Role of N–Oxide Moieties in Tuning Supramolecular Gel-State Properties


Abstract
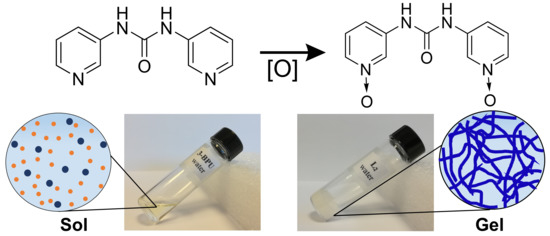
:
1. Introduction
2. Results and Discussion
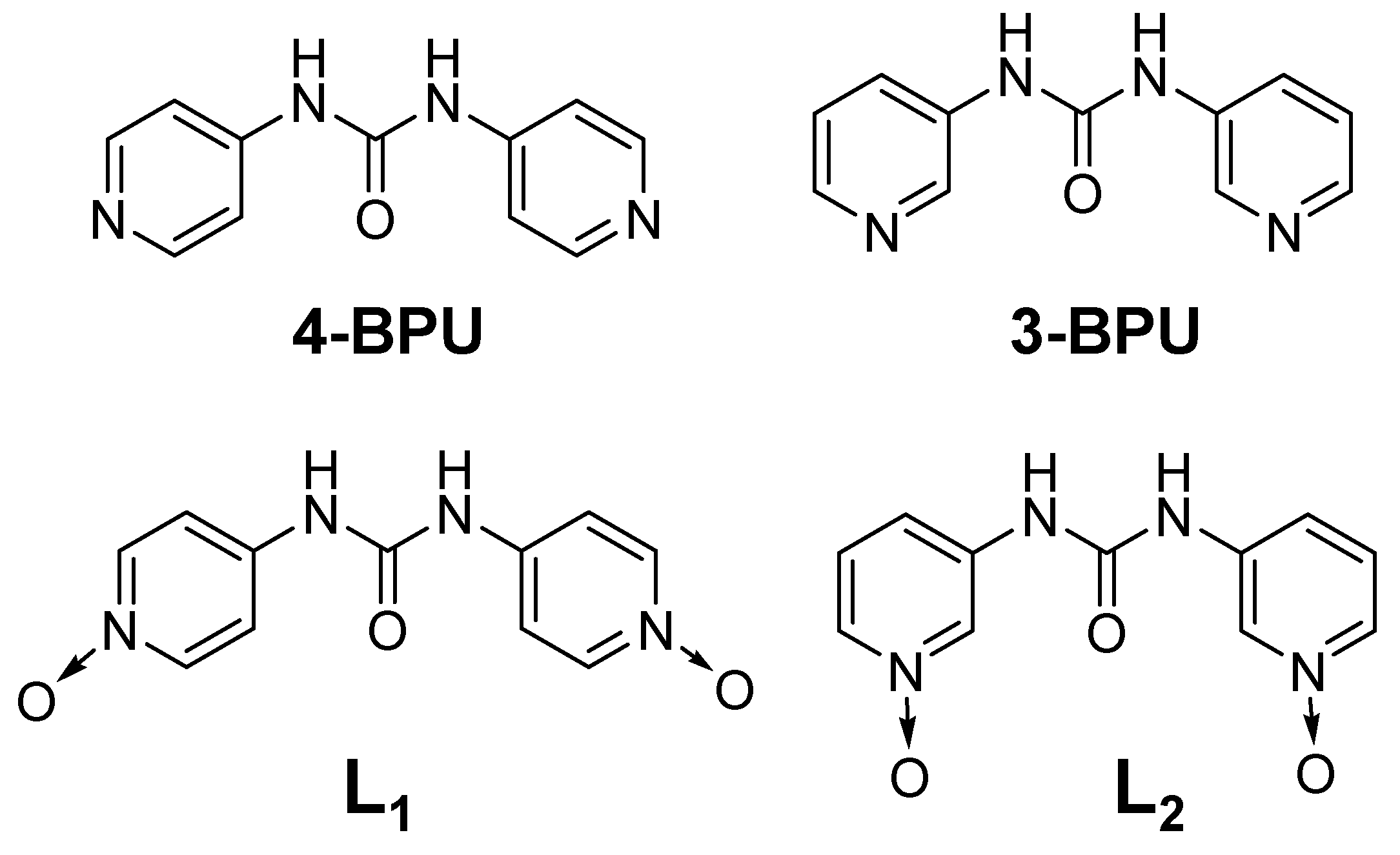
2.1. Design and Synthesis
2.2. Gelation Experiments
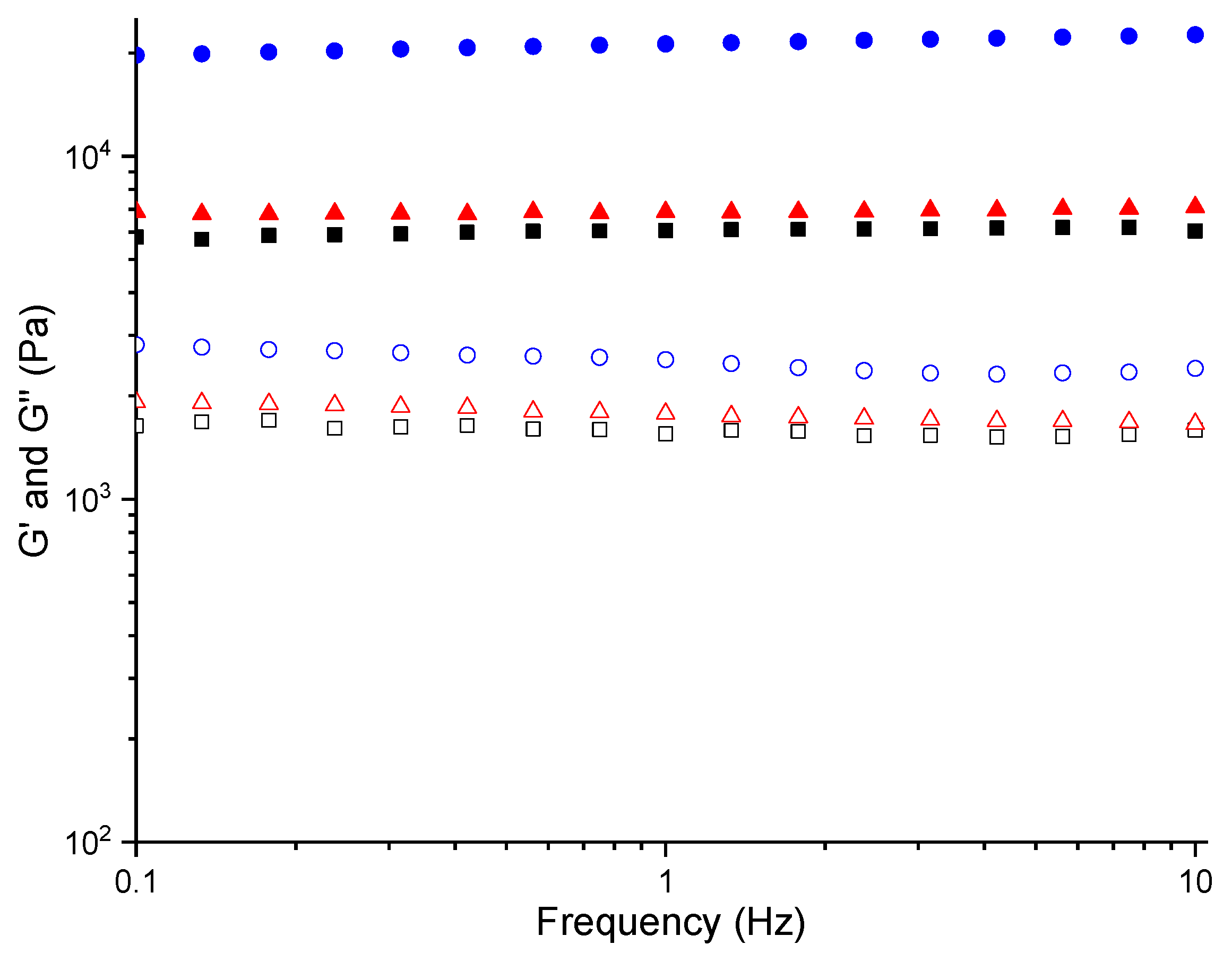
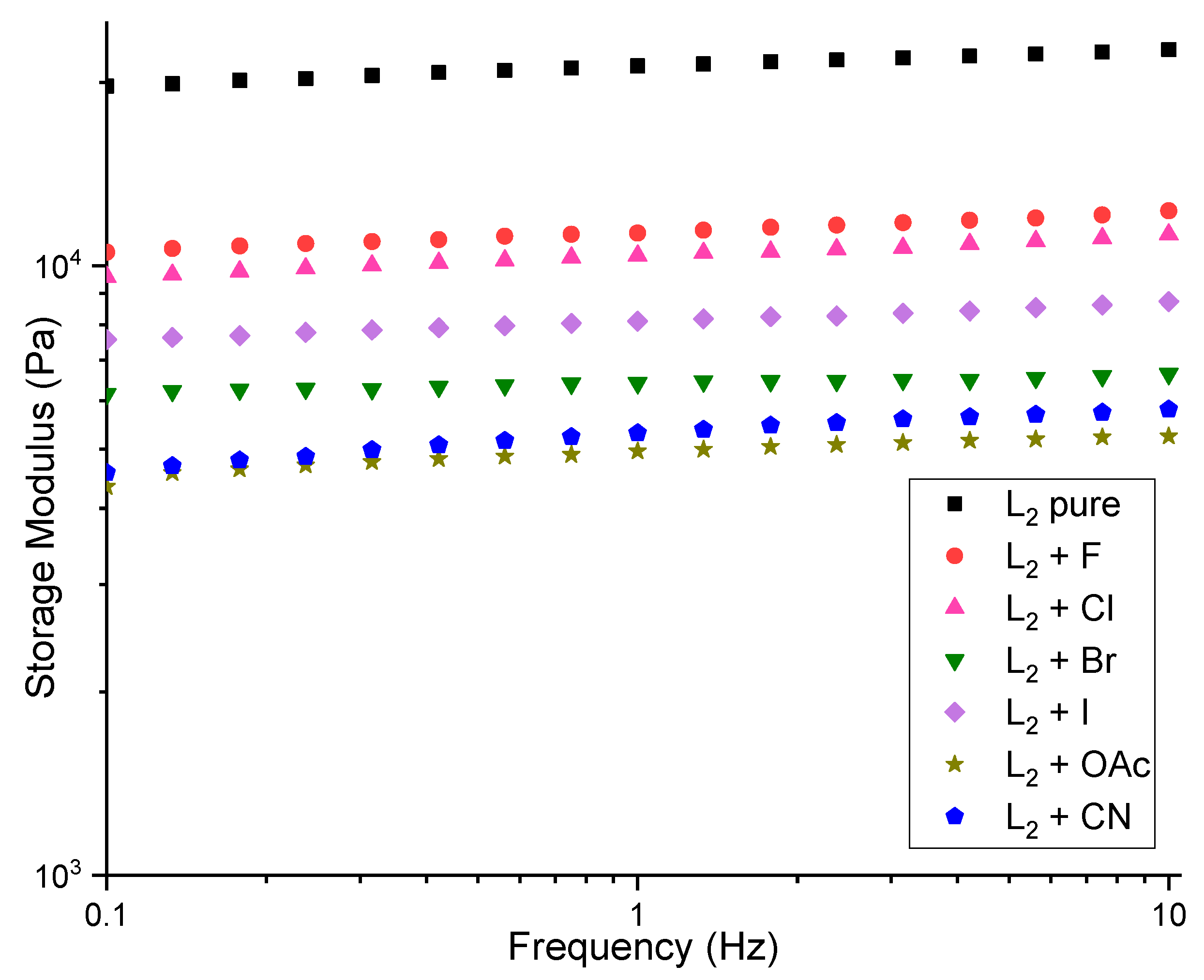
2.3. Rheology
2.4. Gel Morphology
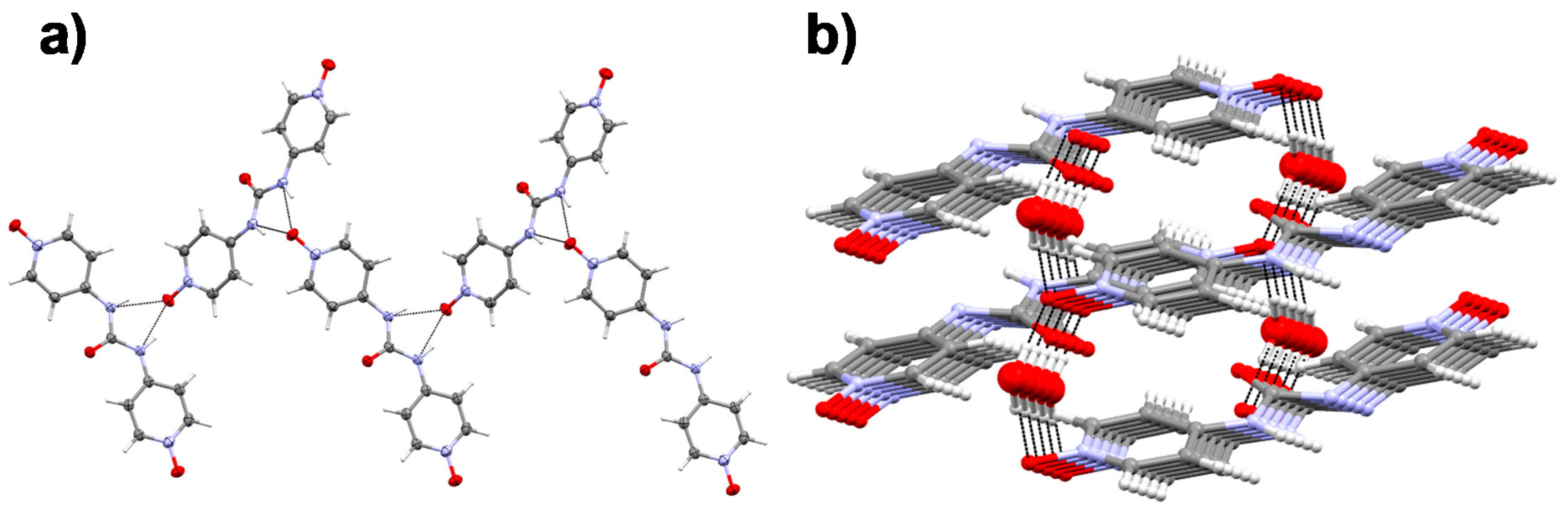
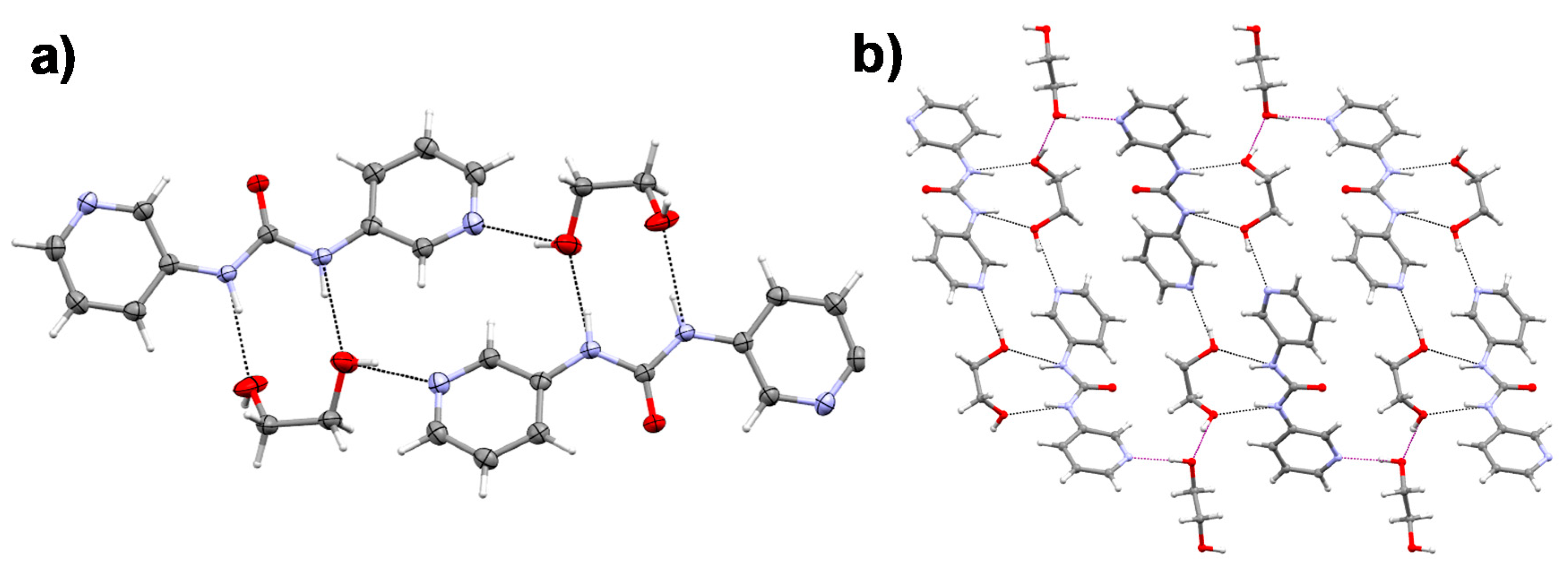
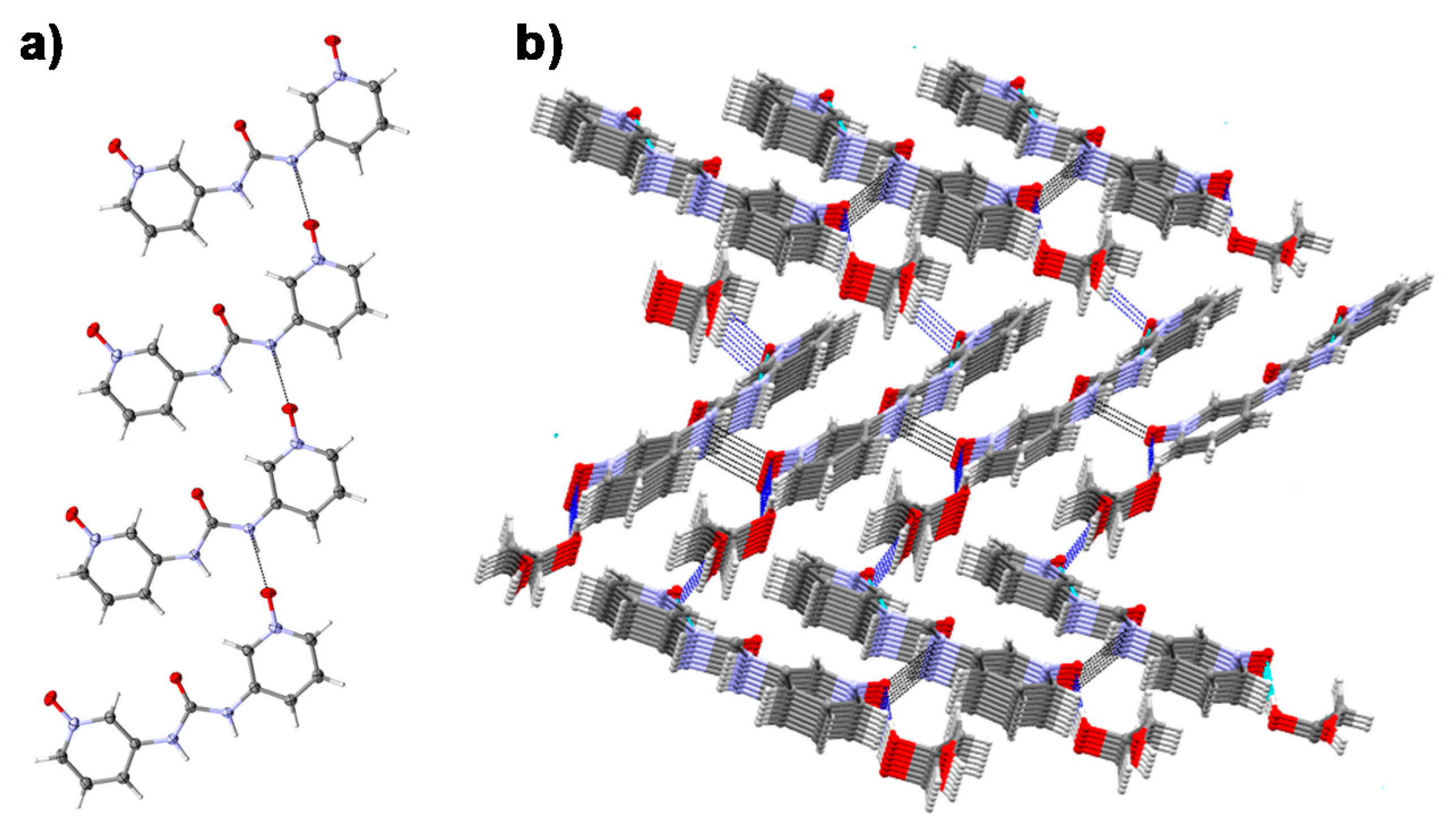
2.5. Single Crystal X-ray Diffraction
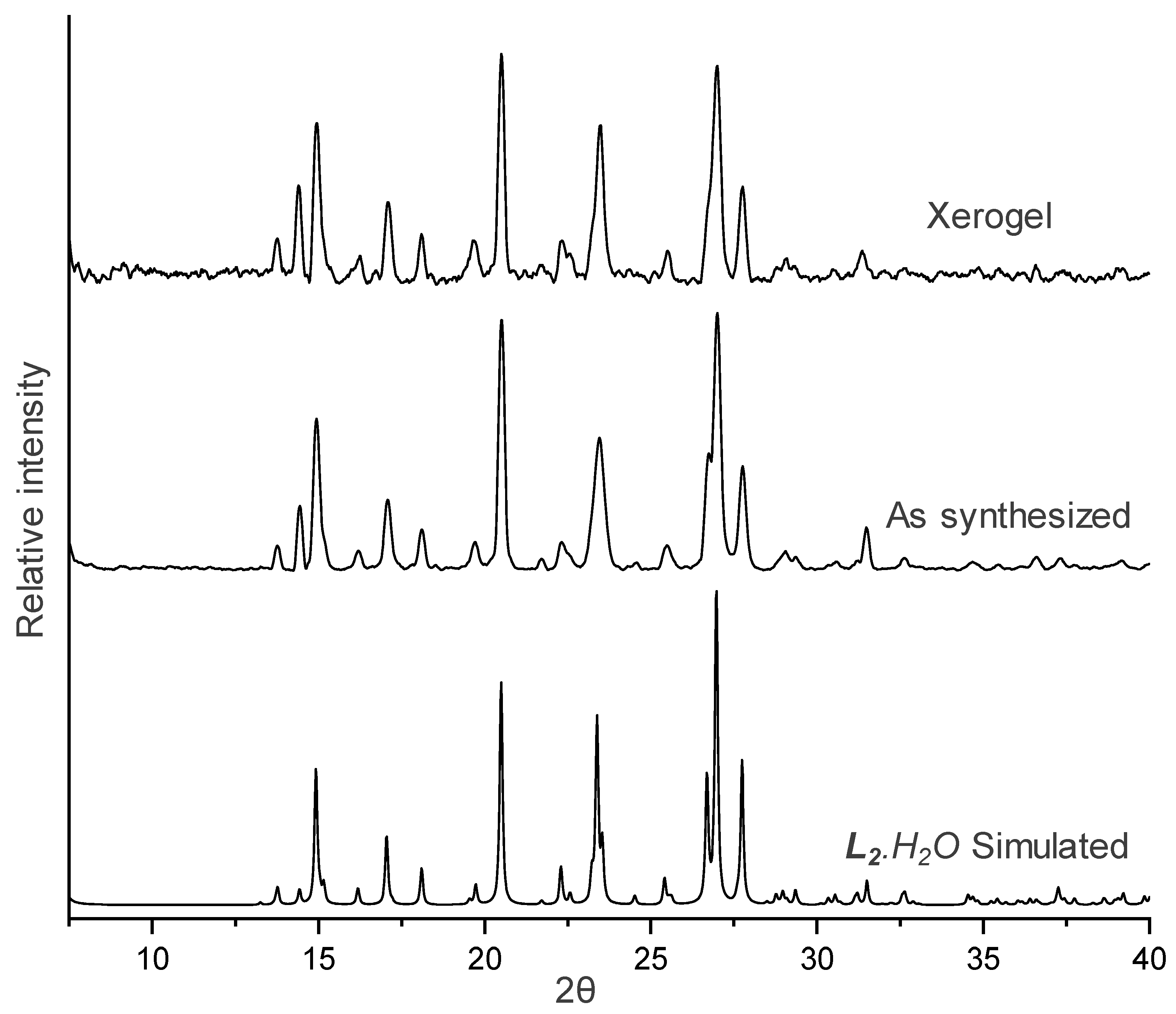
2.6. X-ray Powder Diffraction (XRPD)
2.7. Stimuli-Responsive Property
2.8. Computational Studies
3. Conclusions
4. Materials and Methods
4.1. Synthesis of the Ligand
4.1.1. 4,4’–(carbonylbis(azanediyl))bis(pyridine 1–oxide) (L1)
4.1.2. 3,3’–(carbonylbis(azanediyl))bis(pyridine 1–oxide) (L2)
4.2. Gelation Studies
4.2.1. Gelation Test
4.2.2. Minimum Gel Concentration
4.2.3. Tgel Experiments
4.3. Rheology
4.4. Scanning Electron Microscopy
4.5. Single Crystal X-ray Diffraction
4.6. X-ray Powder Diffraction
4.7. Quantum Chemical Calculations
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flory, P.J. Introductory lecture. Faraday Discuss. 1974, 57, 7–18. [Google Scholar] [CrossRef]
- Estroff, L.A.; Hamilton, A.D. Water Gelation by Small Organic Molecules. Chem. Rev. 2004, 104, 1201–1218. [Google Scholar] [CrossRef] [PubMed]
- Dastidar, P. Supramolecular gelling agents: Can they be designed? Chem. Soc. Rev. 2008, 37, 2699–2715. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.D.; Steed, J.W. Gels with sense: Supramolecular materials that respond to heat, light and sound. Chem. Soc. Rev. 2016, 45, 6546–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steed, J.W. Anion-tuned supramolecular gels: A natural evolution from urea supramolecular chemistry. Chem. Soc. Rev. 2010, 39, 3686–3699. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.; Steed, J.W. Supramolecular gel phase crystallization: Orthogonal self-assembly under non-equilibrium conditions. Chem. Soc. Rev. 2014, 43, 2080–2088. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Yan, X.; Han, C.; Huang, F. Characterization of supramolecular gels. Chem. Soc. Rev. 2013, 42, 6697–6722. [Google Scholar] [CrossRef]
- Fang, W.; Zhang, Y.; Wu, J.; Liu, C.; Zhu, H.; Tu, T. Recent Advances in Supramolecular Gels and Catalysis. Chem. Asian J. 2018, 13, 712–729. [Google Scholar] [CrossRef]
- Smith, D.K. Applications of Supramolecular Gels. In Molecular Gels: Structure and Dynamics; Weiss, R., Ed.; Royal Society of Chemistry: Cambridge, UK, 2018; pp. 300–371. [Google Scholar]
- Foster, J.A.; Damodaran, K.K.; Maurin, A.; Day, G.M.; Thompson, H.P.G.; Cameron, G.J.; Bernal, J.C.; Steed, J.W. Pharmaceutical polymorph control in a drug-mimetic supramolecular gel. Chem. Sci. 2017, 8, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Mooney, D.J. Hydrogels for Tissue Engineering. Chem. Rev. 2001, 101, 1869–1880. [Google Scholar] [CrossRef]
- Van Bommel, K.J.C.; Stuart, M.C.A.; Feringa, B.L.; van Esch, J. Two-stage enzyme mediated drug release from LMWG hydrogels. Org. Biomol. Chem. 2005, 3, 2917–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhang, G.; Zhang, D. Stimuli responsive gels based on low molecular weight gelators. J. Mater. Chem. 2012, 22, 38–50. [Google Scholar] [CrossRef]
- Meazza, L.; Foster, J.A.; Fucke, K.; Metrangolo, P.; Resnati, G.; Steed, J.W. Halogen-bonding-triggered supramolecular gel formation. Nat. Chem. 2013, 5, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.G.; Terech, P. (Eds.) Molecular Gels: Materials with Self-Assembled Fibrillar Networks; Springer: Dordrecht, The Netherlands, 2006; p. 978. [Google Scholar]
- Steed, J.W. Supramolecular gel chemistry: Developments over the last decade. Chem. Commun. 2011, 47, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Chivers, P.R.A.; Smith, D.K. Shaping and structuring supramolecular gels. Nat. Rev. Mater. 2019, 4, 463–478. [Google Scholar] [CrossRef] [Green Version]
- Fages, F.; Voegtle, F.; Zinic, M. Systematic design of amide- and urea-type gelators with tailored properties. Top. Curr. Chem. 2005, 256, 77–131. [Google Scholar] [PubMed]
- Van Esch, J.; Kellogg, R.M.; Feringa, B.L. Di-urea compounds as gelators for organic solvents. Tetrahedron Lett. 1997, 38, 281–284. [Google Scholar] [CrossRef] [Green Version]
- Piepenbrock, M.O.M.; Lloyd, G.O.; Clarke, N.; Steed, J.W. Gelation is crucially dependent on functional group orientation and may be tuned by anion binding. Chem. Commun. 2008, 2644–2646. [Google Scholar] [CrossRef]
- Bastiat, G.; Leroux, J.C. Pharmaceutical organogels prepared from aromatic amino acid derivatives. J. Mater. Chem. 2009, 19, 3867–3877. [Google Scholar] [CrossRef] [Green Version]
- George, M.; Tan, G.; John, V.T.; Weiss, R.G. Urea and Thiourea Derivatives as Low Molecular-Mass Organogelators. Chem. Eur. J. 2005, 11, 3243–3254. [Google Scholar] [CrossRef]
- Poolman, J.M.; Maity, C.; Boekhoven, J.; van der Mee, L.; le Sage, V.A.A.; Groenewold, G.J.M.; van Kasteren, S.I.; Versluis, F.; van Esch, J.H.; Eelkema, R. A toolbox for controlling the properties and functionalisation of hydrazone-based supramolecular hydrogels. J. Mater. Chem. B 2016, 4, 852–858. [Google Scholar] [CrossRef] [PubMed]
- McAulay, K.; Dietrich, B.; Su, H.; Scott, M.T.; Rogers, S.; Al-Hilaly, Y.K.; Cui, H.; Serpell, L.C.; Seddon, A.M.; Draper, E.R.; et al. Using chirality to influence supramolecular gelation. Chem. Sci. 2019, 10, 7801–7806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Zurcher, D.M.; Adhia, Y.J.; Romero, J.D.; McNeil, A.J. Modifying a known gelator scaffold for nitrite detection. Chem. Commun. 2014, 50, 7813–7816. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.K.; Jose, D.A.; Dastidar, P.; Das, A. Nonpolymeric Hydrogelators Derived from Trimesic Amides. Chem. Mater. 2004, 16, 2332–2335. [Google Scholar] [CrossRef]
- Ghosh, D.; Ferfolja, K.; Drabavičius, Ž.; Steed, J.W.; Damodaran, K.K. Crystal habit modification of Cu(ii) isonicotinate–N-oxide complexes using gel phase crystallisation. New J. Chem. 2018, 42, 19963–19970. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.K.; Jose, D.A.; Dastidar, P.; Das, A. Nonpolymeric Hydrogelator Derived from N-(4-Pyridyl)isonicotinamide. Langmuir 2004, 20, 10413–10418. [Google Scholar] [CrossRef]
- Ghosh, D.; Mulvee, M.T.; Damodaran, K.K. Tuning Gel State Properties of Supramolecular Gels by Functional Group Modification. Molecules 2019, 24, 3472. [Google Scholar] [CrossRef] [Green Version]
- Tomasini, C.; Castellucci, N. Peptides and peptidomimetics that behave as low molecular weight gelators. Chem. Soc. Rev. 2013, 42, 156–172. [Google Scholar] [CrossRef]
- Kumar, D.K.; Jose, D.A.; Das, A.; Dastidar, P. First snapshot of a nonpolymeric hydrogelator interacting with its gelling solvents. Chem. Commun. 2005, 4059–4061. [Google Scholar] [CrossRef]
- Abraham, S.; Lan, Y.; Lam, R.S.H.; Grahame, D.A.S.; Kim, J.J.H.; Weiss, R.G.; Rogers, M.A. Influence of Positional Isomers on the Macroscale and Nanoscale Architectures of Aggregates of Racemic Hydroxyoctadecanoic Acids in Their Molecular Gel, Dispersion, and Solid States. Langmuir 2012, 28, 4955–4964. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kang, C.; Chen, Y.; Bian, Z.; Qiu, X.; Gao, L.; Meng, Q. In Situ Gel-to-Crystal Transition and Synthesis of Metal Nanoparticles Obtained by Fluorination of a Cyclic β-Aminoalcohol Gelator. Chem. Eur. J. 2012, 18, 16955–16961. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, L.; Yu, J. Investigation of Spontaneous Transition from Low-Molecular-Weight Hydrogel into Macroscopic Crystals. Cryst. Growth Des. 2008, 8, 884–889. [Google Scholar] [CrossRef]
- Braga, D.; d’Agostino, S.; D’Amen, E.; Grepioni, F. Polymorphs from supramolecular gels: Four crystal forms of the same silver(i) supergelator crystallized directly from its gels. Chem. Commun. 2011, 47, 5154–5156. [Google Scholar] [CrossRef]
- Byrne, P.; Lloyd, G.O.; Applegarth, L.; Anderson, K.M.; Clarke, N.; Steed, J.W. Metal-induced gelation in dipyridyl ureas. New J. Chem. 2010, 34, 2261–2274. [Google Scholar] [CrossRef]
- Dastidar, P.; Roy, R.; Parveen, R.; Sarkar, K. Supramolecular Synthon Approach in Designing Molecular Gels for Advanced Therapeutics. Adv. Ther. 2019, 2, 1800061. [Google Scholar] [CrossRef] [Green Version]
- Chandran, S.K.; Nath, N.K.; Cherukuvada, S.; Nangia, A. N–H…N(pyridyl) and N–H…O(urea) hydrogen bonding and molecular conformation of N-aryl-N′-pyridylureas. J. Mol. Struct. 2010, 968, 99–107. [Google Scholar] [CrossRef]
- Byrne, P.; Turner, D.R.; Lloyd, G.O.; Clarke, N.; Steed, J.W. Gradual Transition from NH···Pyridyl Hydrogen Bonding to the NH···O Tape Synthon in Pyridyl Ureas. Cryst. Growth Des. 2008, 8, 3335–3344. [Google Scholar] [CrossRef]
- Todd, A.M.; Anderson, K.M.; Byrne, P.; Goeta, A.E.; Steed, J.W. Helical or Polar Guest-Dependent Z‘ = 1.5 or Z‘ = 2 Forms of a Sterically Hindered Bis(urea) Clathrate. Cryst. Growth Des. 2006, 6, 1750–1752. [Google Scholar] [CrossRef]
- Reddy, L.S.; Basavoju, S.; Vangala, V.R.; Nangia, A. Hydrogen Bonding in Crystal Structures of N,N’-Bis(3-pyridyl)urea. Why Is the N−H···O Tape Synthon Absent in Diaryl Ureas with Electron-Withdrawing Groups? Cryst. Growth Des. 2006, 6, 161–173. [Google Scholar] [CrossRef]
- Ghosh, D.; Lebedyte, I.; Yufit, D.S.; Damodaran, K.K.; Steed, J.W. Selective gelation of N-(4-pyridyl)nicotinamide by copper(ii) salts. CrystEngComm 2015, 17, 8130–8138. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Deepa; Damodaran, K.K. Metal complexation induced supramolecular gels for the detection of cyanide in water. Supramol. Chem. 2020, 32, 276–286. [Google Scholar] [CrossRef]
- Ghosh, D.; Farahani, A.D.; Martin, A.D.; Thordarson, P.; Damodaran, K.K. Unraveling the Self-Assembly Modes in Multicomponent Supramolecular Gels Using Single-Crystal X-ray Diffraction. Chem. Mater. 2020, 32, 3517–3527. [Google Scholar] [CrossRef]
- Adams, D.J. Does Drying Affect Gel Networks? Gels 2018, 4, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piepenbrock, M.O.M.; Lloyd, G.O.; Clarke, N.; Steed, J.W. Metal- and Anion-Binding Supramolecular Gels. Chem. Rev. 2010, 110, 1960–2004. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Cryst. Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. ωB97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J. Chem. Phys. 2016, 144, 214110. [Google Scholar] [CrossRef] [Green Version]
- Najibi, A.; Goerigk, L. The Nonlocal Kernel in van der Waals Density Functionals as an Additive Correction: An Extensive Analysis with Special Emphasis on the B97M-V and ωB97M-V Approaches. J. Chem. Theory Comput. 2018, 14, 5725–5738. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Hansen, A.; Becker, U. Efficient, approximate and parallel Hartree–Fock and hybrid DFT calculations. A ‘chain-of-spheres’ algorithm for the Hartree–Fock exchange. Chem. Phys. 2009, 356, 98–109. [Google Scholar] [CrossRef]
- Izsák, R.; Neese, F. An overlap fitted chain of spheres exchange method. J. Chem. Phys. 2011, 135, 144105. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | L1 | L2 |
|---|---|---|
| Water | Gel | Gel |
| THF/water | Colloidal | Gel |
| EtOH/water | Colloidal | Gel |
| MeOH/water | Colloidal | Gel |
| Acetonitrile/water | Colloidal | Gel |
| DMF/water | Gel | Gel |
| DMA/water | Gel | Gel |
| DMSO/water | Gel | Gel |
| EG/water † | Gel | Gel * |
| DME/water † | Gel | Gel * |
| Water: EG | 4–BPU * | L1 * | 3–BPU ** | L2 ** |
|---|---|---|---|---|
| 10:0 | 98.1 | 95.2 | Crystal | 88.2 |
| 9:1 | 85.6 | 94.5 | Crystal | 83.6 |
| 8:2 | 81.2 | 91.7 | 57.1 | 79.1 |
| 7:3 | 74.0 | 91.0 | 56.4 | 74.5 |
| 6:4 | 71.6 | 89.4 | Crystal | 72.3 |
| 5:5 | 69.5 | 84.1 | Crystal | 71.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghosh, D.; Bjornsson, R.; Damodaran, K.K. Role of N–Oxide Moieties in Tuning Supramolecular Gel-State Properties. Gels 2020, 6, 41. https://doi.org/10.3390/gels6040041
Ghosh D, Bjornsson R, Damodaran KK. Role of N–Oxide Moieties in Tuning Supramolecular Gel-State Properties. Gels. 2020; 6(4):41. https://doi.org/10.3390/gels6040041
Chicago/Turabian StyleGhosh, Dipankar, Ragnar Bjornsson, and Krishna K. Damodaran. 2020. "Role of N–Oxide Moieties in Tuning Supramolecular Gel-State Properties" Gels 6, no. 4: 41. https://doi.org/10.3390/gels6040041
APA StyleGhosh, D., Bjornsson, R., & Damodaran, K. K. (2020). Role of N–Oxide Moieties in Tuning Supramolecular Gel-State Properties. Gels, 6(4), 41. https://doi.org/10.3390/gels6040041