
Physical Aspects of Organogelation: A Point of View

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
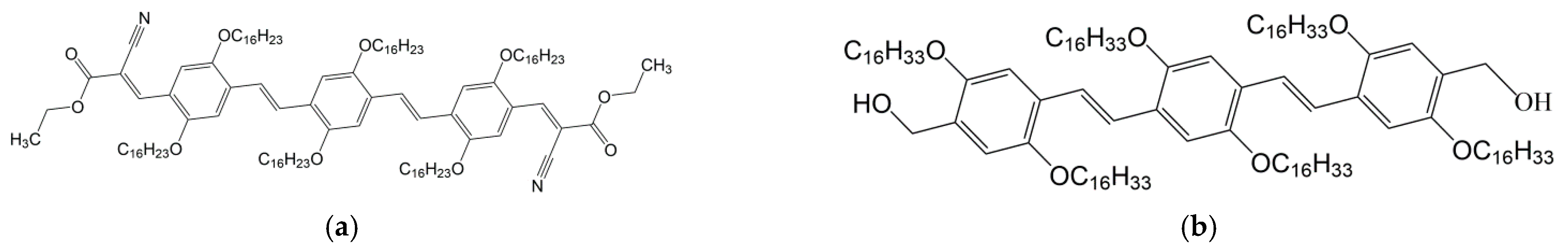{kind=link}
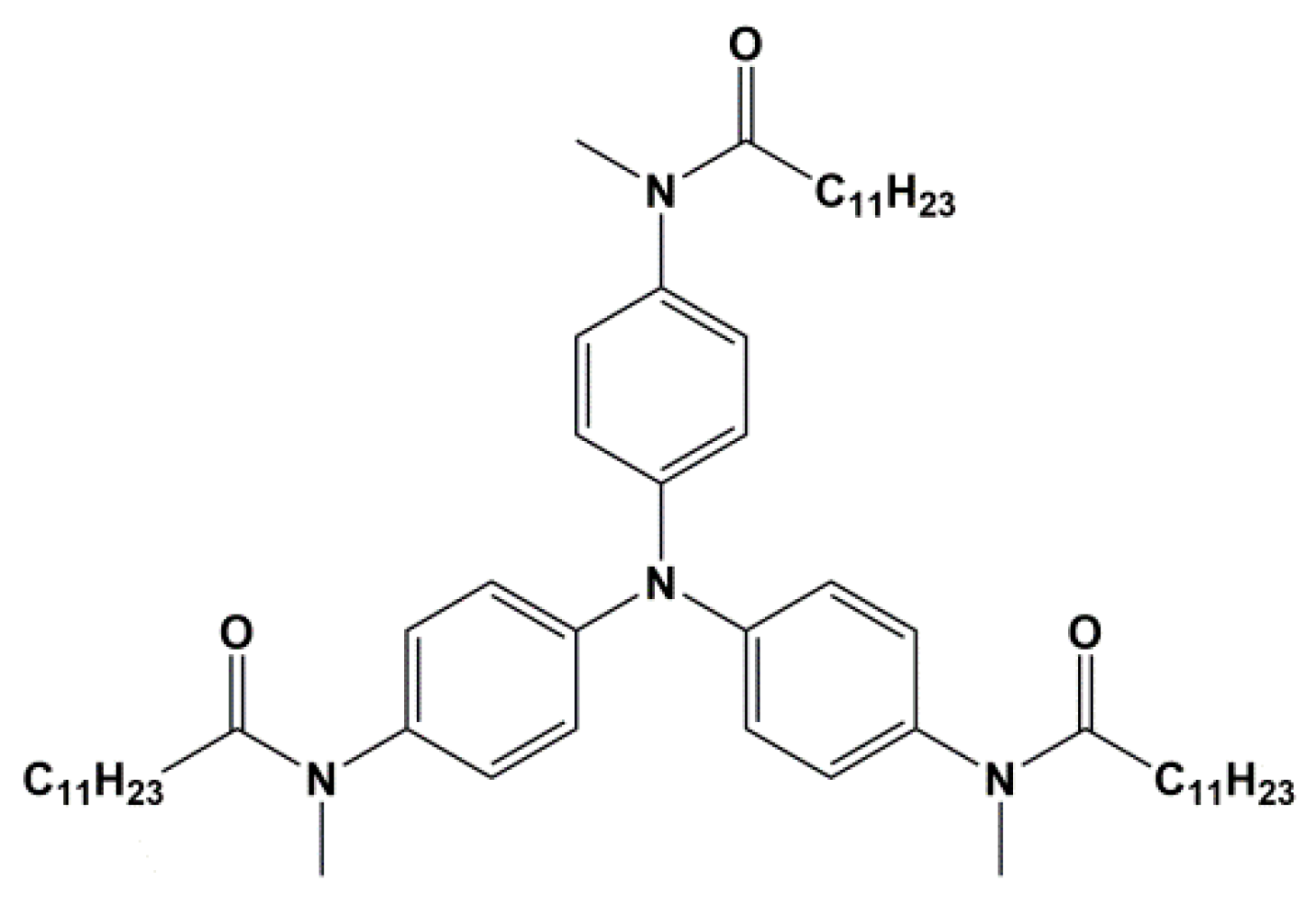{kind=link}
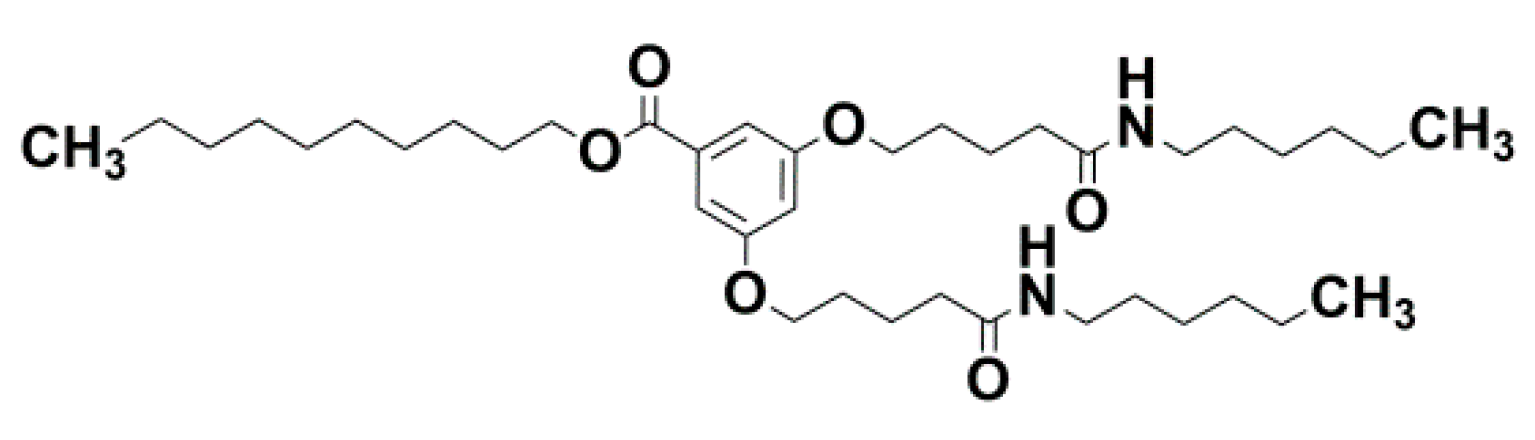{kind=link}
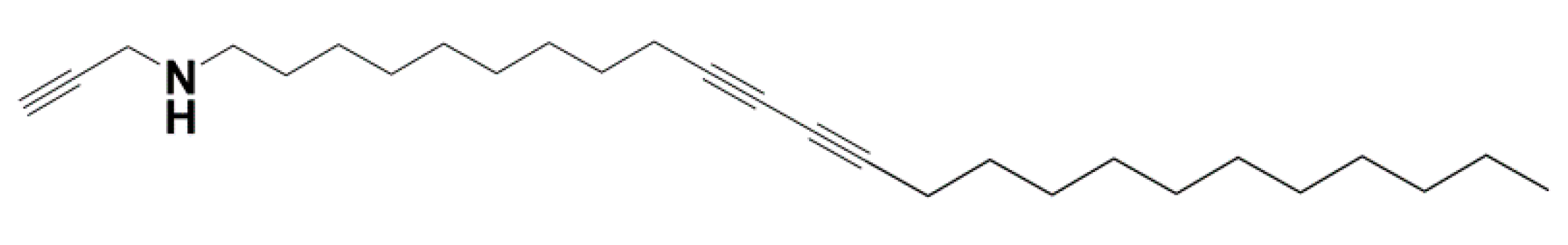{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. A Definition
2.2. Thermodynamics
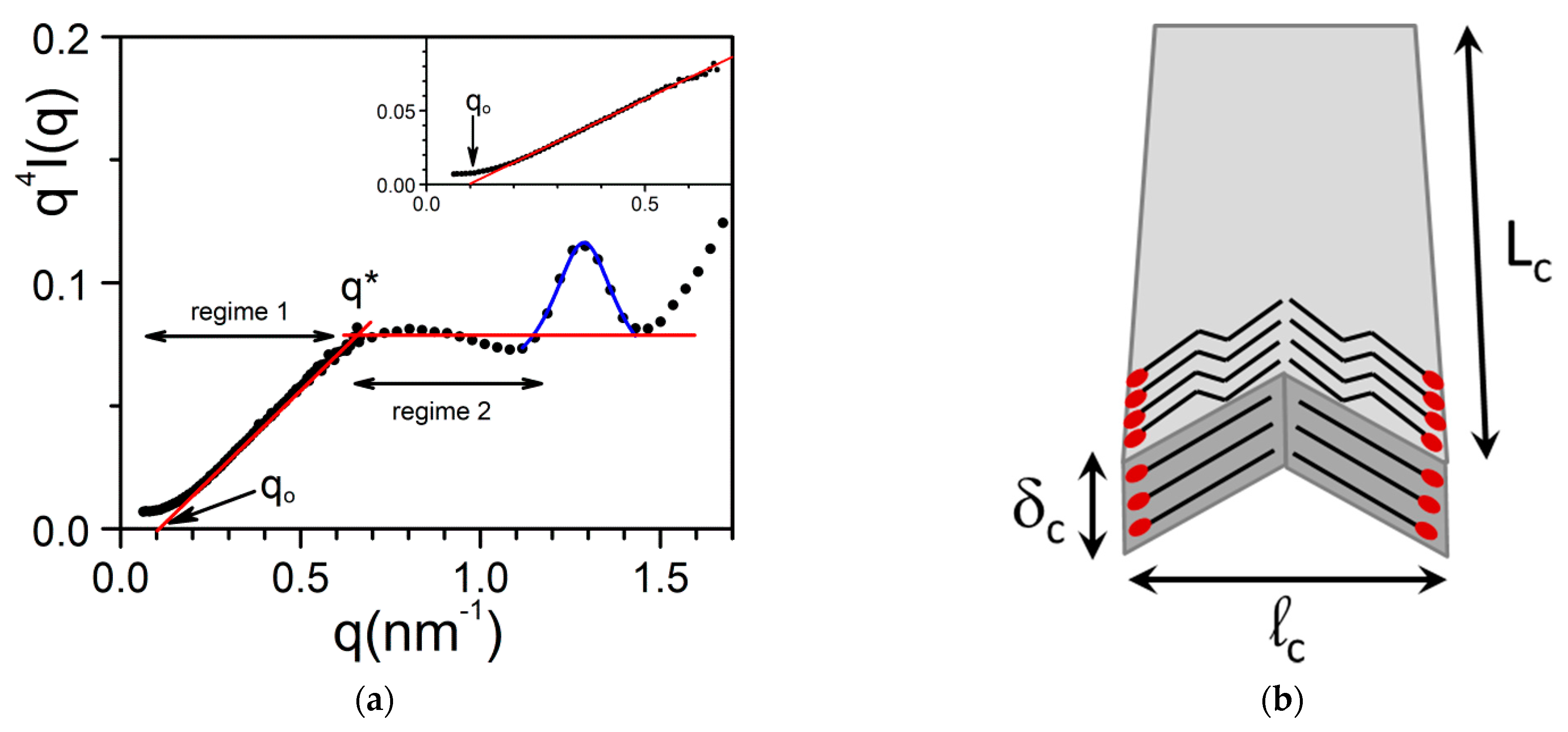
2.2.1. Fibrils Growth
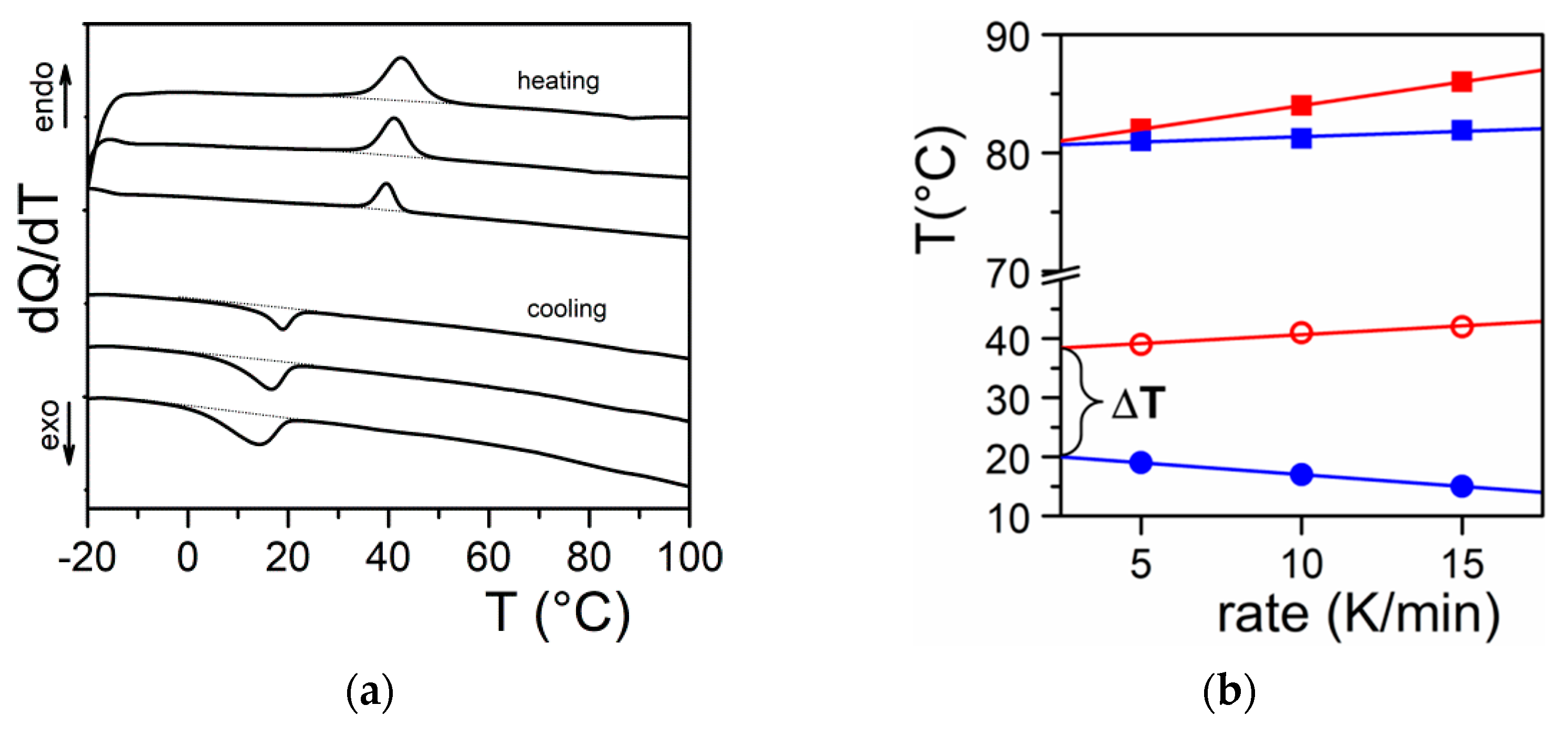
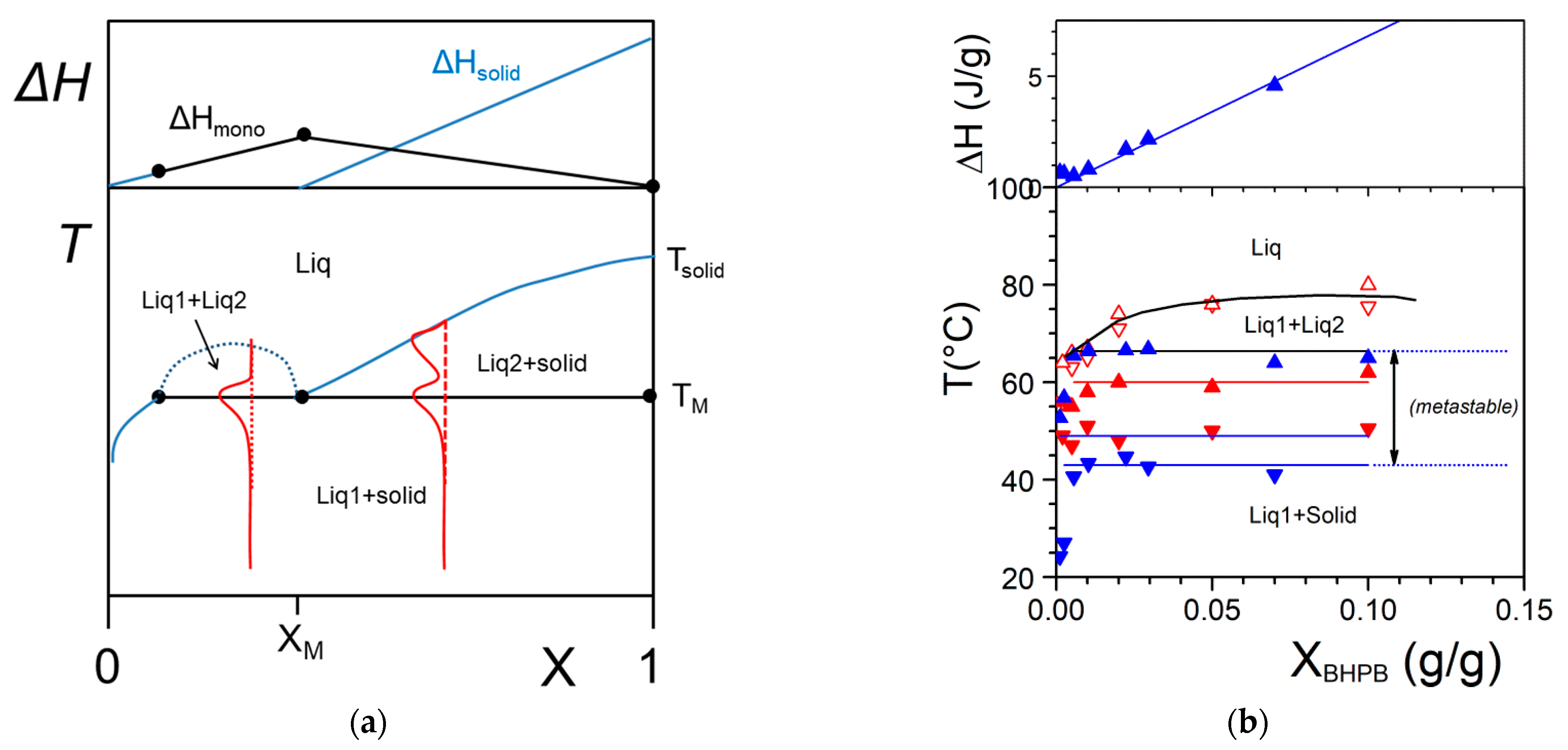
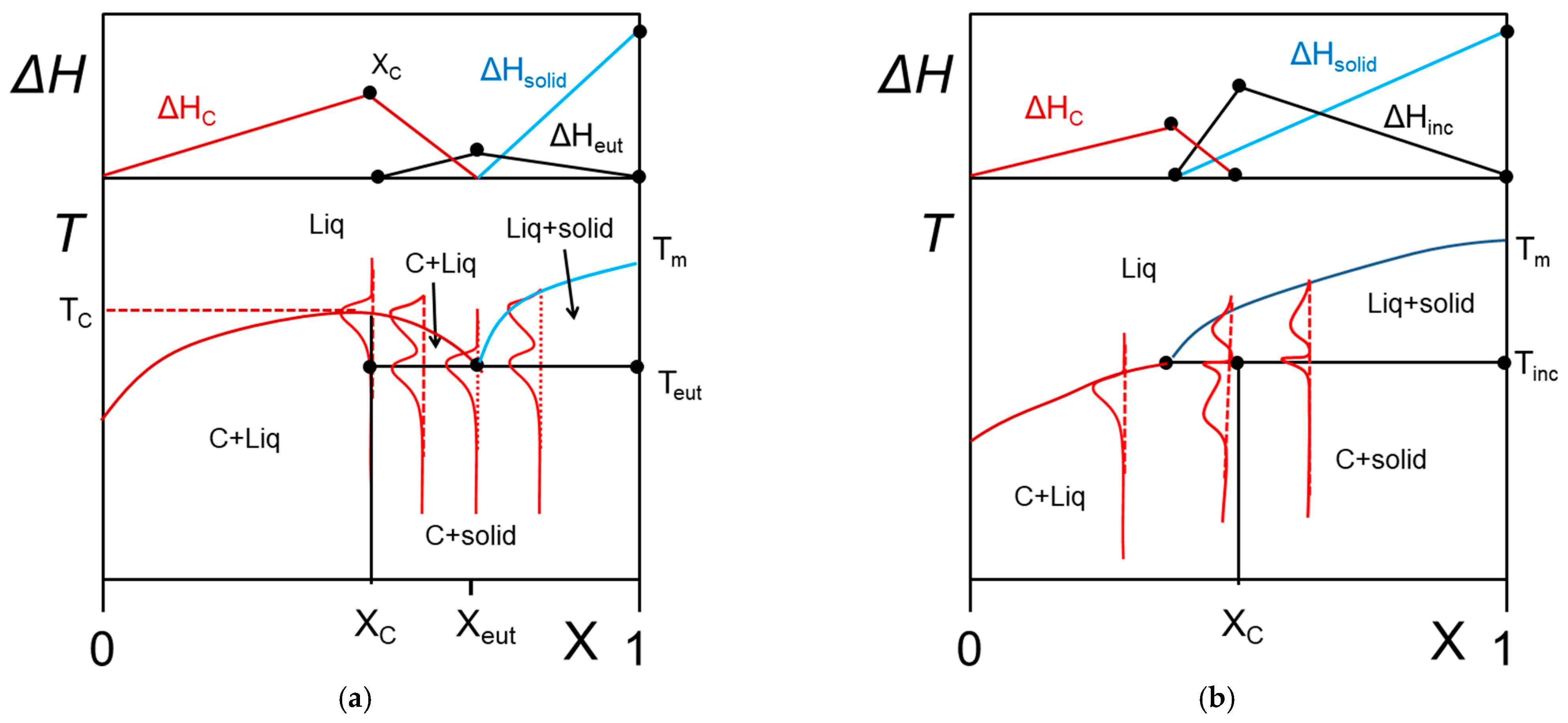
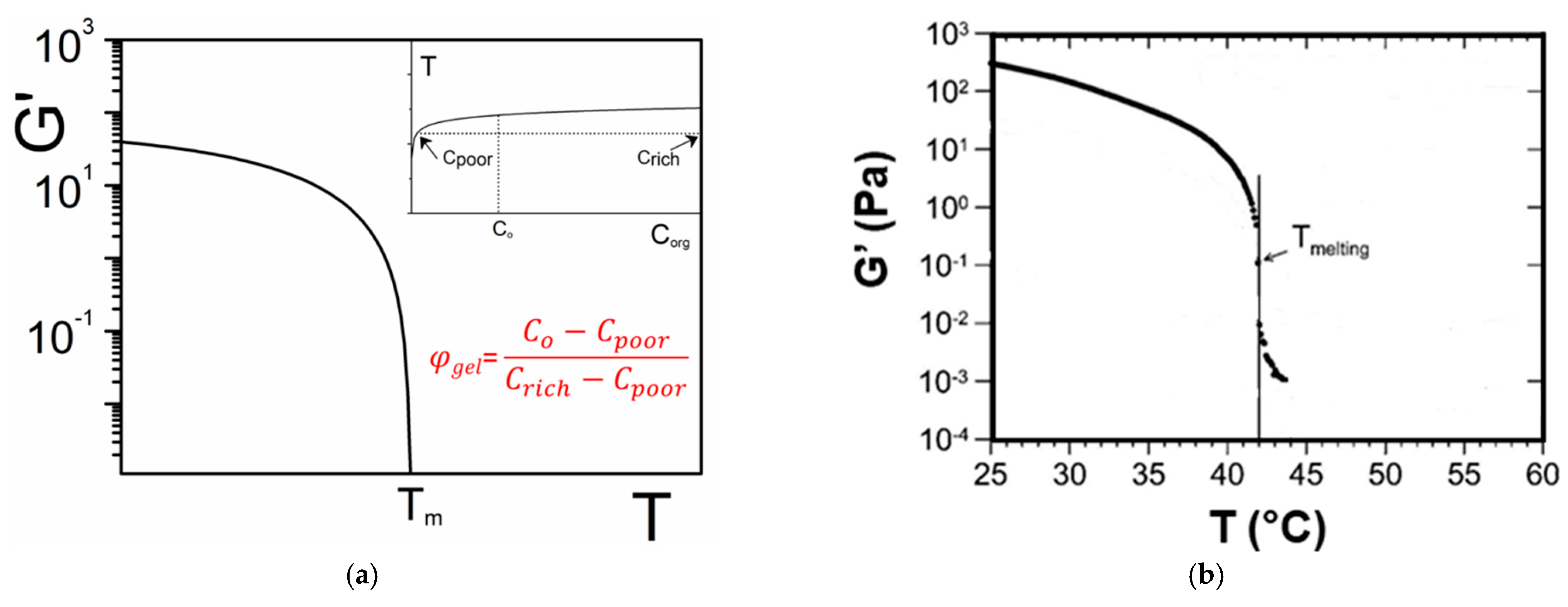
2.2.2. Phase Diagrams
- the variance of the system v, which stands for the number of variables that can be changed without altering the state of the systems. When only the temperature is varied (all other external stimuli being kept constant), the variance reads:where N is the number of components and φ the number of phases. For two components, the number of phases cannot exceed φ = 3 as the variance is v = 0 under these conditions. This implies that the co-existence of the three phases is restricted to a point. The notion of variance possesses another key outcome, the occurrence of non-variant thermal events, namely, the transition temperature remains constant in large range of composition. Note that the composition is always given in w/w or mol/mol so that it does not depend upon temperature, while the concentration expressed in g/cm3 does.
- the lever rule which allows one to calculated the different proportions of the phases.
- 1.
- Solid–liquid transformation
- 2.
- Liquid–Liquid phase separation prior to gelation
- 3.
- Molecular compounds organogelator/solvent
- 4.
- Metatectic tranformation
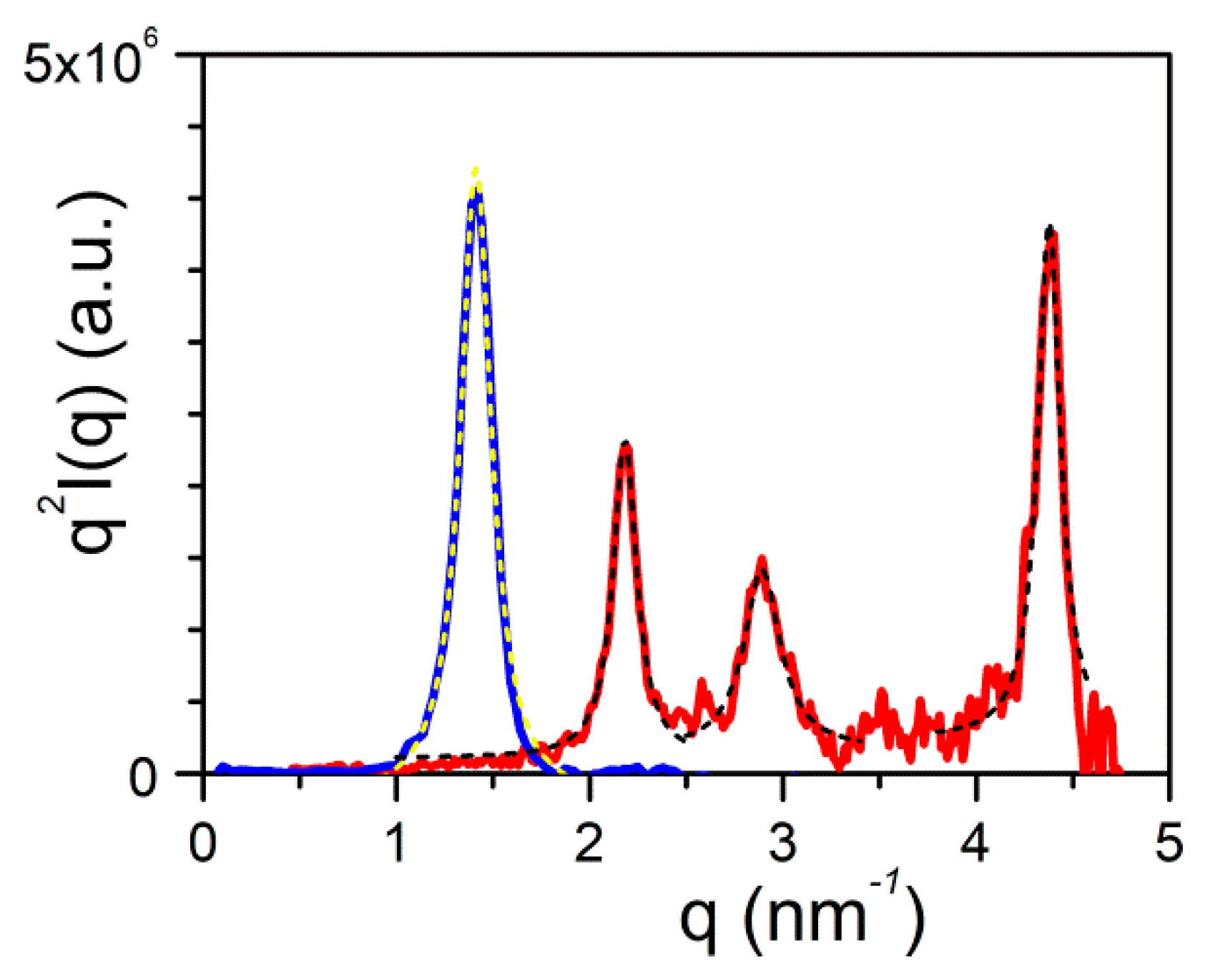
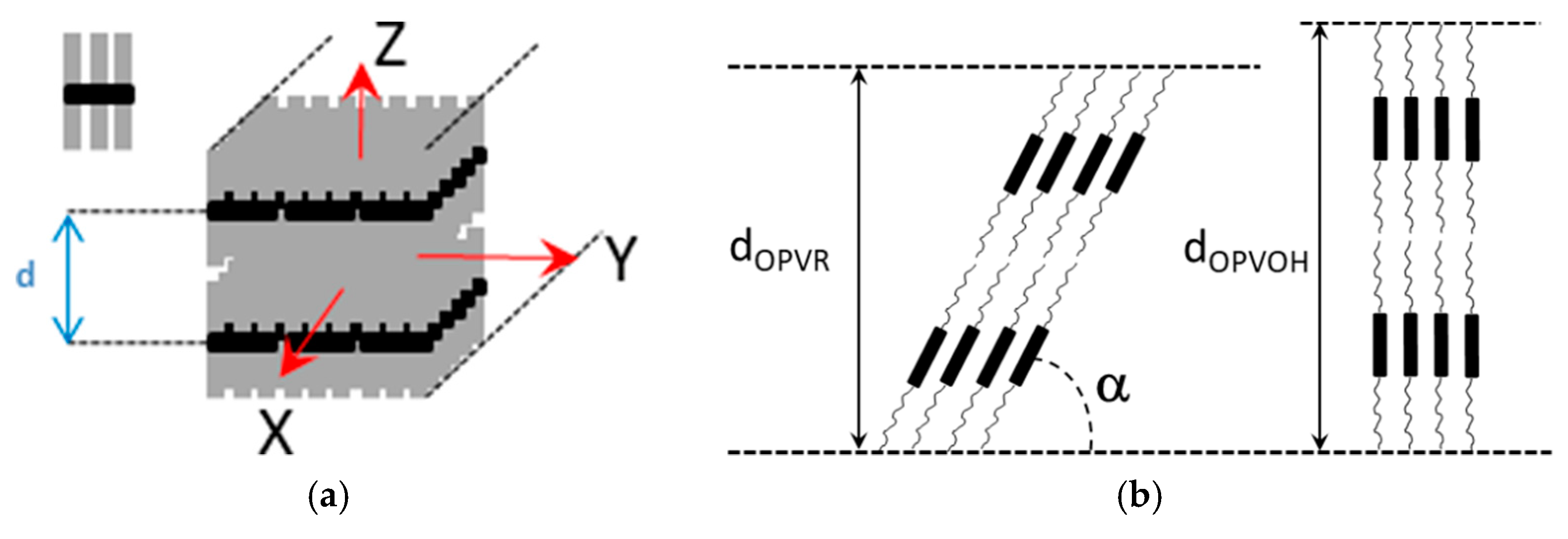
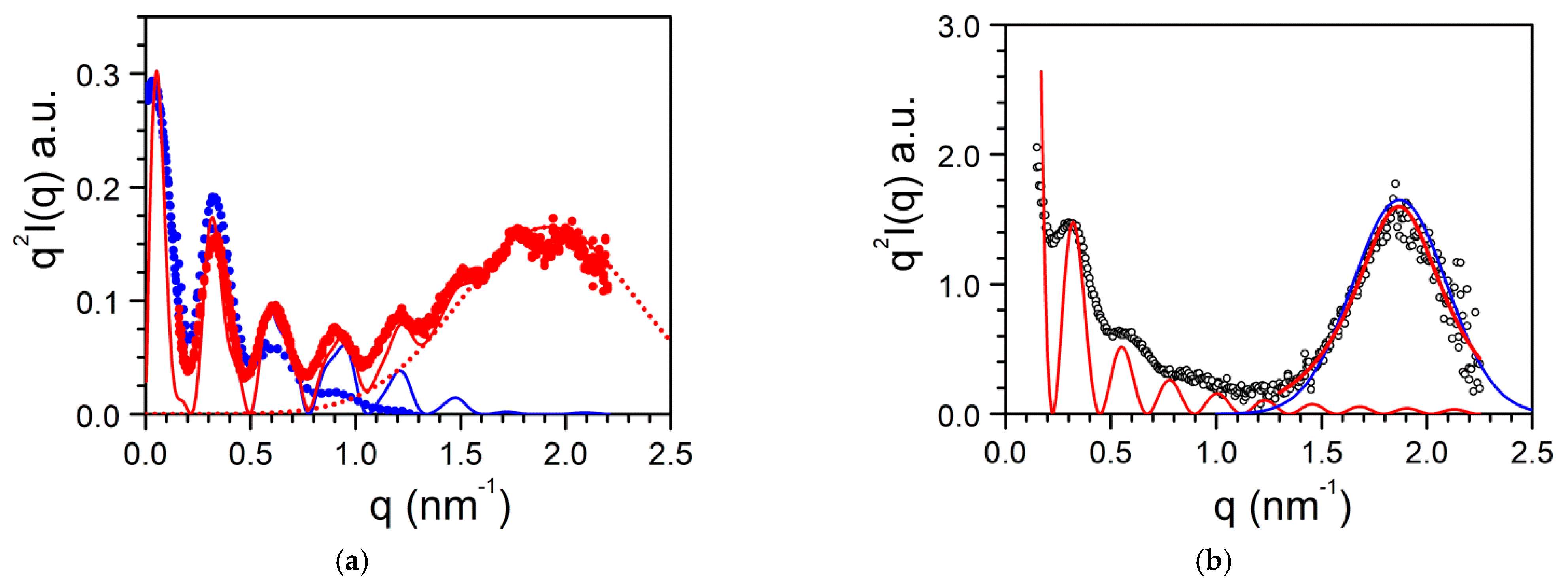
2.3. Morphology, Molecular Structure
2.4. Rheology
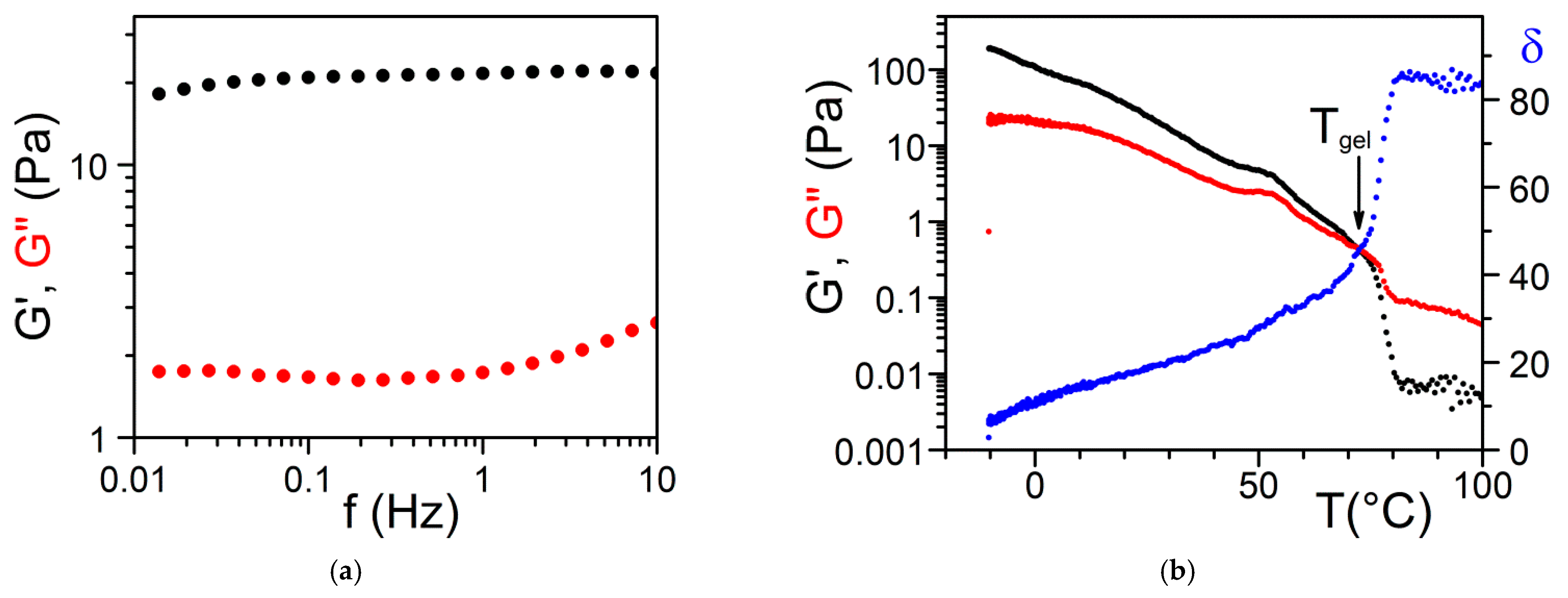
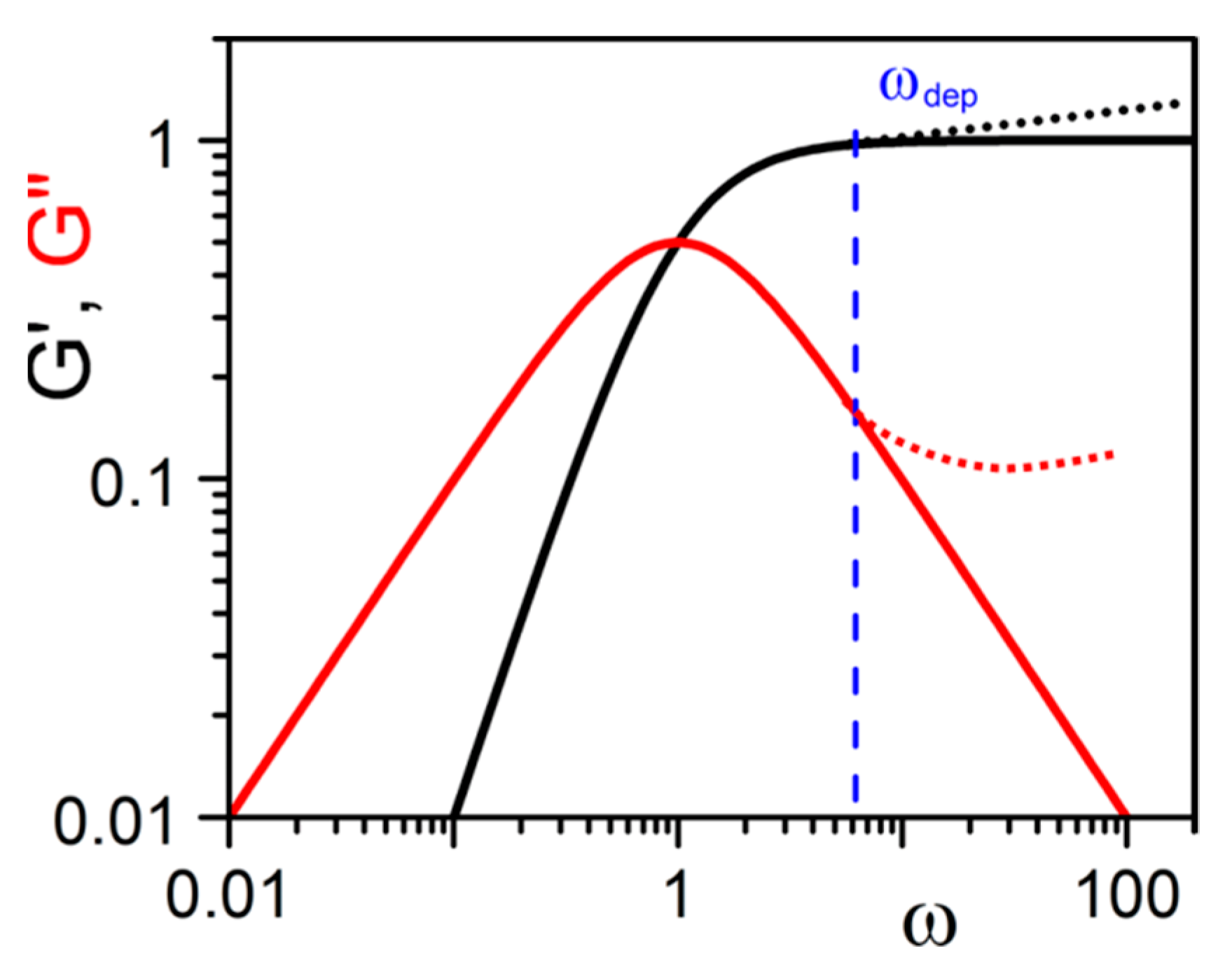
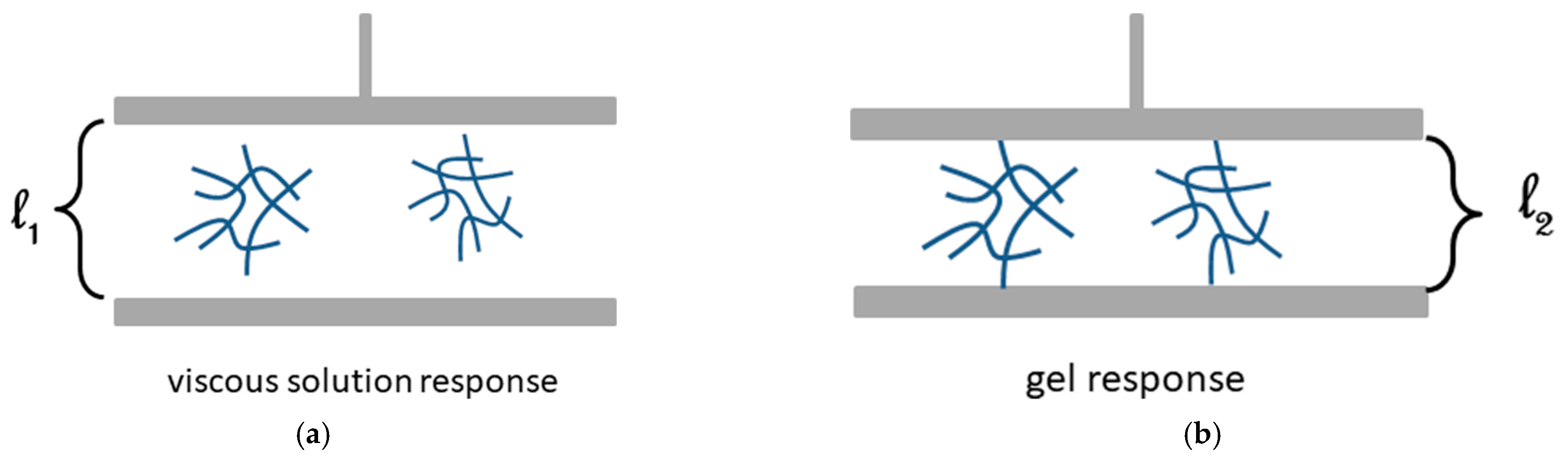
2.4.1. Origin of Elasticity
2.4.2. The Gel Point: Onset Gelation Concentration
2.4.3. Modulus vs. T-C Phase Diagram
3. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





References
- Terech, P.; Weiss, R.G. Low molecular mass gelators of organic liquids and the properties of their gels. Chem. Rev. 1997, 97, 3133–3159. [Google Scholar] [CrossRef]
- Terech, P.; Weiss, R.G. (Eds.) Molecular Gels: Materials with Self-Assembled Fibrillar Networks; Springer: New York, NY, USA, 2006. [Google Scholar]
- Liu, X.L.; Li, J.L. Soft Fibrillar Materials: Fabrication and Applications; Wiley-VCH: Basle, Switzerland, 2013. [Google Scholar]
- Babu, S.S.; Praveen, V.K.; Ajayaghosh, A. Functional π-Gelators and Their Applications. Chem. Rev. 2014, 114, 1973–2129. [Google Scholar] [CrossRef] [PubMed]
- Guenet, J.M. Organogels: Thermodynamics, Structure, Solvent Role and Properties; Springer: New York, NY, USA, 2016. [Google Scholar]
- Data from ISI Web of Science. Available online: https://isiknowledge.com (accessed on 1 December 2020).
- Lloyd, D.J. Colloid Chemistry: Theoretical and Applied; Alexander, J., Ed.; The Chemical Catalog Co.: New York, NY, USA, 1926; Volume 1, p. 7. [Google Scholar]
- Clark, A.H.; Ross-Murphy, S.B. Structural and mechanical properties of biopolymer gels. In Biopolymers. Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 1987; Volume 83. [Google Scholar]
- Guenet, J.M.; Mckenna, G.B. The concentration dependence of the compression modulus of iPS/cis-decalin gels. J. Polym. Sci. Polym. Phys. Ed. 1986, 24, 2499–2508. [Google Scholar] [CrossRef]
- Daniel, C.; Dammer, C.; Guenet, J.M. On the Definition of Thermoreversible Gels: Case of Syndiotactic Polystyrene. Polymer 1994, 35, 4243–4246. [Google Scholar] [CrossRef]
- Guenet, J.M. Thermoreversible Gelation of Polymers and Biopolymers; Academic Press: London, UK, 1992. [Google Scholar]
- Collin, D.; Covis, R.; Allix, F.; Jamart-Grégoire, B.; Martinoty, P. Jamming transition in solutions containing organogelator molecules of amino-acid type: Rheological and calorimetry experiments. Soft Matter 2013, 9, 2947–2958. [Google Scholar] [CrossRef]
- Dasgupta, D.; Thierry, A.; Rochas, C.; Ajayaghosh, A.; Guenet, J.M. Key role of solvent type in organogelation. Soft Matter 2012, 8, 8714–8721. [Google Scholar] [CrossRef]
- Dasgupta, D.; Srinivasan, S.; Rochas, C.; Ajayaghosh, A.; Guenet, J.M. Solvent-mediated fiber growth in organogels. Soft Matter 2011, 7, 9311–9315. [Google Scholar] [CrossRef]
- Guenet, J.M. Polymer-Solvent Molecular Compounds; Elsevier: London, UK, 2008. [Google Scholar]
- Daniel, C.; Longo, S.; Cardea, S.; Vitillo, J.G.; Guerra, G. Monolithic nanoporous-crystalline aerogels based on PPO. RSC Adv. 2012, 2, 12011–12018. [Google Scholar] [CrossRef]
- Lifshitz, I.; Slyozov, V.J. The kinetics of precipitation from supersaturated solid solutions. Phys. Chem. Solids. 1961, 19, 35–50. [Google Scholar] [CrossRef]
- Watzky, M.A.; Finke, R.G. Nanocluster Size-Control and “Magic Number” Investigations. Experimental Tests of the “Living-Metal Polymer” Concept and of Mechanism-Based Size-Control Predictions Leading to the Syntheses of Iridium(0) Nanoclusters Centering about Four Sequential Magic Numbers. Chem. Mater. 1997, 9, 3083–3095. [Google Scholar]
- Thanh, N.T.K.; Maclean, N.; Mahiddine, S. Mechanisms of Nucleation and Growth of Nanoparticles in Solution. Chem. Rev. 2014, 114, 7610–7630. [Google Scholar] [CrossRef]
- Gibbs, J.W. Elementary Principles in Statistical Mechanics; Charles Scribner’s Sons: New York, NY, USA, 1902. [Google Scholar]
- van ‘t Hoff, J.H. Études de Dynamique Chimique; Frederik Muller & Co.: Amsterdam, The Netherlands, 1884; pp. 4–5. [Google Scholar]
- Murata, K.; Aoki, M.; Suzuki, T.; Harada, T.; Kawabata, H.; Komri, T.; Ohrseto, F.; Ueda, K.; Shinkai, S. Thermal and Light Control of the Sol-Gel Phase Transition in Cholesterol-Based Organic Gels. Novel Helical Aggregation Modes As Detected by Circular Dichroism and Electron Microscopic Observation. J. Am. Chem. Soc. 1994, 116, 6664–6676. [Google Scholar] [CrossRef]
- Feng, L.; Cavicchi, K.A. Investigation of the relationships between the thermodynamic phase behavior and gelation behavior of a series of tripodal trisamide compounds. Soft Matter 2012, 8, 6483–6492. [Google Scholar] [CrossRef] [Green Version]
- Reisman, A. Phase Equilibria; Academic Press: New York, NY, USA, 1970. [Google Scholar]
- Carbonnel, L.; Rosso, J.C. Les clathrates des ethers cycliques: Leur stoichiométrie déduite des diagrammes de phases eau-éthers cycliques. J. Solid. State Chem. 1973, 8, 304–311. [Google Scholar] [CrossRef]
- Rie, E. Über die Einfluss der Oberflächenspannung auf Schmelzen und Gefrieren. Z. Phys. Chem. 1923, 104, 354–362. [Google Scholar]
- Moulin, E.; Niess, F.; Maaloum, M.; Buhler, E.; Nyrkova, L.; Giuseppone, N. The hierarchical self-assembly of charge nanocarriers: A highly cooperative process promoted by visible light Angew. Chem. Int. Ed. 2010, 49, 6974–6978. [Google Scholar] [CrossRef]
- Kiflemariam, B.; Collin, D.; Gavat, O.; Carvalho, A.; Moulin, E.; Giuseppone, N.; Guenet, J.M. Hybrid materials from tri-aryl amine organogelators and poly [vinyl chloride] networks. Polymer 2020, 207, 122814. [Google Scholar] [CrossRef]
- Cahn, J.W.; Hilliard, J.E. Free energy of a nonuniform system. I. Interfacial free energy. J. Chem. Phys. 1958, 28, 258–267. [Google Scholar] [CrossRef]
- Christ, E.; Blanc, C.; Al Ouahabi, A.; Maurin, D.; Le Parc, R.; Bantignies, J.L.; Guenet, J.M.; Collin, D.; Mésini, P.J. Origin of Invariant Gel Melting Temperatures in the c−T Phase Diagram of an Organogel. Langmuir 2016, 32, 4975–4982. [Google Scholar] [CrossRef]
- George, S.J.; Tomovic, Z.; Albertus; Schenning, P.H.J.; Meijer, E.W. Insight into the chiral induction in supramolecular stacks through preferential chiral solvation. Chem. Commun. 2011, 47, 3451–3452. [Google Scholar] [CrossRef]
- Kartha, K.K.; Babu, S.S.; Srinivasan, S.; Ajayaghosh, A. Attogram Sensing of Trinitrotolu-ene with a Self-Assembled Molecular Gelator. J. Am. Chem. Soc. 2012, 134, 4834–4841. [Google Scholar] [CrossRef]
- Dasgupta, D.; Kamar, Z.; Rochas, C.; Dahamani, M.; Mésini, P.J.; Guenet, J.M. Design of hybrid networks by sheathing polymer fibrils with self-assembled Nanotubules. Soft Matter 2010, 6, 3573–3581. [Google Scholar] [CrossRef]
- Morin, E.; Guenet, J.M.; Dıaz, D.D.; Remy, J.S.; Wagner, A. Fine-Tuning the Morphology of Self-Assembled Nanostructures of Propargyl Ammonium-Based Amphiphiles. J. Phys. Chem. B 2010, 114, 12495–12500. [Google Scholar] [CrossRef] [Green Version]
- Lowell, R.; Mitchell, G.R. Molecular orientation distribution derived from an arbitrary reflection. Acta Cryst. 1981, A37, 135–137. [Google Scholar]
- Diaz, N.; Simon, F.-X.; Schmutz, M.; Rawiso, M.; Decher, G.; Jestin, J.; Mesini, P.J. Self-Assembled Diamide Nanotubes in Organic Solvents. Angew. Chem. Int. Ed. 2005, 44, 3260–3264. [Google Scholar] [CrossRef]
- Mittelbach, P.; Porod, G. Zur Röntgenkleinwinkelstreuung verdünnter kolloiden Systeme. Acta Phys. Austriaca 1961, 14, 185–211. [Google Scholar]
- Khan, A.N.; Nguyen, T.T.T.; Dobircau, L.; Schmutz, M.; Mesini, P.J.; Guenet, J.M. Investigation of the Interactions Involved in the Formation of Nanotubes from Organogelators. Langmuir 2013, 29, 16127–16134. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, D.; Srinivasan, S.; Rochas, C.; Ajayaghosh, A.; Guenet, J.M. Hybrid the-moreversible gels from covalent polymers and organogels. Langmuir 2009, 25, 8593–8598. [Google Scholar] [CrossRef]
- Porod, G. Die Rontgenkleinwinkelstreuung Von Dichtgepackten Kolloiden Systemen. Kolloid-Zeitschrift 1951, 125, 51–57. [Google Scholar] [CrossRef]
- Jones, J.L.; Marques, C.M. Rigid polymer network models. J. Phys. 1990, 51, 1113–1127. [Google Scholar] [CrossRef] [Green Version]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University Press: Oxford, UK, 1986. [Google Scholar]
- Cates, M.E. Reptation of living polymers: Dynamics of entangled polymers in the presence of reversible chain-scission reactions. Macromolecules 1987, 20, 2289–2296. [Google Scholar] [CrossRef]
- Cates, M.E. Dynamics of living polymers and flexible surfactant micelles: Scaling laws for dilution. J. Phys. France 1988, 49, 1593–1600. [Google Scholar] [CrossRef]
- Lescanne, M.; Colin, A.; Mondain-Monval, O.; Fages, F.; Pozzo, J.L. Structural Aspects of the Gelation Process Observed with Low Molecular Mass Organogelators. Langmuir 2003, 19, 2013–2020. [Google Scholar] [CrossRef]
- Terech, P.; Pasquier, D.; Bordas, V.; Rossat, C. Rheological Properties and Structural Correlations in Molecular Organogels. Langmuir 2000, 16, 4485–4494. [Google Scholar] [CrossRef]
- de Gennes, P.G. On a relation between percolation theory and the elasticity of gels. J. Phys. Fr. Lett. 1976, 37, 1. [Google Scholar] [CrossRef]
- Stauffer, D. Introduction to Percolation Theory; Taylor and Francis: London, UK, 1985. [Google Scholar]
- Guenet, J.M. Structure versus rheological properties in fibrillary thermoreversible gels from polymers and biopolymers. J. Rheol. 2000, 44, 947–960. [Google Scholar] [CrossRef]
- Collin, D.; Martinoty, P. Dynamic macroscopic heterogeneities in a flexible linear polymer melt. Phys. A Stat. Mech. Its Appl. 2003, 320, 235–248. [Google Scholar] [CrossRef]
- De Gennes, P.G. Scaling Concepts in Polymer Physics; Cornell University Press: Ithaca, NY, USA, 1979. [Google Scholar]
- Li, L.; Aoki, Y. Rheological Images of Poly (vinyl chloride) Gels. 1. The Dependence of Sol.-Gel Transition on Concentation. Macromolecules 1997, 30, 7835–7841. [Google Scholar] [CrossRef]
- Li, L.; Uchida, H.; Aoki, Y. Rheological Images of Poly (vinyl chloride) Gels. 2. Divergence ofViscosity and the Scaling Law before the Sol.-Gel Transition. Macromolecules 1997, 30, 7842–7848. [Google Scholar] [CrossRef]
- Lan, Y.; Corradini, M.G.; Weiss, R.G.; Raghavan, S.R.; Rogers, M.A. To gel or not to gel: Correlating molecular gelation with solvent parameters. Chem. Soc. Rev. 2015, 44, 6035–6058. [Google Scholar] [CrossRef]
- Dastidar, P. Supramolecular gelling agents: Can they be designed? Chem. Soc. Rev. 2008, 37, 2699–2715. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Adam, F.B.; Michel, M.; Schmutz, M.; Decher, G.; Mesini, P.J. New bisamides gelators: Relationship between chemical structure and fiber morphology. Tetrahedron Let. 2003, 44, 3171–3174. [Google Scholar] [CrossRef]


















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guenet, J.-M. Physical Aspects of Organogelation: A Point of View. Gels 2021, 7, 65. https://doi.org/10.3390/gels7020065
Guenet J-M. Physical Aspects of Organogelation: A Point of View. Gels. 2021; 7(2):65. https://doi.org/10.3390/gels7020065
Chicago/Turabian StyleGuenet, Jean-Michel. 2021. "Physical Aspects of Organogelation: A Point of View" Gels 7, no. 2: 65. https://doi.org/10.3390/gels7020065
APA StyleGuenet, J. -M. (2021). Physical Aspects of Organogelation: A Point of View. Gels, 7(2), 65. https://doi.org/10.3390/gels7020065