
Preparation, Properties and Cell Biocompatibility of Room Temperature LCST-Hydrogels Based on Thermoresponsive PEO Stars


Abstract
:
1. Introduction
2. Results and Discussion
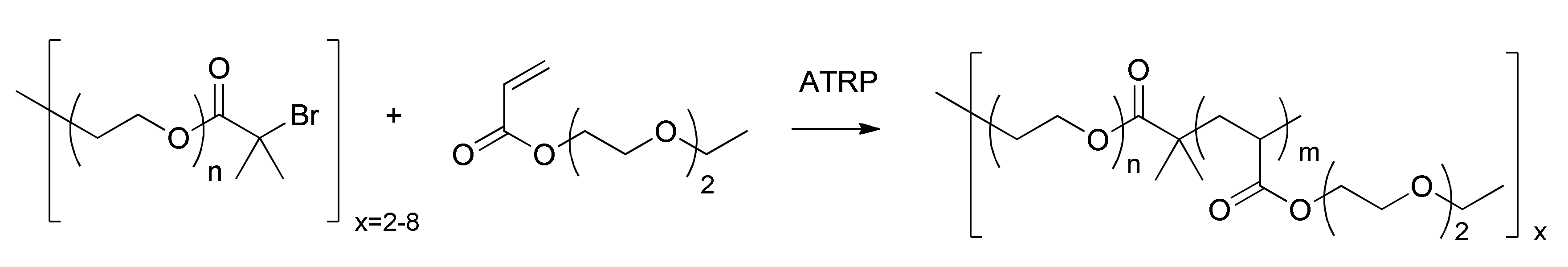
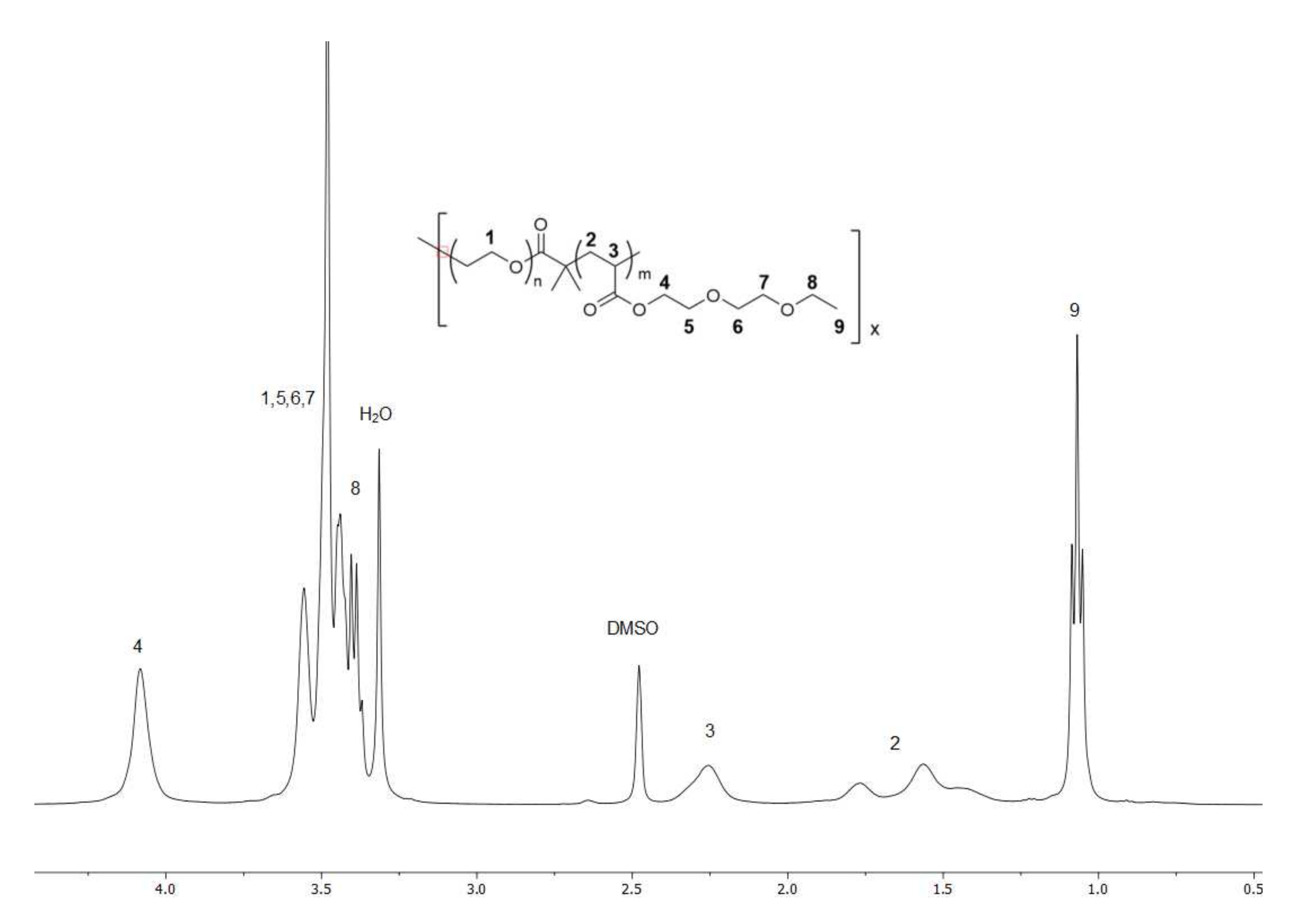
2.1. Synthesis
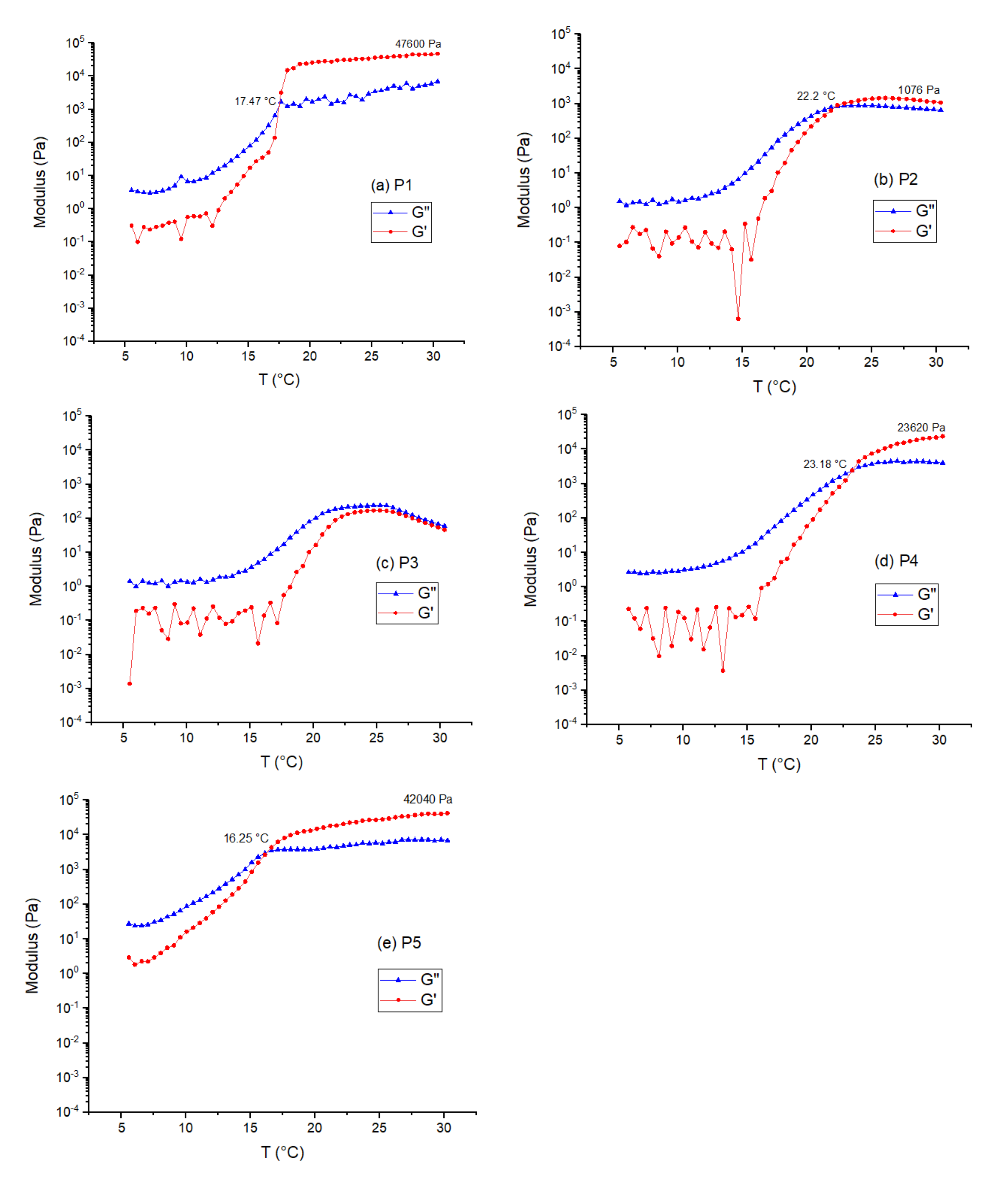
2.2. Gelation Studies
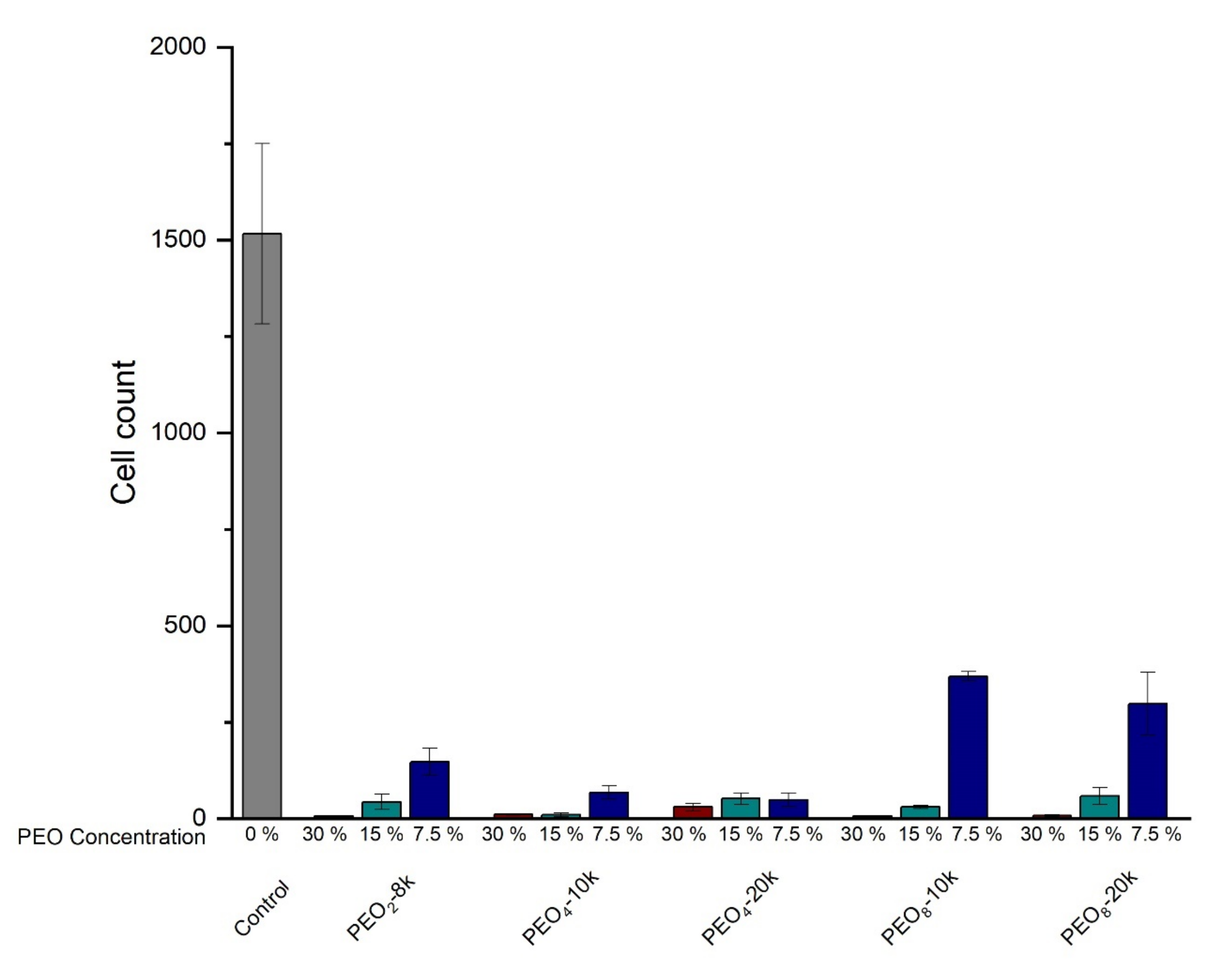
2.3. Cell Studies
3. Conclusions
4. Materials and Methods
4.1. Characterisation
4.2. Synthetic Procedures
4.2.1. General Synthesis of PEO-Star ATRP Initiators
4.2.2. General ATRP Procedure
4.2.3. SARA ATRP Procedure
4.2.4. Cell Studies
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wichterle, O.; Lim, D. Hydrophilic Gels for Biological Use. Nature 1960, 185, 117–118. [Google Scholar] [CrossRef]
- Curcio, M.; Picci, N. Polymer in agriculture: A review. Am. J. Agric. Biol. Sci. 2008, 3, 299–314. [Google Scholar]
- Liu, B.; Hu, J. The application of temperature-sensitive hydrogels to textiles: A review of Chinese and Japanese investigations. Fibres Tex. East. Eur. 2005, 13, 45–49. [Google Scholar]
- Chen, X.; Martin, B.D.; Neubauer, T.K.; Linhardt, R.J.; Dordick, J.S.; Rethwisch, D.G. Enzymatic and chemoenzymatic approaches to synthesis of sugar-based polymer and hydrogels. Carb. Polym. 1995, 28, 15–21. [Google Scholar] [CrossRef]
- Goering, R.V. Pulsed field gel electrophoresis: A review of application and interpretation in the molecular epidemiology of infectious disease. Infect. Genet. Evol. 2010, 10, 866–875. [Google Scholar] [CrossRef]
- Yang, J.; Bai, R.; Chen, B.; Suo, Z. Hydrogel Adhesion: A Supramolecular Synergy of Chemistry, Topology, and Mechanics. Adv. Funct. Mat. 2019, 1901693. [Google Scholar] [CrossRef]
- Roy, D.; Brooks, W.L.A.; Sumerlin, B.S. New directions in thermoresponsive polymers. Chem. Soc. Rev. 2013, 42, 7214–7243. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S. “Intelligent” polymers in medicine and biotechnology. Artif. Organs 1995, 19, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Kamoun, E.A.; Kenawy, E.-R.S.; Chen, X. A review on polymeric hydrogel membranes for wound dressing applications: PVA-based hydrogel dressings. J. Adv. Res. 2017, 8, 217–233. [Google Scholar] [CrossRef]
- Zele, D.V.; Heymans, O. Breast implants a review. Acta Chir. Belg. 2004, 104, 158–165. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Vermani, K.; Garg, S. Hydrogels: From controlled release to pH-responsive drug delivery. Drug Discov. Today 2002, 7, 569–579. [Google Scholar] [CrossRef]
- Krsko, P.; McCann, T.E.; Thach, T.-T.; Laabs, T.L.; Geller, H.M.; Libera, M.R. Length-scale mediated adhesion and directed growth of neural cells by surface-patterned poly (ethylene glycol) hydrogels. Biomaterials 2009, 30, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef] [Green Version]
- Calvert, P. Hydrogels for Soft Machines. Adv. Mater. 2009, 21, 743–756. [Google Scholar] [CrossRef]
- Koetting, M.C.; Peters, J.T.; Steichen, S.D.; Peppas, N.A. Stimulus-responsive hydrogels: Theory, modern advances, and applications. Mater. Sci. Eng. R Rep. 2015, 93, 1–49. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, R.; Sakai, K.; Okano, T.; Sakurai, Y. Modulating the phase transition temperature and thermosensitivity in N- Amphiphilic poly (N-isopropylacrylamides) prepared by isopropylacrylamide copolymer gels. J. Biomater. Sci. Polym. Ed. 1994, 6, 585–598. [Google Scholar] [CrossRef]
- Jeong, B.; Bae, Y.H.; Lee, D.S.; Kim, S.W. Biodegradable block copolymers as injectable drug-delivery systems. Nature 1997, 388, 860. [Google Scholar] [CrossRef] [PubMed]
- Scherlund, M.; Brodin, A.; Malmsten, M. Nonionic cellulose ethers as potential drug delivery systems for periodontal anesthesia. J. Colloid Interface Sci. 2000, 229, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Ratner, B.D.; Hoffman, A.S. Synthetic Hydrogels for Biomedical Applications. J. Am. Chem. Soc. 1976, 31, 1–36. [Google Scholar]
- Tan, H.; Ramirez, C.M.; Miljkovic, N.; Li, H.; Rubin, J.P.; Marra, K.G. Thermosensitive injectable hyaluronic acid hydrogel for adipose tissue engineering. Biomaterials 2009, 30, 6844–6853. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.H.; Park, K.; Han, D.K. Preparation of TGF-β1-conjugated biodegradable pluronic F127 hydrogel and its application with adipose-derived stem cells. J. Control. Release 2010, 147, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H.; Yamamoto, K.; Matsushima, Y.; Takai, N.; Kikuchi, A.; Sakurai, Y.; Okano, T. Temperature-responsive chromatography using poly (N-isopropylacrylamide)-modified silica. Anal. Chem. 1996, 68, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, H.; Yamamoto, K.; Kashiwase, Y.; Matsushima, Y.; Takai, N.; Kikuchi, A.; Sakurai, Y.; Okano, T. Analysis of peptides and proteins by temperature-responsive chromatographic system using N-isopropylacrylamide polymer-modified columns. J. Pharm. Biomed. Anal. 1997, 15, 1545–1550. [Google Scholar] [CrossRef]
- Kushida, A.; Yamato, M.; Konno, C.; Kikuchi, A.; Sakurai, Y.; Okano, T. Decrease in culture temperature releases monolayer endothelial cell sheets together with deposited fibronectin matrix from temperature-responsive culture surfaces. J. Biomed. Mater. Res. 1999, 45, 355–362. [Google Scholar] [CrossRef]
- Ebara, M.; Yamato, M.; Aoyagi, T.; Kikuchi, A.; Sakai, K.; Okano, T. Immobilization of cell-adhesive peptides to temperature-responsive surfaces facilitates both serum-free cell adhesion and noninvasive cell harvest. Tissue Eng. 2004, 10, 1125–1135. [Google Scholar] [CrossRef]
- Klouda, L.; Mikos, A.G. Thermoresponsive hydrogels in biomedical applications. Eur. J. Pharm. Biopharm. 2008, 68, 34–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef]
- Yoshida, R.; Okano, T. Stimuli-Responsive Hydrogels and Their Application to Functional Materials. In Biomedical Applications of Hydrogels Handbook; Ottenbrite, R.M., Park, K., Okano, T., Eds.; Springer: New York, NY, USA, 2010; pp. 19–43. [Google Scholar]
- Aseyev, V.; Tenhu, H.; Winnik, F.M. Non-ionic Thermoresponsive Polymers in Water. In Self Organized Nanostructures of Amphiphilic Block Copolymers II; Müller, H.E.A., Borisov, O., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 29–89. [Google Scholar]
- Liu, M.; Song, X.; Wen, Y.; Zhu, J.-L.; Li, J. Injectable Thermoresponsive Hydrogel Formed by Alginate-g-Poly(N-isopropylacrylamide) That Releases Doxorubicin-Encapsulated Micelles as a Smart Drug Delivery System. ACS Appl. Mater. Interfaces 2017, 9, 35673–35682. [Google Scholar] [CrossRef] [PubMed]
- Hemp, S.T.; Smith, A.E.; Bunyard, W.C.; Rubinstein, M.H.; Long, T.E. RAFT polymerization of temperature- and salt-responsive block copolymers as reversible hydrogels. Polymer 2014, 55, 2325–2331. [Google Scholar] [CrossRef] [Green Version]
- Vancoillie, G.; Frank, D.; Hoogenboom, R. Thermoresponsive poly(oligo ethylene glycol acrylates). Progr. Polym. Sci. 2014, 39, 1074–1095. [Google Scholar] [CrossRef]
- Skrabania, K.; Kristen, J.; Laschewsky, A.; Akdemir, Ö.; Hoth, A.; Lutz, J.-F. Design, Synthesis, and Aqueous Aggregation Behavior of Nonionic Single and Multiple Thermoresponsive Polymers. Langmuir 2007, 23, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. Poly (ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. 2010, 49, 6288–6308. [Google Scholar] [CrossRef]
- Boyer, C.; Whittaker, M.R.; Luzon, M.; Davis, T.P. Design and synthesis of dual thermoresponsive and antifouling hybrid polymer/gold nanoparticles. Macromolecules 2009, 42, 6917–6926. [Google Scholar] [CrossRef]
- De, P.; Sumerlin, B.S. Precision control of temperature response by copolymerization of di (ethylene glycol) acrylate and an acrylamide comonomer. Macromol. Chem. Phys. 2013, 214, 272–279. [Google Scholar] [CrossRef]
- Semenov, A.; Joanny, J.-F.; Khokhlov, A. Associating polymers: Equilibrium and linear viscoelasticity. Macromolecules 1995, 28, 1066–1075. [Google Scholar] [CrossRef]
- Neugebauer, D.; Zhang, Y.; Pakula, T.; Sheiko, S.S.; Matyjaszewski, K. Densely-Grafted and Double-Grafted PEO Brushes via ATRP. A Route to Soft Elastomers. Macromolecules 2003, 36, 6746–6755. [Google Scholar] [CrossRef]
- Aksakal, R.; Resmini, M.; Becer, C.R. SET-LRP of acrylates catalyzed by a 1 penny copper coin. Polym. Chem. 2016, 7, 6564–6569. [Google Scholar] [CrossRef] [Green Version]
- Goswami, S.K.; McAdam, C.J.; Hanton, L.R.; Moratti, S.C. Hyperelastic Tough Gels through Macrocross-Linking. Macromol. Rapid Commun. 2017, 38, 1700103. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, C.V.; Luo, Y.-Z.; Deng, N.-J.; Attwood, D.; Collett, J.H.; Price, C.; Booth, C. Effect of chain length on the micellization and gelation of block copoly(oxyethylene/oxybutylene/oxyethylene). Polymer 1993, 34, 138–144. [Google Scholar] [CrossRef]
- Fechler, N.; Badi, N.; Schade, K.; Pfeifer, S.; Lutz, J.-F. Thermogelation of PEG-based macromolecules of controlled architecture. Macromolecules 2008, 42, 33–36. [Google Scholar] [CrossRef]
- Kirkland, S.E.; Hensarling, R.M.; McConaughy, S.D.; Guo, Y.; Jarrett, W.L.; McCormick, C.L. Thermoreversible hydrogels from RAFT-synthesized BAB triblock copolymers: Steps toward biomimetic matrices for tissue regeneration. Biomacromolecules 2007, 9, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Lui, G.; Li, Y.; Yang, L.; Wei, Y.; Wang, Y.; Wang, X.; Tao, L. Cytotoxicity study of polyethylene glycol derivatives. RSC Adv. 2017, 30, 18252–18259. [Google Scholar]
- Webster, R.; Elliot, V.; Park, K.B.; Walker, D.; Hankin, M.; Taupin, P. PEG and PEG conjugates toxicity: Towards an understanding of the toxicity of PEG and its relevance to PEGylated biologicals. In PEGylated Protein Drugs: Basic Science and Clinical Applications; Milestones in Drug Therapy; Veronese, F.M., Ed.; Birkhäuser: Basel, Switzerland, 2009; pp. 127–146. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NMR a | GPC b | Rheometry c | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Polymer | Initiator | No. of Arms | Mw of PEGDA | Total Mw | Mn (Da) | Mw (Da) | G′max (kPa) | G″max (kPa) | Tgel (°C) |
| P1 | PEO2-8k | 2 | 15,700 | 23,600 | 14,500 | 15,300 | 47.5 | 6.0 | 12.2 |
| P2 | PEO4-10k | 4 | 41,700 | 51,700 | 20,600 | 23,900 | 1.1 | 0.68 | 13.6 |
| P3 | PEO8-10k | 8 | 50,700 | 60,700 | 23,500 | 26,900 | 0.17 | 0.24 | - |
| P4 | PEO8-20k | 8 | 72,700 | 92,700 | 27,000 | 29,600 | 23.6 | 3.9 | 12.6 |
| P5 | PEO8-40k | 8 | 84,500 | 124,500 | 40,400 | 41,500 | 42.0 | 7.2 | 11.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoso, B.; Turner, P.R.; Hanton, L.R.; Moratti, S.C. Preparation, Properties and Cell Biocompatibility of Room Temperature LCST-Hydrogels Based on Thermoresponsive PEO Stars. Gels 2021, 7, 84. https://doi.org/10.3390/gels7030084
Santoso B, Turner PR, Hanton LR, Moratti SC. Preparation, Properties and Cell Biocompatibility of Room Temperature LCST-Hydrogels Based on Thermoresponsive PEO Stars. Gels. 2021; 7(3):84. https://doi.org/10.3390/gels7030084
Chicago/Turabian StyleSantoso, Bagus, Paul R. Turner, Lyall R. Hanton, and Stephen C. Moratti. 2021. "Preparation, Properties and Cell Biocompatibility of Room Temperature LCST-Hydrogels Based on Thermoresponsive PEO Stars" Gels 7, no. 3: 84. https://doi.org/10.3390/gels7030084
APA StyleSantoso, B., Turner, P. R., Hanton, L. R., & Moratti, S. C. (2021). Preparation, Properties and Cell Biocompatibility of Room Temperature LCST-Hydrogels Based on Thermoresponsive PEO Stars. Gels, 7(3), 84. https://doi.org/10.3390/gels7030084