Yu, H.; Ma, L.-J. Accessory Chromosomes in Fusarium oxysporum. Phytopathology 2020, 110, 1488–1496. [Google Scholar] [CrossRef]
- Dean, R.; Van Kan, J.A.L.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology: Top 10 fungal pathogens. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.Z.; Ahmad, K.; Siddiqui, Y.; Saad, N.; Hun, T.G.; Mohd Hata, E.; Rashed, O.; Hossain, M.I.; Kutawa, A.B. First Report of Fusarium wilt disease on Watermelon Caused by Fusarium oxysporum f. sp. niveum (FON) in Malaysia. Plant Dis. 2021; Online ahead of print. [Google Scholar]
- Viljoen, A.; Mostert, D.; Chiconela, T.; Beukes, I.; Fraser, C.; Dwyer, J.; Murray, H.; Amisse, J.; Matabuana, E.L.; Tazan, G.; et al. Occurrence and spread of the banana fungus Fusarium oxysporum f. sp. cubense TR4 in Mozambique. S. Afr. J. Sci. 2020, 116, 1–11. [Google Scholar] [CrossRef]
- Halpern, H.C.; Bell, A.A.; Wagner, T.A.; Liu, J.; Nichols, R.L.; Olvey, J.; Woodward, J.E.; Sanogo, S.; Jones, C.A.; Chan, C.T.; et al. First Report of Fusarium Wilt of Cotton Caused by Fusarium oxysporum f. sp. vasinfectum Race 4 in Texas, USA. Plant Disease 2018, 102, 446. [Google Scholar] [CrossRef]
- Yu, H.; Ayhan, D.H.; Martínez-Soto, D.; Cochavi, S.M.; Ma, L.-J. Accessory Chromosomes of the Fusarium Oxysporum Species Complex and Their Contribution to Host Niche Adaptation. In Plant Relationships, The Mycota; Scott, B., Mesarich, C., Eds.; Springer International Publishing: Cham, Switzerland, 2023; pp. 371–388. [Google Scholar]
- Armstrong, G.M.; Armstrong, J.K. Formae speciales and races of Fusarium oxysporum causing wilt disease. In Fusarium: Disease, Biology, and Taxonomy; Nelson, P.E., Toussoun, T.A., Cook, R.J., Eds.; Pennsylvania State University Press: University Park, PA, USA, 1981; pp. 391–399. [Google Scholar]
- Kredics, L.; Narendran, V.; Shobana, C.S.; Vágvölgyi, C.; Manikandan, P.; Indo-Hungarian Fungal Keratitis Working Group. Filamentous fungal infections of the cornea: A global overview of epidemiology and drug sensitivity. Mycoses 2015, 58, 243–260. [Google Scholar] [CrossRef]
- Hassan, A.S.; Al-Hatmi AM, S.; Shobana, C.S.; van Diepeningen, A.D.; Kredics, L.; Vágvölgyi, C.; Homa, M.; Meis, J.F.; de Hoog, G.S.; Narendran, V.; et al. Antifungal Susceptibility and Phylogeny of Opportunistic Members of the Genus Fusarium Causing Human Keratomycosis in South India. Med. Myco. 2016, 54, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Michielse, C.B.; van Wijk, R.; Reijnen, L.; Manders EM, M.; Boas, S.; Olivain, C.; Alabouvette, C.; Rep, M. The Nuclear Protein Sge1 of Fusarium oxysporum Is Required for Parasitic Growth. PLoS Pathog. 2009, 5, e1000637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Does, H.C.; Fokkens, L.; Yang, A.; Schmidt, S.M.; Langereis, L.; Lukasiewicz, J.M.; Hughes, T.R.; Rep, M. Transcription Factors Encoded on Core and Accessory Chromosomes of Fusarium oxysporum Induce Expression of Effector Genes. PLoS Genet. 2016, 12, e1006401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yang, H.; Turra, D.; Zhou, S.; Ayhan, D.H.; DeIulio, G.A.; Guo, L.; Broz, K.; Wiederhold, N.; Coleman, J.J.; et al. The genome of opportunistic fungal pathogen Fusarium oxysporum carries a unique set of lineage-specific chromosomes. Commun. Biol. 2020, 3, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; Riley, R.; Salamov, A.; Zhao, X.; Korzeniewski, F.; et al. MycoCosm portal: Gearing up for 1000 fungal genomes. Nucl. Acids Res. 2014, 42, D699–D704. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5, genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Park, J.; Jang, S.; Kim, S.; Kong, S.; Choi, J.; Ahn, K.; Kim, J.; Lee, S.; Kim, S.; et al. FTFD: An informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 2008, 24, 1024–1025. [Google Scholar] [CrossRef] [Green Version]
- Shelest, E. Transcription Factors in Fungi: TFome Dynamics, Three Major Families, and Dual-Specificity TFs. Front. Genet. 2017, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Yu, H.; Wang, B.; Vescio, K.; DeIulio, G.A.; Yang, H.; Berg, A.; Zhang, L.; Edel-Hermann, V.; Steinberg, C.; et al. Metatranscriptomic Comparison of Endophytic and Pathogenic Fusarium–Arabidopsis Interactions Reveals Plant Transcriptional Plasticity. MPMI 2021, 34, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Redkar, A.; Sabale, M.; Schudoma, C.; Zechmann, B.; Gupta, Y.K.; López-Berges, M.S.; Venturini, G.; Gimenez-Ibanez, S.; Turrà, D.; Solano, R.; et al. Conserved secreted effectors contribute to endophytic growth and multihost plant compatibility in a vascular wilt fungus. Plant Cell. 2022, 34, 3214–3232. [Google Scholar] [CrossRef]
- Cheng, C.-Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11, a complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017, 89, 789–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokkens, L.; Guo, L.; Dora, S.; Wang, B.; Ye, K.; Sánchez-Rodríguez, C.; Croll, D. A Chromosome-Scale Genome Assembly for the Fusarium oxysporum Strain Fo5176 To Establish a Model Arabidopsis-Fungal Pathosystem. G3 Genes|Genomes|Genet 2020, 7, 3549–3555. [Google Scholar] [CrossRef]
- Wang, B.; Yu, H.; Jia, Y.; Dong, Q.; Steinberg, C.; Alabouvette, C.; Edel-Hermann, V.; Kistler, H.C.; Ye, K.; Ma, L.-J.; et al. Chromosome-Scale Genome Assembly of Fusarium oxysporum Strain Fo47, a Fungal Endophyte and Biocontrol Agent. MPMI 2020, 33, 1108–1111. [Google Scholar] [CrossRef]
- Ma, L.-J.; van der Does, H.C.; Borkovich, K.A.; Coleman, J.J.; Daboussi, M.-J.; Di Pietro, A.; Dufresne, M.; Freitag, M.; Grabherr, M.; Henrissat, B.; et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 2010, 464, 367–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y. Evolution of The Pathogenic Fusarium oxysporum through the Lens of Comparative Genomics. Ph.D. Thesis, University of Massachusetts Amherst, Amherst, MA, USA, 2019. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7, Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2, New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2, Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5, an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 Genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef] [Green Version]
- Wood, V.; Gwilliam, R.; Rajandream, M.-A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002, 415, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.-J.; Wortman, J.R.; Batzoglou, S.; Lee, S.-I.; Baştürkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesth, T.C.; Nybo, J.L.; Theobald, S.; Frisvad, J.C.; Larsen, T.O.; Nielsen, K.F.; Hoof, J.B.; Brandl, J.; Salamov, A.; Riley, R.; et al. Investigation of inter- and intraspecies variation through genome sequencing of Aspergillus section Nigri. Nat. Genet. 2018, 50, 1688–1695. [Google Scholar] [CrossRef] [Green Version]
- Galagan, J.E.; Calvo, S.E.; Borkovich, K.A.; Selker, E.U.; Read, N.D.; Jaffe, D.; FitzHugh, W.; Ma, L.-J.; Smirnov, S.; Purcell, S.; et al. The genome sequence of the filamentous fungus Neurospora crassa. Nature 2003, 422, 859–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, R.A.; Talbot, N.J.; Ebbole, D.J.; Farman, M.L.; Mitchell, T.K.; Orbach, M.J.; Thon, M.; Kulkarni, R.; Xu, J.-R.; Pan, H.; et al. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 2005, 434, 980–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesny, F.; Miyauchi, S.; Thiergart, T.; Pickel, B.; Atanasova, L.; Karlsson, M.; Hüttel, B.; Barry, K.W.; Haridas, S.; Chen, C.; et al. Genetic determinants of endophytism in the Arabidopsis root mycobiome. Nat. Commun. 2021, 12, 7227. [Google Scholar] [CrossRef]
- Gardiner, D.M.; McDonald, M.C.; Covarelli, L.; Solomon, P.S.; Rusu, A.G.; Marshall, M.; Kazan, K.; Chakraborty, S.; McDonald, B.A.; Manners, J.M. Comparative Pathogenomics Reveals Horizontally Acquired Novel Virulence Genes in Fungi Infecting Cereal Hosts. PLoS Pathog. 2012, 8, e1002952. [Google Scholar] [CrossRef] [Green Version]
- Cuomo, C.A.; Guldener, U.; Xu, J.-R.; Trail, F.; Turgeon, B.G.; Di Pietro, A.; Walton, J.D.; Ma, L.-J.; Baker, S.E.; Rep, M.; et al. The Fusarium graminearum Genome Reveals a Link Between Localized Polymorphism and Pathogen Specialization. Science 2007, 317, 1400–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiemann, P.; Sieber CM, K.; Bargen KW von Studt, L.; Niehaus, E.-M.; Espino, J.J.; Huß, K.; Michielse, C.B.; Albermann, S.; Wagner, D.; Bergner, S.V.; et al. Deciphering the Cryptic Genome: Genome-wide Analyses of the Rice Pathogen Fusarium fujikuroi Reveal Complex Regulation of Secondary Metabolism and Novel Metabolites. PLoS Pathog. 2013, 9, e1003475. [Google Scholar] [CrossRef] [Green Version]
- DeIulio, G.A.; Guo, L.; Zhang, Y.; Goldberg, J.M.; Kistler, H.C.; Ma, L.-J. Kinome Expansion in the Fusarium oxysporum Species Complex Driven by Accessory Chromosomes; Mitchell, A.P., Ed.; mSphere: Washington, DC, USA, 2018; Volume 3, p. e00231-18. [Google Scholar]
- Yang, H. Accessory Genes Contribute to Rewiring the Transcriptional Network in Fusarium oxysporum. Ph.D. Thesis, University of Massachusetts Amherst, Amhert, MA, USA, 2020. [Google Scholar]
- Yu, H.; Ayhan, D.H.; Diener, A.C.; Ma, L.-J. Genome Sequence of Fusarium oxysporum f. sp. matthiolae, a Brassicaceae Pathogen. MPMI 2020, 33, 569–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, A.H.; Sharma, M.; Thatcher, L.F.; Azam, S.; Hane, J.K.; Sperschneider, J.; Kidd, B.N.; Anderson, J.P.; Ghosh, R.; Garg, G.; et al. Comparative genomics and prediction of conditionally dispensable sequences in legume–infecting Fusarium oxysporum formae speciales facilitates identification of candidate effectors. BMC Genom. 2016, 17, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latrick, C.M.; Marek, M.; Ouararhni, K.; Papin, C.; Stoll, I.; Ignatyeva, M.; Obri, A.; Ennifar, E.; Dimitrov, S.; Romier, C.; et al. Molecular basis and specificity of H2A.Z–H2B recognition and deposition by the histone chaperone YL1. Nat. Struct. Mol. Biol. 2016, 23, 309–316. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Narita, T.; Inukai, N.; Wada, T.; Handa, H. SPT Genes: Key Players in the Regulation of Transcription, Chromatin Structure and Other Cellular Processes. J. Biochem. 2001, 129, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.M.; Winston, F. SPT20/ADA5 encodes a novel protein functionally related to the TATA-binding protein and important for transcription in Saccharomyces cerevisiae. Mol. Cell Biol. 1996, 16, 3206–3213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.R. TBP-associated factors (TAF II s): Multiple, selective transcriptional mediators in common complexes. Trends Biochem. Sci. 2000, 25, 59–63. [Google Scholar] [CrossRef]
- Fribourg, S.; Kellenberger, E.; Rogniaux, H.; Poterszman, A.; Van Dorsselaer, A.; Thierry, J.-C.; Egly, J.-M.; Moras, D.; Kieffer, B. Structural Characterization of the Cysteine-rich Domain of TFIIH p44 Subunit. J. Biol. Chem. 2000, 275, 31963–31971. [Google Scholar] [CrossRef] [Green Version]
- Goppelt, A.; Stelzer, G.; Lottspeich, F.; Meisterernst, M. A mechanism for repression of class II gene transcription through specific binding of NC2 to TBP-promoter complexes via heterodimeric histone fold domains. EMBO J. 1996, 15, 3105–3116. [Google Scholar] [CrossRef]
- Chalabi Hagkarim, N.; Grand, R.J. The Regulatory Properties of the Ccr4–Not Complex. Cells 2020, 9, 2379. [Google Scholar] [CrossRef]
- Liang, X.; Shan, S.; Pan, L.; Zhao, J.; Ranjan, A.; Wang, F.; Zhang, Z.; Huang, Y.; Feng, H.; Wei, D.; et al. Structural basis of H2A.Z recognition by SRCAP chromatin-remodeling subunit YL1. Nat. Struct. Mol. Biol. 2016, 23, 317–323. [Google Scholar] [CrossRef]
- Kamura, T.; Burian, D.; Khalili, H.; Schmidt, S.L.; Sato, S.; Liu, W.J.; Conrad, M.N.; Conaway, R.C.; Conaway, J.W.; Shilatifard, A. Cloning and characterization of ELL-associated proteins EAP45 and EAP20. a role for yeast EAP-like proteins in regulation of gene expression by glucose. J. Biol. Chem. 2001, 276, 16528–16533. [Google Scholar] [CrossRef]
- Vuorio, T.; Maity, S.N.; de Crombrugghe, B. Purification and molecular cloning of the “A” chain of a rat heteromeric CCAAT-binding protein. Sequence identity with the yeast HAP3 transcription factor. J. Biol. Chem. 1990, 265, 22480–22486. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.M.; Fikes, J.D.; Guarente, L. A cDNA encoding a human CCAAT-binding protein cloned by functional complementation in yeast. Proc. Natl. Acad. Sci. USA 1991, 88, 1968–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milkereit, P.; Gadal, O.; Podtelejnikov, A.; Trumtel, S.; Gas, N.; Petfalski, E.; Tollervey, D.; Mann, M.; Hurt, E.; Tschochner, H. Maturation and Intranuclear Transport of Pre-Ribosomes Requires Noc Proteins. Cell 2001, 105, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromont-Racine, M.; Senger, B.; Saveanu, C.; Fasiolo, F. Ribosome assembly in eukaryotes. Gene 2003, 313, 17–42. [Google Scholar] [CrossRef]
- Edskes, H.K.; Ohtake, Y.; Wickner, R.B. Mak21p of Saccharomyces cerevisiae, a Homolog of Human CAATT-binding Protein, Is Essential for 60 S Ribosomal Subunit Biogenesis. J. Biol. Chem. 1998, 273, 28912–28920. [Google Scholar] [CrossRef] [Green Version]
- Xin, C.; Zhang, J.; Nian, S.; Wang, G.; Wang, Z.; Song, Z.; Ren, G. Analogous and Diverse Functions of APSES-Type Transcription Factors in the Morphogenesis of the Entomopathogenic Fungus Metarhizium rileyi. Appl. Environ. Microbiol. 2020, 86, e02928-19. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Heitman, J. Sok2 Regulates Yeast Pseudohyphal Differentiation via a Transcription Factor Cascade That Regulates Cell-Cell Adhesion. Mol. Cell Biol. 2000, 20, 8364–8372. [Google Scholar] [CrossRef] [Green Version]
- Gimeno, C.J.; Fink, G.R. Induction of Pseudohyphal Growth by Overexpression of PHD1, a Saccharomyces cerevisiae Gene Related to Transcriptional Regulators of Fungal Developmentt. Mol. Cell. Biol. 1994, 14, 13. [Google Scholar]
- Lysøe, E.; Pasquali, M.; Breakspear, A.; Kistler, H.C. The Transcription Factor FgStuAp Influences Spore Development, Pathogenicity, and Secondary Metabolism in Fusarium graminearum. MPMI 2011, 24, 54–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, C.; Moll, T.; Neuberg, M.; Ahorn, H.; Nasmyth, K. A Role for the Transcription Factors Mbp1 and Swi4 in Progression from G1 to S Phase. Science 1993, 261, 1551–1557. [Google Scholar] [CrossRef]
- Son, M.; Lee, Y.; Kim, K.-H. The Transcription Cofactor Swi6 of the Fusarium graminearum Is Involved in Fusarium Graminearum Virus 1 Infection-Induced Phenotypic Alterations. Plant Pathol. J. 2016, 32, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikashige, Y.; Yamane, M.; Okamasa, K.; Tsutsumi, C.; Kojidani, T.; Sato, M.; Haraguchi, T.; Hiraoka, Y. Membrane proteins Bqt3 and -4 anchor telomeres to the nuclear envelope to ensure chromosomal bouquet formation. J. Cell Biol. 2009, 187, 413–427. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, M.; Nikawa, J.-I. The Saccharomyces cerevisiae Isw2p-Itc1p Complex Represses INO1 Expression and Maintains Cell Morphology. J. Bacteriol. 2001, 183, 4985–4993. [Google Scholar] [CrossRef] [Green Version]
- Lubelsky, Y.; Reuven, N.; Shaul, Y. Autorepression of Rfx1 Gene Expression: Functional Conservation from Yeast to Humans in Response to DNA Replication Arrest. Mol. Cell. Biol. 2005, 25, 10665–10673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, K.; Son, H.; Lim, J.Y.; Choi, G.J.; Kim, J.-C.; Harris, S.D.; Lee, Y.-W. Transcription Factor RFX1 Is Crucial for Maintenance of Genome Integrity in Fusarium graminearum. Eukaryot. Cell. 2014, 13, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Cairns, B.R.; Lorch, Y.; Li, Y.; Zhang, M.; Lacomis, L.; Erdjument-Bromage, H.; Tempst, P.; Du, J.; Laurent, B.; Kornberg, R.D. RSC, an Essential, Abundant Chromatin-Remodeling Complex. Cell 1996, 87, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.; Huang, J.; Meluh, P.B.; Laurent, B.C. The Yeast RSC Chromatin-Remodeling Complex Is Required for Kinetochore Function in Chromosome Segregation. Mol. Cell Biol. 2003, 23, 3202–3215. [Google Scholar] [CrossRef] [Green Version]
- Olesen, J.T.; Fikes, J.D.; Guarente, L. The Schizosaccharomyces pombe homolog of Saccharomyces cerevisiae HAP2 reveals selective and stringent conservation of the small essential core protein domain. Mol. Cell Biol. 1991, 11, 9. [Google Scholar]
- Ridenour, J.B.; Bluhm, B.H. The HAP complex in Fusarium verticillioides is a key regulator of growth, morphogenesis, secondary metabolism, and pathogenesis. Fungal Genet. Biol. 2014, 69, 52–64. [Google Scholar] [CrossRef]
- Jung, U.S.; Sobering, A.K.; Romeo, M.J.; Levin, D.E. Regulation of the yeast Rlm1 transcription factor by the Mpk1 cell wall integrity MAP kinase: Reporters for cell wall integrity signalling. Mol. Microbiol. 2002, 46, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, C.S.; Shim, W.-B. The role of MADS-box transcription factors in secondary metabolism and sexual development in the maize pathogen Fusarium verticillioides. Microbiology 2013, 159, 2259–2268. [Google Scholar] [CrossRef] [Green Version]
- Martin, T.; Lu, S.-W.; Tilbeurgh, H.; van Ripoll, D.R.; Dixelius, C.; Turgeon, B.G.; Debuchy, R. Tracing the Origin of the Fungal α1 Domain Places Its Ancestor in the HMG-Box Superfamily: Implication for Fungal Mating-Type Evolution. PLoS ONE 2010, 5, e15199. [Google Scholar] [CrossRef] [Green Version]
- Arie, T.; Kaneko, I.; Yoshida, T.; Noguchi, M.; Nomura, Y.; Yamaguchi, I. Mating-type genes from asexual phytopathogenic ascomycetes Fusarium oxysporum and Alternaria alternata. Mol. Plant Microbe. Interact. 2000, 13, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- Albert, T.K.; Hanzawa, H.; Legtenberg, Y.I.; de Ruwe, M.J.; Heuvel, F.A.V.D.; Collart, M.A.; Boelens, R.; Timmers, H. Identification of a ubiquitin-protein ligase subunit within the CCR4-NOT transcription repressor complex. EMBO J. 2002, 21, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messenguy, F.; Dubois, E. Role of MADS box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 2003, 316, 1–21. [Google Scholar] [CrossRef]
- Ding, Z.; Xu, T.; Zhu, W.; Li, L.; Fu, Q. A MADS-box transcription factor FoRlm1 regulates aerial hyphal growth, oxidative stress, cell wall biosynthesis and virulence in Fusarium oxysporum f. sp. cubense. Fungal. Biol. 2020, 124, 183–193. [Google Scholar] [CrossRef]
- Feder, M.E.; Hofmann, G.E. Heat-Shock Proteins, Molecular Chaperones, and the Stress Response: Evolutionary and Ecological Physiology. Annu. Rev. Physiol. 1999, 61, 243–282. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhou, X.; Kong, L.; Wang, Y.; Zhang, H.; Zhu, H.; Mitchell, T.K.; Dean, R.A.; Xu, J.-R. MoSfl1 Is Important for Virulence and Heat Tolerance in Magnaporthe oryzae. PLoS ONE 2011, 6, e19951. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Zhang, S.; Zhang, Q.; Tao, Y.; Wang, C.; Xu, J.-R. FgSKN 7 and FgATF 1 have overlapping functions in ascosporogenesis, pathogenesis and stress responses in Fusarium graminearum: Overlapping functions between FgSKN7 and FgATF1. Environ. Microbiol. 2015, 17, 1245–1260. [Google Scholar] [CrossRef]
- Paré, A.; Kim, M.; Juarez, M.T.; Brody, S.; McGinnis, W. The Functions of Grainy Head-Like Proteins in Animals and Fungi and the Evolution of Apical Extracellular Barriers. PLoS ONE 2012, 7, e36254. [Google Scholar] [CrossRef] [Green Version]
- Schleif, R. AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS Microbiol. Rev. 2010, 34, 779–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallegos, M.-T.; Michán, C.; Ramos, J.L. The XylS/AraC family of regulators. Nucl. Acids Res. 1993, 21, 807–810. [Google Scholar] [CrossRef] [Green Version]
- Bustos, S.A.; Schleif, R.F. Functional domains of the AraC protein. Proc. Natl. Acad. Sci. USA 1993, 90, 5638–5642. [Google Scholar] [CrossRef] [Green Version]
- Siegmund, T.; Lehmann, M. The Drosophila Pipsqueak protein defines a new family of helix-turn-helix DNA-binding proteins. Dev. Genes Evol. 2002, 212, 152–157. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, S.; Larochelle, M.; Turcotte, B. A Fungal Family of Transcriptional Regulators: The Zinc Cluster Proteins. Microbiol. Mol. Biol. Rev. 2006, 70, 583–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niño-Sánchez, J.; Casado-Del Castillo, V.; Tello, V.; De Vega-Bartol, J.J.; Ramos, B.; Sukno, S.A.; Díaz Mínguez, J.M. The FTF gene family regulates virulence and expression of SIX effectors in Fusarium oxysporum. Mol. Plant Pathol. 2016, 17, 1124–1139. [Google Scholar] [CrossRef] [Green Version]
- Ramos, B.; Alves-Santos, F.M.; García-Sánchez, M.A.; Martín-Rodrigues, N.; Eslava, A.P.; Díaz-Mínguez, J.M. The gene coding for a new transcription factor (ftf1) of Fusarium oxysporum is only expressed during infection of common bean. Fungal Genet. Biol. 2007, 44, 864–876. [Google Scholar] [CrossRef]
- Zuriegat, Q.; Zheng, Y.; Liu, H.; Wang, Z.; Yun, Y. Current progress on pathogenicity-related transcription factors in Fusarium oxysporum. Mol. Plant Pathol. 2021, 22, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; An, B.; Guo, Y.; Hou, X.; Luo, H.; He, C.; Wang, Q. Label free proteomics and systematic analysis of secretome reveals effector candidates regulated by SGE1 and FTF1 in the plant pathogen Fusarium oxysporum f. sp. cubense tropical race 4. BMC Genom. 2020, 21, 275. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.L.M.; Di Pietro, A.; Ruiz-Roldán, C.; Roncero, M.I.G. Ctf1, a transcriptional activator of cutinase and lipase genes in Fusarium oxysporum is dispensable for virulence. Mol. Plant Pathol. 2008, 9, 293–304. [Google Scholar] [CrossRef]
- Imazaki, I.; Kurahashi, M.; Iida, Y.; Tsuge, T. Fow2, a Zn(II)2Cys6-type transcription regulator, controls plant infection of the vascular wilt fungus Fusarium oxysporum. Mol. Microbiol. 2007, 63, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Calero-Nieto, F.; Di Pietro, A.; Roncero MI, G.; Hera, C. Role of the Transcriptional Activator XlnR of Fusarium oxysporum in Regulation of Xylanase Genes and Virulence. MPMI 2007, 20, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Jonkers, W.; Xayamongkhon, H.; Haas, M.; Olivain, C.; van der Does, H.C.; Broz, K.; Rep, M.; Alabouvette, C.; Steinberg, C.; Kistler, H.C. EBR1 genomic expansion and its role in virulence of Fusarium species: EBR1 in virulence of Fusarium species. Environ. Microbiol. 2014, 16, 1982–2003. [Google Scholar] [CrossRef]
- Fedotova, A.A.; Bonchuk, A.N.; Mogila, V.A.; Georgiev, P.G. C2H2 Zinc Finger Proteins: The Largest but Poorly Explored Family of Higher Eukaryotic Transcription Factors. Acta Nat. 2017, 9, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Yun, Y.; Zhou, X.; Yang, S.; Wen, Y.; You, H.; Zheng, Y.; Norvienyeku, J.; Shim, W.-B.; Wang, Z. Fusarium oxysporum f. sp. lycopersici C2H2 transcription factor FolCzf1 is required for conidiation, fusaric acid production, and early host infection. Curr. Genet. 2019, 65, 773–783. [Google Scholar] [CrossRef]
- Ruiz-Roldán, C.; Pareja-Jaime, Y.; González-Reyes, J.A.; GRoncero, M.I. The Transcription Factor Con7-1 Is a Master Regulator of Morphogenesis and Virulence in Fusarium oxysporum. MPMI 2015, 28, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Caracuel, Z.; Roncero, M.I.G.; Espeso, E.A.; González-Verdejo, C.I.; García-Maceira, F.I.; Di Pietro, A. The pH signalling transcription factor PacC controls virulence in the plant pathogen Fusarium oxysporum: PacC controls virulence in Fusarium. Mol. Microbiol. 2003, 48, 765–779. [Google Scholar] [CrossRef]
- López-Berges, M.S. ZafA-mediated regulation of zinc homeostasis is required for virulence in the plant pathogen Fusarium oxysporum. Mol. Plant Pathol. 2020, 21, 244–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asunción García-Sánchez, M.; Martín-Rodrigues, N.; Ramos, B.; de Vega-Bartol, J.J.; Perlin, M.H.; Díaz-Mínguez, J.M. fost12, the Fusarium oxysporum homolog of the transcription factor Ste12, is upregulated during plant infection and required for virulence. Fungal Genet. Biol. 2010, 47, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Rispail, N.; Di Pietro, A. Fusarium oxysporum Ste12 Controls Invasive Growth and Virulence Downstream of the Fmk1 MAPK Cascade. MPMI 2009, 22, 830–839. [Google Scholar] [CrossRef] [Green Version]
- Bader, A.G.; Vogt, P.K. Leucine Zipper Transcription Factors: bZIP Proteins. In Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine; Springer: Berlin/Heidelberg, Germany, 2006; pp. 964–967. [Google Scholar]
- Li, X.; Han, X.; Liu, Z.; He, C. The function and properties of the transcriptional regulator COS1 in Magnaporthe oryzae. Fungal. Biol. 2013, 117, 239–249. [Google Scholar] [CrossRef]
- López-Berges, M.S.; Capilla, J.; Turrà, D.; Schafferer, L.; Matthijs, S.; Jöchl, C.; Cornelis, P.; Guarro, J.; Haas, H.; Di Pietro, A. HapX-Mediated Iron Homeostasis Is Essential for Rhizosphere Competence and Virulence of the Soilborne Pathogen Fusarium oxysporum. Plant Cell 2012, 24, 3805–3822. [Google Scholar] [CrossRef] [Green Version]
- López-Berges, M.S.; Rispail, N.; Prados-Rosales, R.C.; Di Pietro, A. A Nitrogen Response Pathway Regulates Virulence Functions in Fusarium oxysporum via the Protein Kinase TOR and the bZIP Protein MeaB. Plant Cell 2010, 22, 2459–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Park, S.-Y.; Kim, K.S.; Rho, H.-S.; Chi, M.-H.; Choi, J.; Park, J.; Kong, S.; Park, J.; Goh, J.; et al. Homeobox Transcription Factors Are Required for Conidiation and Appressorium Development in the Rice Blast Fungus Magnaporthe oryzae. PLoS Genet. 2009, 5, e1000757. [Google Scholar] [CrossRef] [Green Version]
- Honjo, M.; Nakayama, A.; Fukazawa, K.; Kawamura, K.; Ando, K.; Hori, M.; Furutani, Y. A novel Bacillus subtilis gene involved in negative control of sporulation and degradative-enzyme production. J. Bacteriol. 1990, 172, 1783–1790. [Google Scholar] [CrossRef] [Green Version]
- Pierce, M.; Benjamin, K.R.; Montano, S.P.; Georgiadis, M.M.; Winter, E.; Vershon, A.K. Sum1 and Ndt80 Proteins Compete for Binding to Middle Sporulation Element Sequences That Control Meiotic Gene Expression. Mol. Cell Biol. 2003, 23, 4814–4825. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, D.; Yang, Y.; Lacefield, S. Positive Feedback of NDT80 Expression Ensures Irreversible Meiotic Commitment in Budding Yeast. PLoS Genet. 2014, 10, e1004398. [Google Scholar] [CrossRef] [Green Version]
- Doyle, C.E.; Kitty Cheung, H.Y.; Spence, K.L.; Saville, B.J. Unh1, an Ustilago maydis Ndt80-like protein, controls completion of tumor maturation, teliospore development, and meiosis. Fungal. Genet. Biol. 2016, 94, 54–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepworth, S.R.; Friesen, H.; Segall, J. NDT80 and the Meiotic Recombination Checkpoint Regulate Expression of Middle Sporulation-Specific Genes in Saccharomyces cerevisiae. Mol. Cell Biol. 1998, 18, 5750–5761. [Google Scholar] [CrossRef] [Green Version]
- Jones, S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004, 6, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahadoor, A.; Brauer, E.K.; Bosnich, W.; Schneiderman, D.; Johnston, A.; Aubin, Y.; Blackwell, B.; Melanson, J.E.; Harris, L.J. Gramillin A and B: Cyclic Lipopeptides Identified as the Nonribosomal Biosynthetic Products of Fusarium graminearum. J. Am. Chem. Soc. 2018, 140, 16783–16791. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, Y.; Wu, Z.; Li, N.; Chen, Y.; Qin, T.; Geng, H.; Xiong, L.; Liu, D. A Novel Sterol Regulatory Element-Binding Protein Gene (sreA) Identified in Penicillium digitatum Is Required for Prochloraz Resistance, Full Virulence and erg11 (cyp51) Regulation. PLoS ONE 2015, 10, e0117115. [Google Scholar] [CrossRef] [Green Version]
- Donczew, R.; Warfield, L.; Pacheco, D.; Erijman, A.; Hahn, S. Two roles for the yeast transcription coactivator SAGA and a set of genes redundantly regulated by TFIID and SAGA. eLife 2020, 9, e50109. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Araki, H. Noncompetitive Counteractions of DNA Polymerase ε and ISW2/yCHRAC for Epigenetic Inheritance of Telomere Position Effect in Saccharomyces cerevisiae. Mol. Cell Biol. 2004, 24, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Braun, M.A.; Costa, P.J.; Crisucci, E.M.; Arndt, K.M. Identification of Rkr1, a Nuclear RING Domain Protein with Functional Connections to Chromatin Modification in Saccharomyces cerevisiae. Mol. Cell Biol. 2007, 27, 2800–2811. [Google Scholar] [CrossRef] [Green Version]
- Iwahara, J. The structure of the Dead ringer-DNA complex reveals how AT-rich interaction domains (ARIDs) recognize DNA. EMBO J. 2002, 21, 1197–1209. [Google Scholar] [CrossRef] [Green Version]
- Breeden, L.; Nasmyth, K. Cell cycle control of the yeast HO gene: Cis- and Trans-acting regulators. Cell 1987, 48, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, J.N.; Brown, S.A.; Clark, C.D.; Winston, F. Evidence that SNF2/SWI2 and SNF5 activate transcription in yeast by altering chromatin structure. Genes Dev. 1992, 6, 2288–2298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rispail, N.; Di Pietro, A. The homeodomain transcription factor Ste12, connecting fungal MAPK signaling to plant pathogenicity. Commun. Integr. Biol. 2010, 3, 327–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gancedo, J.M. Control of pseudohyphae formation in Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2001, 25, 107–123. [Google Scholar] [CrossRef]
- Song, Z.; Krishna, S.; Thanos, D.; Strominger, J.L.; Ono, S.J. A novel cysteine-rich sequence-specific DNA-binding protein interacts with the conserved X-box motif of the human major histocompatibility complex class II genes via a repeated Cys-His domain and functions as a transcriptional repressor. J. Exp. Med. 1994, 180, 1763–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddock, J.R.; Weidenhammer, E.M.; Adams, C.C.; Lunz, R.L.; Woolford, J.L. Extragenic suppressors of Saccharomyces cerevisiae prp4 mutations identify a negative regulator of PRP genes. Genetics 1994, 136, 833–847. [Google Scholar] [CrossRef]
- Braglia, P.; Dugas, S.L.; Donze, D.; Dieci, G. Requirement of Nhp6 Proteins for Transcription of a Subset of tRNA Genes and Heterochromatin Barrier Function in Saccharomyces cerevisiae. Mol. Cell Biol. 2007, 27, 1545–1557. [Google Scholar] [CrossRef] [Green Version]
- Vizoso-Vázquez, Á.; Lamas-Maceiras, M.; Becerra, M.; González-Siso, M.I.; Rodríguez-Belmonte, E.; Cerdán, M.E. Ixr1p and the control of the Saccharomyces cerevisiae hypoxic response. Appl. Microbiol. Biotechnol. 2012, 94, 173–184. [Google Scholar] [CrossRef]
- Kim, H.-J.; Lee, K.-L.; Kim, K.-D.; Roe, J.-H. The iron uptake repressor Fep1 in the fission yeast binds Fe-S cluster through conserved cysteines. Biochem. Biophys. Res. Commun. 2016, 478, 187–192. [Google Scholar] [CrossRef]
- Takemaru, K.; Li, F.-Q.; Ueda, H.; Hirose, S. Multiprotein bridging factor 1 (MBF1) is an evolutionarily conserved transcriptional coactivator that connects a regulatory factor and TATA element-binding protein. Proc. Natl. Acad. Sci. USA 1997, 94, 7251–7256. [Google Scholar] [CrossRef] [Green Version]
- Ohara, T.; Inoue, I.; Namiki, F.; Kunoh, H.; Tsuge, T. REN1 Is Required for Development of Microconidia and Macroconidia, but Not of Chlamydospores, in the Plant Pathogenic Fungus Fusarium oxysporum. Genetics 2004, 166, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Li, Y.; Yue, X.; Wang, C.; Que, Y.; Kong, D.; Ma, Z.; Talbot, N.J.; Wang, Z. Two Novel Transcriptional Regulators Are Essential for Infection-related Morphogenesis and Pathogenicity of the Rice Blast Fungus Magnaporthe oryzae. PLoS Pathog. 2011, 7, e1002385. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Que, Y.; Xu, L.; Deng, S.; Peng, Y.; Talbot, N.J.; Wang, Z. ZNF1 Encodes a Putative C2H2 Zinc-Finger Protein Essential for Appressorium Differentiation by the Rice Blast Fungus Magnaporthe oryzae. Mol. Plant Microbe. Interact. 2016, 29, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Ruiz, G.; Ruiz-Roldán, C.; Roncero, M.I.G. Lipolytic system of the tomato pathogen Fusarium oxysporum f. sp. lycopersici. Mol. Plant Microbe Interact. 2013, 26, 1054–1067. [Google Scholar] [CrossRef] [Green Version]
- Wight, W.D.; Kim, K.-H.; Lawrence, C.B.; Walton, J.D. Biosynthesis and role in virulence of the histone deacetylase inhibitor depudecin from Alternaria brassicicola. Mol. Plant Microbe. Interact. 2009, 22, 1258–1267. [Google Scholar] [CrossRef] [Green Version]
- Jones DA, B.; John, E.; Rybak, K.; Phan HT, T.; Singh, K.B.; Lin, S.-Y.; Solomon, P.S.; Oliver, R.P.; Tan, K.-C. A specific fungal transcription factor controls effector gene expression and orchestrates the establishment of the necrotrophic pathogen lifestyle on wheat. Sci. Rep. 2019, 9, 15884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, M.; Son, H.; Choi, G.J.; Lee, C.; Kim, J.-C.; Kim, H.; Lee, Y.-W. Transcription factor ART1 mediates starch hydrolysis and mycotoxin production in Fusarium graminearum and F. verticillioides. Mol. Plant Pathol. 2016, 17, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Dufresne, M.; Perfect, S.; Pellier, A.-L.; Bailey, J.A.; Langin, T. A GAL4-like Protein Is Involved in the Switch between Biotrophic and Necrotrophic Phases of the Infection Process of Colletotrichum lindemuthianum on Common Bean. Plant Cell 2000, 12, 1579–1590. [Google Scholar] [CrossRef] [Green Version]
- Son, H.; Fu, M.; Lee, Y.; Lim, J.Y.; Min, K.; Kim, J.-C.; Choi, G.J.; Lee, Y.-W. A novel transcription factor gene FHS1 is involved in the DNA damage response in Fusarium graminearum. Sci. Rep. 2016, 6, 21572. [Google Scholar] [CrossRef] [Green Version]
- Ridenour, J.B.; Bluhm, B.H. The novel fungal-specific gene FUG1 has a role in pathogenicity and fumonisin biosynthesis in Fusarium verticillioides. Mol. Plant Pathol. 2017, 18, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Zhang, C.; Wu, C.; Sun, P.; Hou, R.; Liu, H.; Wang, C.; Xu, J.-R. TRI6 and TRI10 play different roles in the regulation of deoxynivalenol (DON) production by cAMP signalling in Fusarium graminearum. Environ. Microbiol. 2016, 18, 3689–3701. [Google Scholar] [CrossRef]
- Schumacher, J.; Simon, A.; Cohrs, K.C.; Viaud, M.; Tudzynski, P. The Transcription Factor BcLTF1 Regulates Virulence and Light Responses in the Necrotrophic Plant Pathogen Botrytis cinerea. PLoS Genet. 2014, 10, e1004040. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Pokhrel, A.; Coleman, J.J. The Extracellular Superoxide Dismutase Sod5 from Fusarium oxysporum Is Localized in Response to External Stimuli and Contributes to Fungal Pathogenicity. Front. Plant Sci. 2021, 12, 608861. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Mendoza, A.; Eskova, A.; Weise, C.; Czajkowski, R.; Kahmann, R. Hap2 regulates the pheromone response transcription factor prf1 in Ustilago maydis. Mol. Microbiol. 2009, 72, 683–698. [Google Scholar] [CrossRef] [PubMed]
- de Vega-Bartol, J.J.; Martín-Dominguez, R.; Ramos, B.; García-Sánchez, M.-A.; Díaz-Mínguez, J.M. New Virulence Groups in Fusarium oxysporum f. sp. phaseoli: The Expression of the Gene Coding for the Transcription Factor ftf1 Correlates with Virulence. Phytopathology 2011, 101, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Li, G.; Lin, C.; He, C. Conidiophore Stalk-less1 Encodes a Putative Zinc-Finger Protein Involved in the Early Stage of Conidiation and Mycelial Infection in Magnaporthe oryzae. MPMI 2009, 22, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Soto, D.; Yu, H.; Allen, K.S.; Ma, L.-J. Differential colonization of the plant vasculature between endophytic versus pathogenic Fusarium oxysporum strains. MPMI 2022, 36, 4–13. [Google Scholar] [CrossRef]
- Baumgart, L.A.; Lee, J.E.; Salamov, A.; Dilworth, D.J.; Na, H.; Mingay, M.; Blow, M.J.; Zhang, Y.; Yoshinaga, Y.; Daum, C.G.; et al. Persistence and plasticity in bacterial gene regulation. Nat. Methods 2021, 18, 1499–1505. [Google Scholar] [CrossRef]
- Guo, L.; Ji, M.; Ye, K. Dynamic network inference and association computation discover gene modules regulating virulence, mycotoxin and sexual reproduction in Fusarium graminearum. BMC Genom. 2020, 21, 179. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Zhao, G.; Xu, J.; Kistler, H.C.; Gao, L.; Ma, L. Compartmentalized gene regulatory network of the pathogenic fungus Fusarium graminearum. New Phytol. 2016, 211, 527–541. [Google Scholar] [CrossRef]
- Gordon, T.R. Fusarium oxysporum and the Fusarium Wilt Syndrome. Annu. Rev. Phytopathol. 2017, 55, 23–39. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fungal Species or Strains | MycoCosm Identifier | Genome Size (MB) | No. of Genes | TFome Size | Host | Reference |
|---|---|---|---|---|---|---|
| Saccharomyces cerevisiae | Sacce1 | 12.07 | 6575 | 284 | [38] | |
| Schizosaccharomyces pombe | Schpo1 | 12.61 | 5134 | 228 | [39] | |
| Aspergillus nidulans | Aspnid1 | 30.48 | 10,680 | 635 | [40] | |
| Aspergillus acristatulus | Aspacri1 | 32.59 | 11,221 | 666 | [41] | |
| Neurospora crassa | Neucr2 | 41.04 | 9730 | 447 | [42] | |
| Magnaporthe oryzae | Magor1 | 40.49 | 12,673 | 520 | Rice | [43] |
| Fusarium solani | Fusso1 | 52.93 | 17,656 | 1137 | broad hosts | [44] |
| F. pseudograminearum | Fusps1 | 36.33 | 12,395 | 627 | Wheat | [45] |
| F. graminearum | Fusgr1 | 36.45 | 13,321 | 608 | Wheat | [46] |
| F. venenatum | Fusven1 | 37.45 | 12,845 | 802 | [44] | |
| F. tricinctum | Fustr1 | 43.69 | 14,106 | 925 | Broad hosts | [44] |
| F. verticillioides | Fusve2 | 41.78 | 15,869 | 917 | Corn | [29] |
| F. fujikuroi | Fusfu1 | 43.83 | 14,813 | 901 | Broad hosts | [47] |
| F. redolens | Fusre1 | 52.56 | 17,051 | 1098 | Broad hosts | [44] |
| F. commune | Fusco1 | 48.37 | 15,731 | 1012 | Broad hosts | [44] |
| F. oxysporum f. sp. cubense (II5) | FoxII5 | 49.43 | 16,048 | 1047 | Banana | [32] |
| F. oxysporum f. sp. radicis-lycopersici (CL57) | Fusoxrad1 | 49.36 | 18,238 | 1151 | Tomato | [48] |
| F. oxysporum Fo47 (Fo47) | FusoxFo47_2 | 50.36 | 16,207 | 1082 | [28] | |
| F. oxysporum f. sp. lycopersici (MN25) | Fusoxlyc1 | 48.64 | 17,931 | 1119 | Tomato | [48] |
| F. oxysporum NRRL26365 (NRRL26365) | Fox26365_1 | 48.46 | 16,047 | 1036 | Human | [49] |
| F. oxysporum f. sp. melonis (FoMelon) | Fusoxmel1 | 54.03 | 19,661 | 1219 | Melon | [2] |
| F. oxysporum f. sp. lycopersici (Fol4287) | Fusox2 | 61.36 | 20,925 | 1292 | Tomato | [29] |
| F. oxysporum NRRL32931 (NRRL32931) | Fusox32931 | 47.91 | 17,280 | 1072 | Human | [18] |
| F. oxysporum MRL8996 (MRL8996) | FoxMRL8996 | 50.07 | 16,631 | 1057 | Human | [18] |
| F. oxysporum f. sp. matthiolae (PHW726) | FoxPHW726_1 | 57.22 | 17,996 | 1157 | Brassica | [50] |
| F. oxysporum f. sp. vasinfectum (FoCotton) | Fusoxvas1 | 52.91 | 19,143 | 1189 | Cotton | [48] |
| F. oxysporum f. sp. pisi (HDV247) | Fusoxpis1 | 55.19 | 19,623 | 1229 | Pea | [51] |
| F. oxysporum f. sp. raphani (PHW815) | Fusoxrap1 | 53.5 | 19,306 | 1132 | Brassica | [48] |
| F. oxysporum f. sp. conglutinans (PHW808) | Fusoxcon1 | 53.58 | 19,854 | 1142 | Brassica | [48] |
| F. oxysporum Fo5176 (Fo5176) | FoxFo5176 | 67.98 | 19,130 | 1236 | Arabidopsis | [27] |
| IPR | Term | EIy |
|---|---|---|
| Group 1 | ||
| IPR000814 | TBP | 1 |
| IPR003228 | TFIID_TAF12 | 1 |
| IPR004595 | TFIIH_C1-like | 1 |
| IPR006809 | TAFII28 | 1 |
| IPR042225 | Ncb2 | 1 |
| IPR008570 | Vps25 | 1 |
| IPR008895 | Vps72/YL1 | 1 |
| IPR007196 | CNOT1 | 1 |
| IPR005612 | CBF | 1 |
| IPR001289 | NFYA | 1 |
| IPR018004 | APSES-type HTH | 1 |
| IPR003150 | RFX | 1 |
| IPR033896 | MADS_MEF2-like | 1 |
| IPR018501 | DDT | 1 |
| Group 2 | ||
| IPR006856 | MATalpha_HMGbox | 0.8 |
| IPR039515 | NOT4 | 0.9 |
| IPR033897 | MADS_SRF-like | 0.95 |
| IPR000232 | HSF | 0.98 |
| Group 3 | ||
| IPR003163 | Tscrpt_reg_HTH_APSES-type | 1.04 |
| IPR001766 | Fork_head | 1.05 |
| IPR011016 | Znf_RING-CH | 1.11 |
| IPR001965 | Znf_PHD | 1.11 |
| IPR009071 | HMG_box | 1.12 |
| IPR004181 | Znf_MIZ | 1.24 |
| IPR001606 | ARID | 1.25 |
| IPR000679 | Znf_GATA | 1.3 |
| IPR001005 | SANT/Myb | 1.32 |
| IPR000818 | TEA/ATTS | 1.33 |
| IPR003120 | Ste12 | 1.33 |
| IPR003958 | CBFA_NFYB | 1.35 |
| IPR001083 | Cu_fist | 1.37 |
| IPR000967 | Znf_NFX1 | 1.4 |
| IPR006565 | Bromodomain | 1.52 |
| IPR001387 | Cro/C1-type_HTH | 1.6 |
| IPR001841 | Znf_RING | 1.64 |
| IPR000571 | Znf_CCCH | 1.74 |
| IPR001878 | Znf_CCHC | 1.83 |
| IPR010666 | Znf_GRF | 2 |
| IPR018060 | HTH_AraC * | 2 |
| IPR001356 | Homeobox | 2.28 |
| IPR007604 | CP2 * | 2.73 |
| IPR007396 | PAI2 | 3.42 |
| IPR024061 | NDT80 | 3.47 |
| IPR011598 | bHLH | 3.48 |
| IPR007889 | HTH_Psq * | 3.53 |
| IPR013087 | Znf_C2H2 | 4.15 |
| IPR004827 | bZIP | 5.8 |
| IPR001138 | Zn2-C6 | 15.09 |
| TF | Reported Species | References | Family | Overlap * | Average_Fo | Average_ Non-Fo | EIf |
|---|---|---|---|---|---|---|---|
| Ftf1/Ftf2 | F. oxysporum | [96] | Zn2-C6 | Yes | 4.80 | 1.11 | 2.75 |
| Ebr1/Ebr2 | F. oxysporum | [103] | Zn2-C6 | Yes | 5.27 | 1.56 | 2.45 |
| Znf1 | M. oryzae | [140] | Zn2-C6 | Yes | 6.47 | 2.78 | 1.98 |
| Ctf2 | F. oxysporum | [141] | Zn2-C6 | Yes | 2.93 | 1.33 | 1.69 |
| Fow2 | F. oxysporum | [101] | Zn2-C6 | Yes | 2.07 | 1.00 | 1.53 |
| Dep6 | A. brassicicola | [142] | Zn2-C6 | Yes | 0.93 | 0.67 | 1.16 |
| Pf2 | A. brassicicola | [143] | Zn2-C6 | Yes | 1.20 | 1.00 | 1.10 |
| Art1 | F. verticilioides | [144] | Zn2-C6 | Yes | 1.00 | 0.89 | 1.06 |
| Clta1 | C. lindemuthianum | [145] | Zn2-C6 | Yes | 1.07 | 1.00 | 1.03 |
| Fhs1 | F. graminearum | [146] | Zn2-C6 | Yes | 1.07 | 1.00 | 1.03 |
| Cos1 | M. oryzae | [112] | Znf_C2H2 | Yes | 1.80 | 0.00 | 2.80 |
| PacC | F. oxysporum | [107] | Znf_C2H2 | Yes | 2.13 | 1.00 | 1.57 |
| Fug1 | F. verticillioides | [147] | AreA_GATA | No | 7.27 | 1.33 | 3.54 |
| Ren1 | F. oxysporum | [138] | disordered | No | 3.00 | 1.00 | 2.00 |
| Tri10 | F. graminearum | [148] | Fun_TF | No | 1.13 | 0.33 | 1.60 |
| Ltf1 | B. cinerea | [149] | Znf_GATA | Yes | 4.00 | 2.44 | 1.45 |
| Ndt80 | U. maydis | [119] | NDT80 | Yes | 1.73 | 1.11 | 1.29 |
| Hap3p | F. verticillioides | [147] | CBFA_NFYB | Yes | 1.33 | 1.00 | 1.17 |
| Sod1 | F. oxysporum | [150] | SOD_Cu_Zn | No | 1.47 | 1.22 | 1.11 |
| Prf1 | F. oxysporum | [151] | HMG_box | Yes | 1.07 | 1.00 | 1.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, H.; Yang, H.; Haridas, S.; Hayes, R.D.; Lynch, H.; Andersen, S.; Newman, M.; Li, G.; Martínez-Soto, D.; Milo-Cochavi, S.; et al. Conservation and Expansion of Transcriptional Factor Repertoire in the Fusarium oxysporum Species Complex. J. Fungi 2023, 9, 359. https://doi.org/10.3390/jof9030359
Yu H, Yang H, Haridas S, Hayes RD, Lynch H, Andersen S, Newman M, Li G, Martínez-Soto D, Milo-Cochavi S, et al. Conservation and Expansion of Transcriptional Factor Repertoire in the Fusarium oxysporum Species Complex. Journal of Fungi. 2023; 9(3):359. https://doi.org/10.3390/jof9030359
Chicago/Turabian StyleYu, Houlin, He Yang, Sajeet Haridas, Richard D. Hayes, Hunter Lynch, Sawyer Andersen, Madison Newman, Gengtan Li, Domingo Martínez-Soto, Shira Milo-Cochavi, and et al. 2023. "Conservation and Expansion of Transcriptional Factor Repertoire in the Fusarium oxysporum Species Complex" Journal of Fungi 9, no. 3: 359. https://doi.org/10.3390/jof9030359
APA StyleYu, H., Yang, H., Haridas, S., Hayes, R. D., Lynch, H., Andersen, S., Newman, M., Li, G., Martínez-Soto, D., Milo-Cochavi, S., Hazal Ayhan, D., Zhang, Y., Grigoriev, I. V., & Ma, L. -J. (2023). Conservation and Expansion of Transcriptional Factor Repertoire in the Fusarium oxysporum Species Complex. Journal of Fungi, 9(3), 359. https://doi.org/10.3390/jof9030359