2. Results and Discussion
Cylindrical (1 mm in diameter and 30 mm in length) and disk-shaped (80 mm in diameter and 20 mm in thickness) acrylamide gels were used for the solvent exchange experiment and for indentation test, respectively. The synthesis procedure of the gel (including the composition of the pre-gel solution) was identical to that in previous investigations [
4,
6]; see
Section 4 for experimental details. The indentation test was done in order to measure the bulk mechanical property of the specimen gel and to check the consistency with the osmotic property; the used indenter is a stainless steel ball with a radius of
.
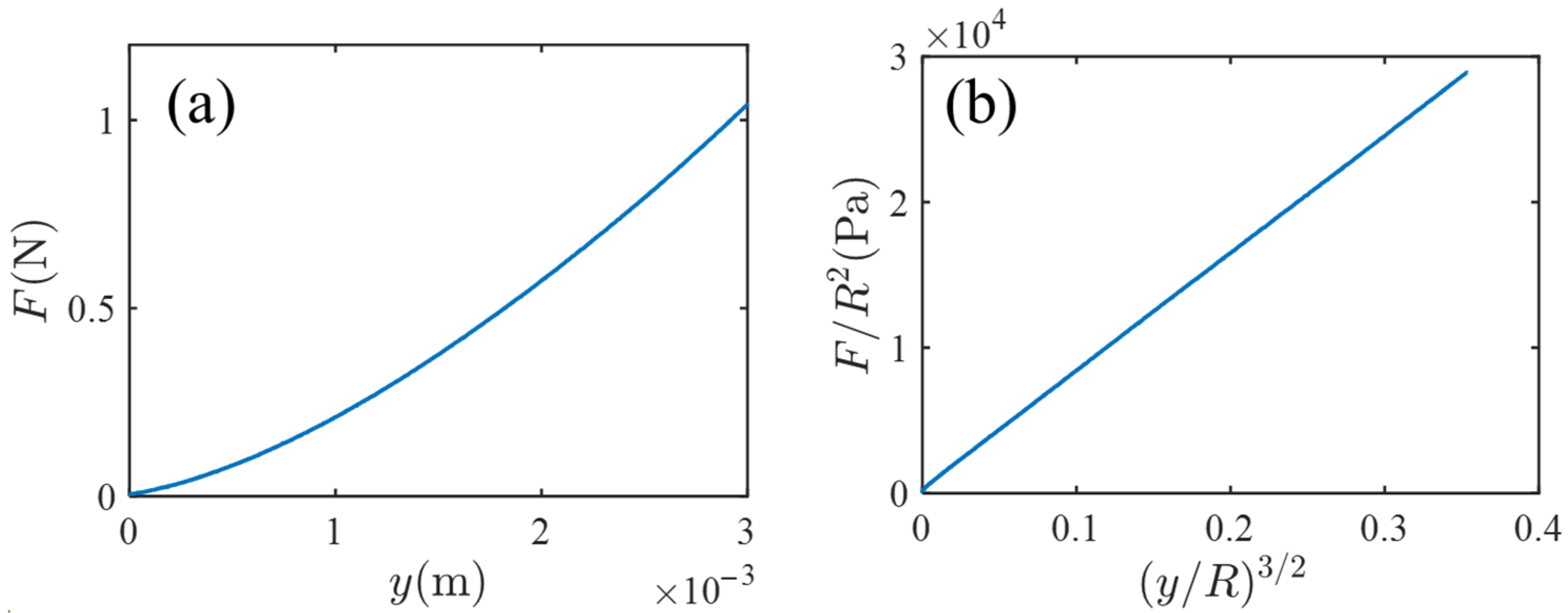
Figure 2 shows the result of the indentation test.
Figure 2a is a plot of the measured indentation force
versus indentation length
.
Figure 2b is the reduced plot of (
versus
; the choice of the quantities of the vertical and horizontal axes is based on the prediction of the Hertz contact theory [
7],
, where
and
are the Young modulus and the Poisson ratio, respectively. In
Figure 2b, the linearity is fine, and from the slope of
Figure 2b (and setting
), the Young modules
is estimated by
Pa.
Blow, we present results of the solvent exchange experiment in which EG-swollen cylindrical gels are immersed into PEG solutions; hereafter, the concentration of PEG is denoted by
.
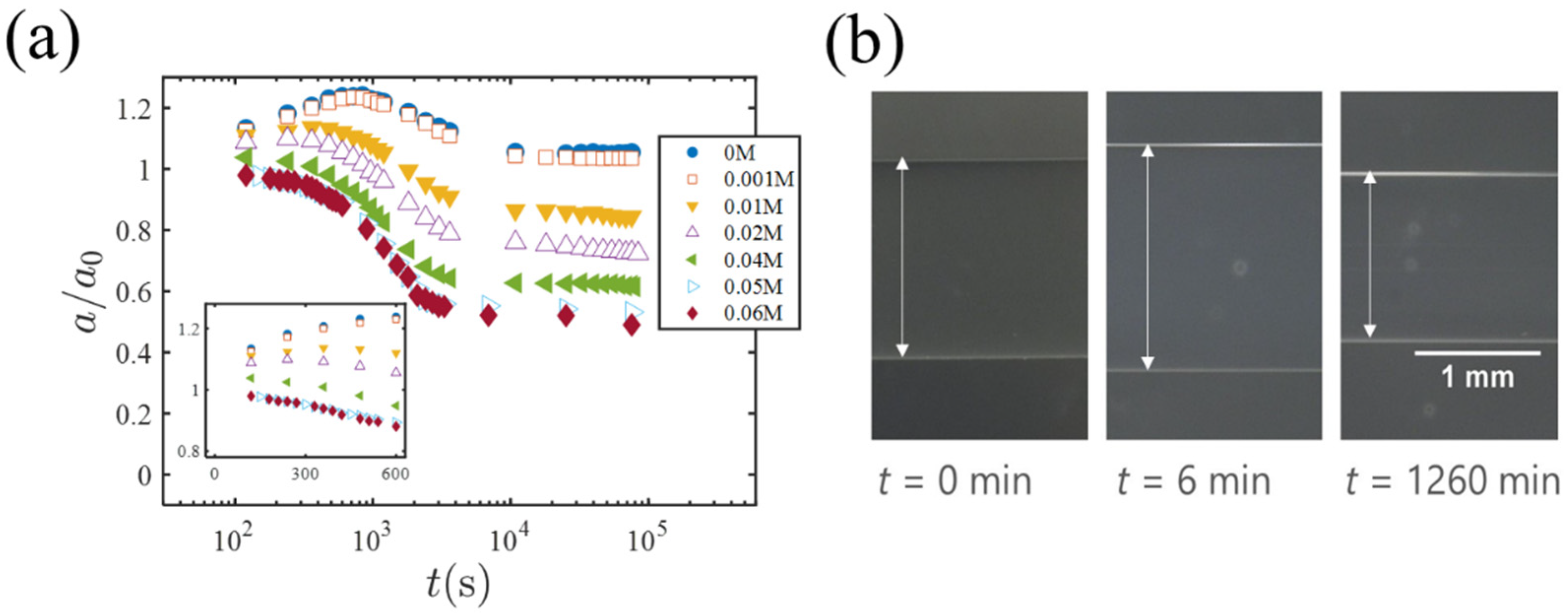
Figure 3a is a plot of the gel radius
(normalized by the initial radius
) versus
(
is the time when the EG-swollen gel is exposed to the PEG solution). The insert shows the short-time behavior.
Figure 3b shows photos of a gel specimen (
0.01 M) at different times. For low PEG concentrations (
= 0 and 0.001 M), the radius
shows a clear peak and then decreases to a final value almost the same as (but slightly larger than)
. With increasing
, the peak becomes lower and almost flat at a higher concentration of
M. For
and
M, it is almost certain that the peak of the gel volume does not occurs (see the insert of
Figure 3a).
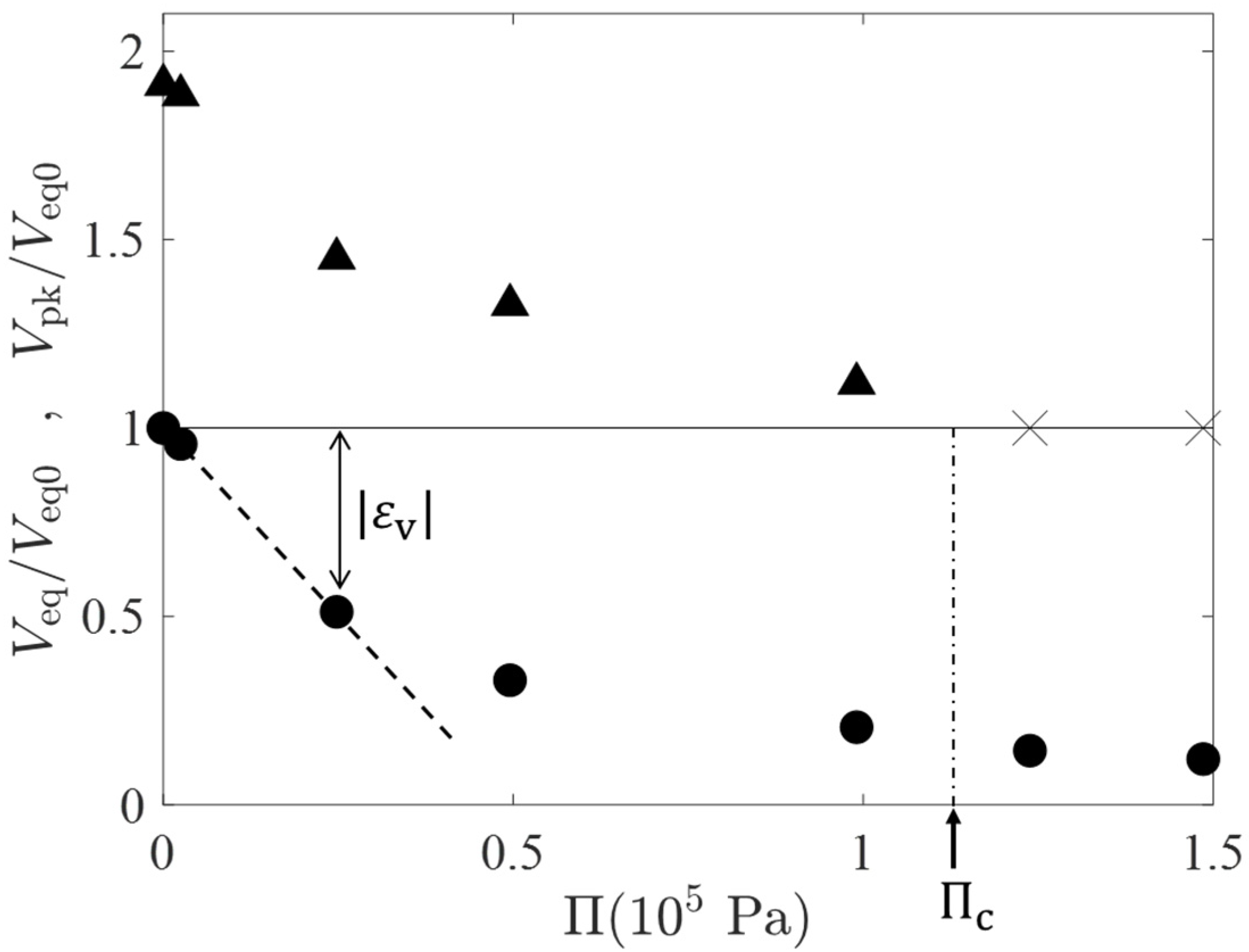
Figure 4 shows a plot of the osmotic pressure
estimated by the van’t Hoff equation (
; the unit of
is converted to mol/m
3) versus the peak volume
(the filled triangles) during the temporal swelling and the equilibrium volume
(filled circles) estimated by
. In
Figure 4, those characteristic volumes are normalized by
, the equilibrium volume for
(i.e.,
). The cross marks in the horizontal line of a height of unity represent that the volume peak was not observed at the
values. From the behavior of the peak volume, we have an estimation for the critical osmotic pressure
at which the volume peak just disappears,
Pa.
represents the volumetric strain at the final equilibrium (see the double-headed arrow in
Figure 4) and the initial slope of
versus
relation (the dashed line) gives an estimation for the osmotic bulk modulus
of
Pa;
is of the same order of
and approximately a half of
. The
-
relation deviates from the linear one when
exceeds 0.5. This is probably due to the strong repulsion between partial chains of the highly compressed gels.
In what follows, we discuss the experimental results based on the theoretical model developed in our previous investigation [
4]. We introduce a simple modification to the theoretical setting, that is, the PEG in the outer solution exerts a constant (i.e., independent of time and position) osmotic pressure
on the gel surface.
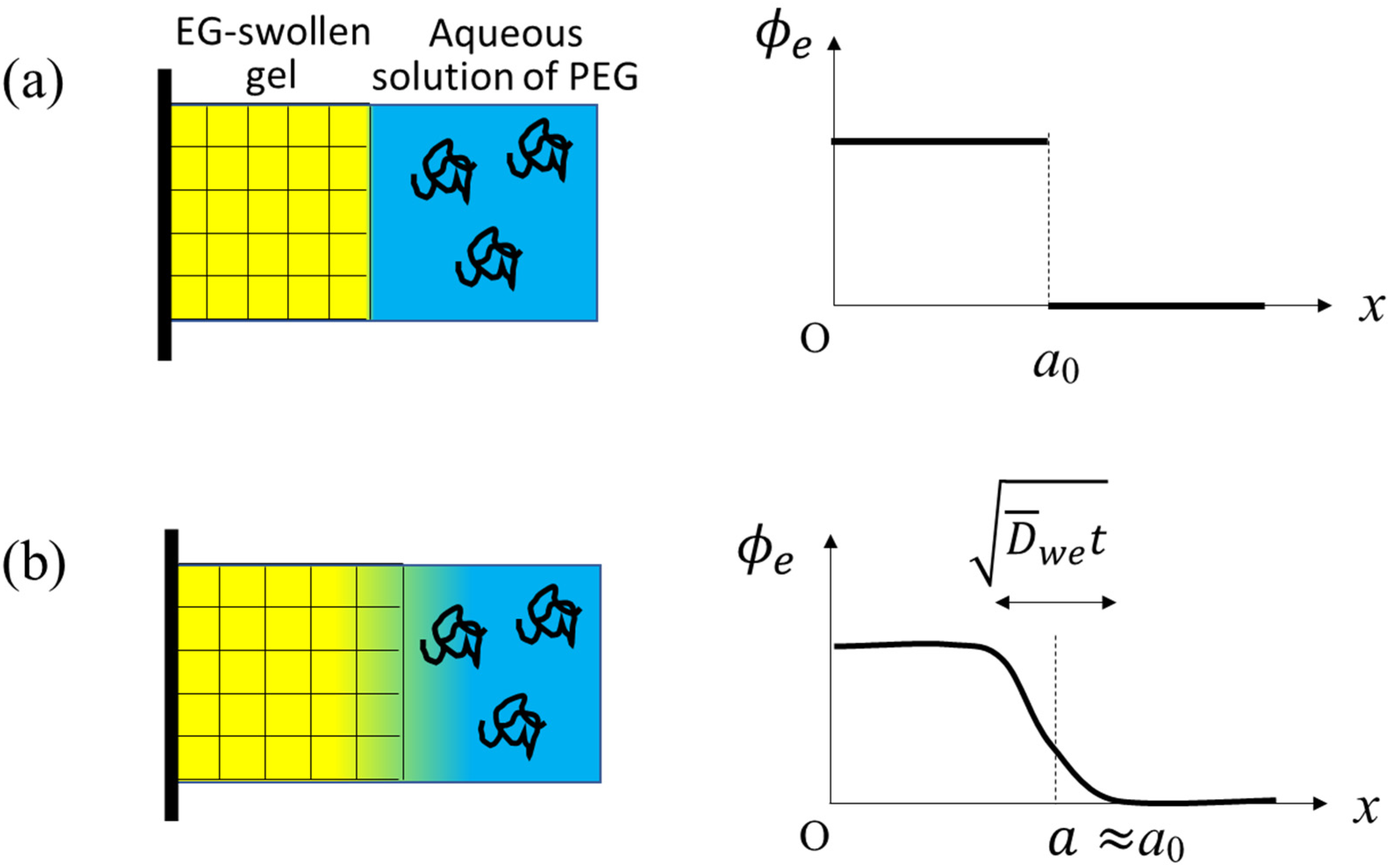
We suppose a 1-dimentional gel (i.e., gel slab) upon the solvent exchange from EG to water (see the illustrations in
Figure 1). The left surface (
) of the gel slab is fixed to a rigid wall and the other surface (
) is exposed to the outer solution. Actual specimens used in the experiment were cylindrical, and at the central axis of the cylinder, the radial components of the displacement of the gel network and solvent fluxes are zero by symmetry. The rigid wall in the present theoretical setting corresponds to the central axis.
The Onsager principle gives a set of time-evolutional equations of the gel dynamics in the binary solvent [
4] with the aid of the conservation laws:
where
and
are the volume fraction and velocity of the
i-th component, respectively (subscripts of “w”, “e” and “p” represent water, EG and polymer (of the gel network), respectively). Note that
, where
is the displacement of the gel network from the initial and equilibrium position (hereafter, we often use the dot notation for time derivatives). The Onsager principle in the present case is symbolically expressed as
, where
is the Rayleighian defined as the sum of a half of the dissipation rate
of the entire system and the time derivative of the system energy
. The above symbolic expression represents the stationary condition for
with respective to a small change of
at fixed
. We employ the following forms of
and
where
is the friction coefficient per volume (and per unit volume fractions) between the
-
combination;
is the mixing free energy density (where
is the mean molecular volume of the solvents; we consider the entropic term only as in [
4], because the equilibrium swelling volume of the gel hardly depends on
, the solvent composition) and
is the elastic modulus of the gel network. The second term of Equation (3), corresponding to the “
pV term” in the enthalpy of gases, comes from the assumption that the effect of PEG in the outer solution is exerting a constant osmotic pressure
on the surface of the gel network at
(and the left surface is fixed to the rigid surface,
). Because of the incompressibility of the system,
, the quantity to be minimized is
, where the Lagrange multiplier
has the physical meaning of pressure. The integral terms contained in
are completely identical to those in [
4]. Thus, a set of time-evolutional equations completely identical to those in [
4] are obtained by the parallel procedure of calculation; that is, (i) replacing
with
by use of the conservation law of Equation (1) and of integration by parts; (ii) calculating the variation of
with respect to
to obtain the relations between the velocities
and the thermodynamical potentials
; and (iii) re-employing Equation (1) to obtain the closed set of equations. When the gel network is dilute but its deformation remains small, the process of the solvent mixing is hardly disturbed by the gel network. In this case, a perturbative consideration allows us to simplify a complicated set of coupled partial differential equation of the gel dynamics into a pair of diffusion equations describing the mixing of the solvents
and the force balance condition for a unit volume of the gel network
where
is the mutual diffusion coefficient of the solvents,
is the volume flux of EG, and
is the polymer volume fraction in the initial and the final equilibrium state, and
is the friction coefficient of the ‘means’ solvent. The term proportional to
in Equation (5) represents the friction force by the solvent fluxes.
On the other hand, the osmotic pressure term
in Equation (3) plays an important role in the boundary condition on the right gel surface. That is, by using integration by parts,
and the variation of the second term leads to the following boundary condition on the gel surface,
Note that in the present simplified (perturbative) treatment, any boundary (or connection) condition of is not imposed on the gel surface, because we assume that the mixing of the solvents occurs as if the gel network does not exist.
Based on Equations (4)–(7), we consider the critical osmotic pressure
at which the peak of the gel volume just disappears. At
, the gel surface is stationary in the early stage of the solvent exchange (or solvent mixing); we may consider that the inside of the gel is also stationary,
for
. With this simplification, the force balance condition of Equation (5) is reduced to
. Integrating both sides from
to
and using Equation (7), we obtain
, where
is the mutual diffusion coefficient in the EG-rich limit. In the first approximative equality, we use
in the early stage of the solvent mixing (see
Figure 1b) and the rightmost inequality comes from the fact that
decreases with
. Because
is 1/2 or so, we have an order estimation for the critical osmotic pressure:
The right-hand side of Equation (8) can be estimated by relating
and
to the cooperative diffusion constants in pure EG and pure water. Setting
and
in Equations (5) and (6), we have
, where
and
are the cooperative diffusion constants of the gel in water and in EG, respectively, and
. Because the equilibrium volume of the gel (or the mesh size of the gel network) is almost the same for water and for EG,
is governed by the ratio of viscosities of water (
) and EG (
). Hence,
. At room temperature, the viscosity ratio is
. Combining the above observation,
and
. Thus, Equation (8) becomes
. According to literature,
m/s
2 [
6] and
m
2/s [
8]. We may consider
Pa. Thus, we have
Pa. This is, however, much (2 digits) larger than the experimental value of
estimated by the van’t Hoff equation. The above theoretical consideration says that the “effective osmotic pressure” in the early stage of the solvent exchange is much higher than the equilibrium one.
Why can the PEG chains in the outer solution suppress the friction-driven volume expansion so effectively? One may suppose that the outward EG flux exerts friction forces on the PEG chains, as well as the gel network, to drive them away from the gel surface (and the osmotic pressure by PEG is screened). This is opposite to what actually occurs. A possible answer for the above question is the affinity (i.e., the enthalpic interactions) among the components, which has not been taken into account in our coupled diffusion model. Because of the similarity in the chemical structure, PEG chains may have a stronger affinity to EG than that to water and be attracted by the EG secreted on the gel surface to form a condensation layer. If the condensation layer is actually formed, it could strongly suppress the friction-driven swelling by localized enhancement of the osmotic pressure around the gel surface and/or by a sort of masking effect that weakens the outward EG flux. To judge the validity of this conjecture, further experimental, theoretical and numerical investigations are needed.


{kind=link}
{kind=link}
{kind=link}
{kind=link}



