

Peptide-Based Supramolecular Hydrogels as Drug Delivery Agents: Recent Advances

 , , , and
, , , and
Abstract
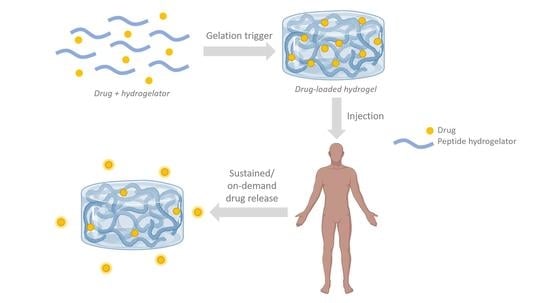
:
1. Introduction
2. ‘Smart’ Peptide Hydrogels: Triggers for Self-Assembly and Drug Release
2.1. pH-Responsive Gels
2.2. Temperature-Responsive Gels
2.3. Redox-Responsive Gels
2.4. Metal Ion-Responsive Gels
2.5. Light-Responsive Gels
2.6. Other Stimuli
3. Hydrogel Mechanical Properties and Stability for Drug Delivery
3.1. β-Sheet Forming Peptides
3.2. β-Hairpin Forming Peptides
3.3. α-Helix Forming Peptides
4. Hydrogel Delivery Routes and Macroscopic Properties for Drug Delivery
4.1. Macroscopic Hydrogels
4.1.1. In Situ-Gelling Hydrogels
4.1.2. Shear-Thinning Hydrogels
4.1.3. Macroporous Hydrogels
4.2. Microgels and Nanogels
5. Bioadhesive Properties and Biofunctionalization of Self-Assembling Peptide Hydrogels
5.1. Peptide-Based Hydrogel Adhesion to Surfaces for Local Drug Delivery
5.2. Biomimetic Functionalization
6. Drug Release Profiles
6.1. Introduction to Drug Release Kinetics of Peptide Hydrogel Matrices
6.2. DOX as a Model Drug: Different Hydrogel Matrices for Anticancer Drug Loading and Release
7. Conclusions and Future Direction
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Fleming, S.; Ulijn, R.V. Design of nanostructures based on aromatic peptide amphiphiles. Chem. Soc. Rev. 2014, 43, 8150–8177. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.G.; Scott, J.G. Peptide-directed self-assembly of hydrogels. Acta Biomater. 2012, 100, 130–134. [Google Scholar]
- Jonker, A.M.; Löwik, D.W.P.M.; Van Hest, J.C.M. Peptide- and protein-based hydrogels. Chem. Mater. 2012, 24, 759–773. [Google Scholar] [CrossRef]
- Feksa, L.R.; Troian, E.A.; Muller, C.D.; Viegas, F.; Machado, A.B.; Rech, V.C. Chapter 11: Hydrogels for biomedical applications. In Nanostructures for the Engineering of Cells, Tissues and Organs: From Design to Applications; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 403–443. [Google Scholar]
- Akhtar, M.F.; Hanif, M.; Ranjha, N.M. Methods of synthesis of hydrogels…A review. Saudi Pharm. J. 2016, 24, 554–559. [Google Scholar] [CrossRef] [Green Version]
- Coviello, T.; Matricardi, P.; Marianecci, C.; Alhaique, F. Polysaccharide hydrogels for modified release formulations. J. Control. Release 2007, 119, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Veloso, S.R.S.; Magalhães, C.A.B.; Rodrigues, A.R.O.; Vilaça, H.; Queiroz, M.-J.R.P.; Martins, J.A.; Coutinho, P.J.G.; Ferreira, P.M.T.; Castanheira, E.M.S. Novel dehydropeptide-based magnetogels containing manganese ferrite nanoparticles as antitumor drug nanocarriers. Phys. Chem. Chem. Phys. 2019, 21, 10377–10390. [Google Scholar] [CrossRef]
- Du, X.; Zhou, J.; Shi, J.; Xu, B. Supramolecular Hydrogelators and Hydrogels: From Soft Matter to Molecular Biomaterials. Chem. Rev. 2015, 115, 13165–13307. [Google Scholar] [CrossRef]
- Langer, R. Drug delivery and targeting. Nature 1998, 392, 5–10. [Google Scholar]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef] [Green Version]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annu. Rev. Chem. Biomol. Eng. 2010, 1, 149–173. [Google Scholar] [CrossRef] [Green Version]
- Florence, A.T.; Jani, P.U. Novel Oral Drug Formulations. Drug Saf. 1994, 10, 233–266. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.P.; Messersmith, P.B.; Israelachvil, J.; Waite, J.H. Mussel-Inspired Adhesives and Coatings. Annu. Rev. Mater. Res. 2011, 41, 99–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brubaker, C.E.; Kissler, H.; Wang, L.J.; Kaufman, D.B.; Messersmith, P.B. Biological performance of mussel-inspired adhesive in extrahepatic islet transplantation. Biomaterials 2010, 31, 420–427. [Google Scholar] [CrossRef] [Green Version]
- Mayr, J.; Saldías, S.; Díaz, D.D. Release of small bioactive molecules from physical gels. Chem. Soc. Rev. 2018, 47, 1484–1515. [Google Scholar] [CrossRef]
- Sis, M.J.; Webber, M.J. Drug Delivery with Designed Peptide Assemblies. Trends Pharmacol. Sci. 2019, 40, 747–762. [Google Scholar] [CrossRef]
- Saboktakin, M.R.; Tabatabaei, R.M. Supramolecular hydrogels as drug delivery systems. Int. J. Biol. Macromol. 2015, 75, 426–436. [Google Scholar] [CrossRef]
- Amin, M.C.I.M.; Ahmad, N.; Pandey, M.; Abeer, M.M.; Mohamad, N. Recent advances in the role of supramolecular hydrogels in drug delivery. Expert Opin. Drug Deliv. 2015, 12, 1149–1161. [Google Scholar] [CrossRef]
- Pepelanova, I. Tunable Hydrogels: Introduction to the World of Smart Materials for Biomedical Applications. In Tunable Hydrogels. Advances in Biochemical Engineering/Biotechnology; Lavrentieva, A., Pepelanova, I., Seliktar, D., Eds.; Springer: Cham, Switzerland, 2021; Volume 178, pp. 1–35. [Google Scholar] [CrossRef]
- Braun, G.A.; Ary, B.E.; Dear, A.J.; Rohn, M.C.H.; Payson, A.M.; Lee, D.S.M.; Parry, R.C.; Friedman, C.; Knowles, T.P.J.; Linse, S.; et al. On the Mechanism of Self-Assembly by a Hydrogel-Forming Peptide. Biomacromolecules 2020, 21, 4781–4794. [Google Scholar] [CrossRef]
- He, S.; Mei, L.; Wu, C.; Tao, M.; Zhai, Z.; Xu, K.; Zhong, W. In situ hydrogelation of bicalutamide-peptide conjugates at prostate tissue for smart drug release based on pH and enzymatic activity. Nanoscale 2019, 11, 5030–5037. [Google Scholar] [CrossRef]
- Pandya, M.J.; Spooner, G.M.; Sunde, M.; Thorpe, J.R.; Rodger, A.; Woolfson, D.N. Sticky-end assembly of a designed peptide fiber provides insight into protein fibrillogenesis. Biochemistry 2000, 39, 8728–8734. [Google Scholar] [CrossRef] [PubMed]
- Aggeli, A.; Nyrkova, I.A.; Bell, M.; Harding, R.; Carrick, L.; McLeish, T.C.B.; Semenov, A.N.; Boden, N. Hierarchical self-assembly of chiral rod-like molecules as a model for peptide β-sheet tapes, ribbons, fibrils, and fibers. Proc. Natl. Acad. Sci. USA 2001, 98, 11857–11862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Ji, Y.; Wang., J.; Wu, L..; Li, W.; Chen, R.; Chen, Z. Self-assembly behaviours of peptide–drug conjugates: Influence of multiple factors on aggregate morphology and potential self-assembly mechanism. R. Soc. Open Sci. 2018, 5, 172040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandit, G.; Roy, K.; Agarwal, U.; Chatterjee, S. Self-Assembly Mechanism of a Peptide-Based Drug Delivery Vehicle. ACS Omega 2018, 3, 3143–3155. [Google Scholar] [CrossRef]
- Gupta, S.; Singh, I.; Sharma, A.K.; Kumar, P. Ultrashort Peptide Self-Assembly: Front-Runners to Transport Drug and Gene Cargos. Front. Bioeng. Biotechnol. 2020, 8, 504. [Google Scholar] [CrossRef]
- Cote, Y.; Fu, I.W.; Dobson, E.T.; Goldberger, J.E.; Nguyen, H.D.; Shen, J.K. Mechanism of the pH-controlled self-assembly of nanofibers from peptide amphiphiles. J. Phys. Chem. C 2014, 118, 16272–16278. [Google Scholar] [CrossRef] [Green Version]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, G.; Barman, R.; Sarkar, J.; Ghosh, S. pH-Responsive Biocompatible Supramolecular Peptide Hydrogel. J. Phys. Chem. B 2019, 123, 5909–5915. [Google Scholar] [CrossRef]
- Raza, F.; Zhu, Y.; Chen, L.; You, X.; Zhang, J.; Khan, A.; Khan, M.W.; Hasnat, M.; Zafar, H.; Wu, J.; et al. Paclitaxel-loaded pH responsive hydrogel based on self-assembled peptides for tumor targeting. Biomater. Sci. 2019, 7, 2023–2036. [Google Scholar] [CrossRef]
- Raza, F.; Zafar, H.; You, X.; Khan, A.; Wu, J.; Ge, L. Cancer nanomedicine: Focus on recent developments and self-assembled peptide nanocarriers. J. Mater. Chem. B. 2019, 7, 7639–7655. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.; Gong, C.; Liu, B.; Li, Y.; Wang, L.; Su, Z.; Wei, G. Recent advances in the fabrication, functionalization, and bioapplications of peptide hydrogels. Soft Matter 2020, 16, 10029–10045. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yokoi, H.; Tanaka, M.; Kinoshita, T.; Tan, T. Self-assembled pH-responsive hydrogels composed of the RATEA16 peptide. Biomacromolecules 2008, 9, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.R.; Ge, R.; Luo, S.Z. Self-assembly of pH and calcium dual-responsive peptide-amphiphilic hydrogel. J. Pept. Sci. 2013, 19, 737–744. [Google Scholar] [CrossRef]
- Kaur, H.; Sharma, P.; Patel, N.; Pal, V.K.; Roy, S. Accessing Highly Tunable Nanostructured Hydrogels in a Short Ionic Complementary Peptide Sequence via pH Trigger. Langmuir 2020, 36, 12107–12120. [Google Scholar] [CrossRef]
- Liu, Y.; Ran, Y.; Ge, Y.; Raza, F.; Li, S.; Zafar, H.; Wu, Y.; Paiva-Santos, A.C.; Yu, C.; Sun, M.; et al. pH-Sensitive Peptide Hydrogels as a Combination Drug Delivery System for Cancer Treatment. Pharmaceutics 2022, 14, 652. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, T.; Tian, Y.; You, L.; Huang, Y.; Wang, S. Novel self-assembling peptide hydrogel with pH-tunable assembly microstructure, gel mechanics and the entrapment of curcumin. Food Hydrocoll. 2022, 124, 107338. [Google Scholar] [CrossRef]
- Yamamoto, S.; Nishimura, K.; Morita, K.; Kanemitsu, S.; Nishida, Y.; Morimoto, T.; Aoi, T.; Tamura, A.; Maruyama, T. Microenvironment pH-Induced Selective Cell Death for Potential Cancer Therapy Using Nanofibrous Self-Assembly of a Peptide Amphiphile. Biomacromolecules 2021, 22, 2524–2531. [Google Scholar] [CrossRef]
- Xian, S.; Webber, M.J. Temperature-responsive supramolecular hydrogels. J. Mater. Chem. B 2020, 8, 9197–9211. [Google Scholar] [CrossRef]
- Takata, K.; Takai, H.; Yoshizaki, Y.; Nagata, T.; Kawahara, K.; Yoshida, Y.; Kuzuya, A.; Ohya, Y. Peptide Drug Release Behavior from Biodegradable Temperature-Responsive Injectable Hydrogels Exhibiting Irreversible Gelation. Gels 2017, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Wang, Y.; Hu, X.; Gong, H.; Li, R.; Cox, H.; Zhang, J.; Waigh, T.A.; Xu, H.; Lu, J.R. Reversible Thermoresponsive Peptide-PNIPAM Hydrogels for Controlled Drug Delivery. Biomacromolecules 2019, 20, 3601–3610. [Google Scholar] [CrossRef] [PubMed]
- De Leon-Rodriguez, L.M.; Hemar, Y.; Mo, G.; Mitra, A.K.; Cornish, J.; Brimble, M.A. Multifunctional thermoresponsive designer peptide hydrogels. Acta Biomater. 2017, 47, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, R.S.C.; Galas, R.J.; Lin, C.Y.; Liu, J.C. Redox-Responsive Resilin-Like Hydrogels for Tissue Engineering and Drug Delivery Applications. Macromol. Biosci. 2019, 19, e1900122. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, L.; Li, Y.; Huang, Z.; Luo, S.; He, Y.; Han, H.; Raza, F.; Wu, J.; Ge, L. Injectable pH and redox dual responsive hydrogels based on self-assembled peptides for anti-tumor drug delivery. Biomater. Sci. 2020, 8, 5415–5426. [Google Scholar] [CrossRef]
- Wu, C.; Li, R.; Yin, Y.; Wang, J.; Zhang, L.; Zhong, W. Redox-responsive supramolecular hydrogel based on 10-hydroxy camptothecin-peptide covalent conjugates with high loading capacity for drug delivery. Mater. Sci. Eng. C. 2017, 76, 196–202. [Google Scholar] [CrossRef]
- Shao, T.; Falcone, N.; Kraatz, H.B. Supramolecular Peptide Gels: Influencing Properties by Metal Ion Coordination and Their Wide-Ranging Applications. ACS Omega 2020, 5, 1312–1317. [Google Scholar] [CrossRef]
- Gallivan, J.P.; Dougherty, D.A. Cation-π interactions in structural biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.; Sabet, Z.F.; Liu, J.; You, M.; Zhou, H.; Wang, Y.; Gao, Y.; Li, J.; Ma, X.; Chen, C. Metal ions modulation of the self-assembly of short peptide conjugated nonsteroidal anti-inflammatory drugs (NSAIDs). Nanoscale 2020, 12, 7960–7968. [Google Scholar] [CrossRef]
- D’Souza, A.; Yoon, J.H.; Beaman, H.; Gosavi, P.; Lengyel-Zhand, Z.; Sternisha, A.; Centola, G.; Marshall, L.R.; Wehrman, M.D.; Schultz, K.M.; et al. Nine-Residue Peptide Self-Assembles in the Presence of Silver to Produce a Self-Healing, Cytocompatible, Antimicrobial Hydrogel. ACS Appl. Mater. Interfaces 2020, 12, 17091–17099. [Google Scholar] [CrossRef]
- Mishra, A.; Chan, K.H.; Reithofer, M.R.; Hauser, C.A.E. Influence of metal salts on the hydrogelation properties of ultrashort aliphatic peptides. RSC Adv. 2013, 3, 9985–9993. [Google Scholar] [CrossRef]
- Tao, M.; Xu, K.; He, S.; Li, H.; Zhang, L.; Luo, X.; Zhong, W. Zinc-ion-mediated self-assembly of forky peptides for prostate cancer-specific drug delivery. Chem. Commun. 2018, 54, 4673–4676. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, C.; Zhang, Q.; Cheng, Y. Near infrared light-responsive and injectable supramolecular hydrogels for on-demand drug delivery. Chem. Commun. 2016, 52, 978–981. [Google Scholar] [CrossRef] [PubMed]
- Roth-Konforti, M.E.; Comune, M.; Halperin-Sternfeld, M.; Grigoriants, I.; Shabat, D.; Adler-Abramovich, L. UV Light–Responsive Peptide-Based Supramolecular Hydrogel for Controlled Drug Delivery. Macromol. Rapid Commun. 2018, 39, e1800588. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, J.; Liu, B.; He, S.; Dai, G.; Xu, B.; Zhong, W. NIR light-responsive short peptide/2D NbSe2 nanosheets composite hydrogel with controlled-release capacity. J. Mater. Chem. B 2019, 7, 3134–3142. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Chang, R.; Duan, H.Z.; Chen, Y.X. Metal ion and light sequentially induced sol-gel-sol transition of a responsive peptide-hydrogel. Soft Matter 2020, 16, 7652–7658. [Google Scholar] [CrossRef]
- Guilbaud-Chéreau, C.; Dinesh, B.; Wagner, L.; Chaloin, O.; Ménard-Moyon, C.; Bianco, A. Aromatic Dipeptide Homologue-Based Hydrogels for Photocontrolled Drug Release. Nanomaterials 2022, 12, 1643. [Google Scholar] [CrossRef]
- Ikeda, M.; Tanida, T.; Yoshii, T.; Hamachi, I. Rational Molecular Design of Stimulus-Responsive Supramolecular Hydrogels Based on Dipeptides. Adv. Mater. 2011, 23, 2819–2822. [Google Scholar] [CrossRef]
- Yoshii, T.; Ikeda, M.; Hamachi, I. Two-Photon-Responsive Supramolecular Hydrogel for Controlling Materials Motion in Micrometer Space. Angew. Chem. Int. Ed. 2014, 53, 7264–7267. [Google Scholar] [CrossRef]
- Jervis, P.J.; Hilliou, L.; Pereira, R.B.; Pereira, D.M.; Martins, J.A.; Ferreira, P.M.T. Evaluation of a model photo-caged dehydropeptide as a stimuli-responsive supramolecular hydrogel. Nanomaterials 2021, 11, 704. [Google Scholar] [CrossRef]
- Chatterjee, S.; Chi-leung Hui, P. Stimuli-Responsive Hydrogels: An Interdisciplinary Overview. In Hydrogels: Smart Materials for Biomedical Applications; Popa, L., Ghica, M., Dinu-Pîrvu, C.-E., Eds.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Carvalho, A.; Gallo, J.; Pereira, D.M.; Valentão, P.; Andrade, P.B.; Hilliou, L.; Ferreira, P.M.T.; Bañobre-López, M.; Martins, J.A. Magnetic Dehydrodipeptide-Based Self-Assembled Hydrogels for Theragnostic Applications. Nanomaterials 2019, 9, 541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gayen, K.; Nandi, N.; Das, K.S.; Hermida-Merino, D.; Hamley, I.W.; Banerjee, A. The aging effect on the enhancement of thermal stability, mechanical stiffness and fluorescence properties of histidine-appended naphthalenediimide based two-component hydrogels. Soft Matter 2020, 16, 10106–10114. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Lu, S.; Wang, N.; Xu, H.; Cox, H.; Li, R.; Waigh, T.A.; Han, Y.; Wang, Y.; Lu, J.R. Enzyme-Triggered Morphological Transition of Peptide Nanostructures for Tumor-Targeted Drug Delivery and Enhanced Cancer Therapy. ACS Appl. Mater. Interfaces 2019, 11, 16357–16366. [Google Scholar] [CrossRef]
- Kulkarni, K.; Minehan, R.L.; Gamot, T.; Coleman, H.A.; Bowles, S.; Lin, Q.; Hopper, D.; Northfield, S.E.; Hughes, R.A.; Widdop, R.E.; et al. Esterase-Mediated Sustained Release of Peptide-Based Therapeutics from a Self-Assembled Injectable Hydrogel. ACS Appl. Mater. Interfaces 2021, 13, 58279–58290. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, L.; Zhao, L.; Xu, B.; Wang, C.; Li, S.; Xu, B. Multi-stimuli-responsive supramolecular hydrogel based on an oxidized glutathione derivative. Dyes Pigm. 2022, 205, 110552. [Google Scholar] [CrossRef]
- Deming, T. (Ed.) Peptide-Based Materials; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar] [CrossRef]
- Zhang, S. Discovery and design of self-assembling peptides. Interface Focus 2017, 7, 20170028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Leon Rodriguez, L.M.; Hemar, Y.; Cornish, J.; Brimble, M.A. Structure-mechanical property correlations of hydrogel forming β-sheet peptides. Chem. Soc. Rev. 2016, 45, 4797–4824. [Google Scholar] [CrossRef]
- Fichman, G.; Schneider, J.P. Utilizing Frémy’s Salt to Increase the Mechanical Rigidity of Supramolecular Peptide-Based Gel Networks. Front. Bioeng. Biotechnol. 2021, 8, 594258. [Google Scholar] [CrossRef]
- Reches, M.; Gazit, E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science 2003, 300, 625–627. [Google Scholar] [CrossRef]
- Adler-Abramovich, L.; Reches, M.; Sedman, V.L.; Allen, S.; Tendler, S.J.B.; Gazit, E. Thermal and chemical stability of diphenylalanine peptide nanotubes: Implications for nanotechnological applications. Langmuir 2006, 22, 1313–1320. [Google Scholar] [CrossRef]
- Smith, A.M.; Williams, R.J.; Tang, C.; Coppo, P.; Collins, R.F.; Turner, M.L.; Ulijn, R.V. Fmoc-diphenylalanine self assembles to a hydrogel via a novel architecture based on π-π interlocked β-sheets. Adv. Mater. 2008, 20, 37–41. [Google Scholar] [CrossRef]
- Diaferia, C.; Morelli, G.; Accardo, A. Fmoc-diphenylalanine as a suitable building block for the preparation of hybrid materials and their potential applications. J. Mater. Chem. B. 2019, 7, 5142–5155. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Gupta, A.; Mehra, R.R.; Khan, N.; Harjit, J.; Ashby, C.R., Jr.; Basu, A.; Tiwari, A.K.; Singh, M.; Konar, A.D. Fluorinated diphenylalanine analogue based supergelators: A stencil that accentuates the sustained release of antineoplastic drugs. Supramol. Chem. 2020, 32, 495–507. [Google Scholar] [CrossRef]
- Boekhoven, J.; Zha, R.H.; Tantakitti, F.; Zhuang, E.; Zandi, R.; Newcomb, C.J.; Stupp, S.I. Alginate-peptide amphiphile core-shell microparticles as a targeted drug delivery system. RSC Adv. 2015, 5, 8753–8756. [Google Scholar] [CrossRef]
- Feger, G.; Angelov, B.; Angelova, A. Prediction of Amphiphilic Cell-Penetrating Peptide Building Blocks from Protein-Derived Amino Acid Sequences for Engineering of Drug Delivery Nanoassemblies. J. Phys. Chem. B 2020, 124, 4069–4078. [Google Scholar] [CrossRef]
- Kaur, K.; Wang, Y.; Matson, J.B. Linker-regulated H2S release from aromatic peptide amphiphile hydrogels. Biomacromolecules 2020, 21, 1171–1178. [Google Scholar] [CrossRef]
- Zhang, S.; Holmes, T.; Lockshin, C.; Rich, A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc. Natl. Acad. Sci. USA 1992, 90, 3334–3338. [Google Scholar] [CrossRef] [Green Version]
- Calvanese, L.; Brun, P.; Messina, G.M.L.; Russo, T.; Zamuner, A.; Falcigno, L.; D’Auria, G.; Gloria, A.; Vitagliano, L.; Marletta, G.; et al. EAK Hydrogels Cross-Linked by Disulfide Bonds: Cys Number and Position Are Matched to Performances. ACS Biomater. Sci. Eng. 2020, 6, 1154–1164. [Google Scholar] [CrossRef]
- Zhang, S.; Lockshin, C.; Cook, R.; Rich, A. Unusually stable beta-sheet formation in an ionic self-complementary oligopeptide. Biopolymers 1994, 34, 663–672. [Google Scholar] [CrossRef]
- Rajagopal, K.; Lamm, M.S.; Haines-Butterick, L.A.; Pochan, D.J.; Schneider, J.P. Tuning the pH responsiveness of β-hairpin peptide folding, self-assembly, and hydrogel material formation. Biomacromolecules 2009, 10, 2619–2625. [Google Scholar] [CrossRef]
- Haines-Butterick, L.; Rajagopal, K.; Branco, M.; Salick, D.; Rughani, R.; Pilarz, M.; Lamm, M.S.; Pochan, D.J.; Schneider, J.P. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. USA 2007, 104, 7791–7796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozbas, B.; Kretsinger, J.; Rajagopal, K.; Schneider, J.P.; Pochan, D.J. Salt-triggered peptide folding and consequent self-assembly into hydrogels with tunable modulus. Macromolecules 2004, 37, 7331–7337. [Google Scholar] [CrossRef]
- Pochan, D.J.; Schneider, J.P.; Kretsinger, J.; Ozbas, B.; Rajagopal, K.; Haines, L. Thermally reversible hydrogels via intramolecular folding and consequent self-assembly of a de novo designed peptide. J. Am. Chem. Soc. 2003, 125, 11802–11803. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.P.; Pochan, D.J.; Ozbas, B.; Rajagopal, K.; Pakstis, L.; Kretsinger, J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002, 124, 15030–15037. [Google Scholar] [CrossRef] [PubMed]
- Haines, L.A.; Rajagopal, K.; Ozbas, B.; Salick, D.A.; Pochan, D.J.; Schneider, J.P. Light-Activated Hydrogel Formation via the Triggered Folding and Self-Assembly of a Designed Peptide. J. Am. Chem. Soc. 2005, 127, 17025–17029. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, T.K.; Ghosh, A.; Kumar, S.K.; Kunwar, A.C. Nucleation of -Hairpin Structures with Cis Amide Bonds in E-Vinylogous Proline-Containing Peptides. J. Org. Chem. 2003, 11, 6459–6462. [Google Scholar] [CrossRef]
- Espinosa, J.F.; Syud, F.A.; Gellman, S.H. Analysis of the factors that stabilize a designed two-stranded antiparallel β-sheet. Protein Sci. 2002, 11, 1492–1505. [Google Scholar] [CrossRef]
- Santiveri, C.M.; Rico, M.; Jiménez, M.A. 13Cα and 13Cβ chemical shifts as a tool to delineate β-hairpin structures in peptides. J Biomol. NMR 2001, 19, 331–345. [Google Scholar] [CrossRef]
- Worthington, P.; Langhans, S.; Pochan, D. Β-Hairpin Peptide Hydrogels for Package Delivery. Adv. Drug. Deliv. Rev. 2017, 110–111, 127–136. [Google Scholar] [CrossRef]
- De Leon-Rodriguez, L.M.; Park, Y.E.; Naot, D.; Musson, D.S.; Cornish, J.; Brimble, M.A. Design, characterization and evaluation of ß-hairpin peptide hydrogels as a support for osteoblast cell growth and bovine lactoferrin delivery. RSC Adv. 2020, 10, 18222–18230. [Google Scholar] [CrossRef]
- Phillips, S.T.; Piersanti, G.; Bartlett, P.A. Quantifying amino acid conformational preferences and side-chain-side-chain interactions in β-hairpins. Proc. Natl. Acad. Sci. USA 2005, 102, 13737–13742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, K.; Brodsky, B. Supercoiled protein motifs: The collagen triple-helix and the α- helical coiled coil. J. Struct. Biol. 1998, 122, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Potekhin, S.; Melnik, T.; Popov, V.; Lanina, N.; Vazina, A.; Rigler, P.; Verdini, A.; Corradin, G.; Kajava, A. De novo design of fibrils made of short α-helical coiled coil peptides. Chem Biol. 2001, 8, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Wagner, D.E.; Phillips, C.L.; Ali, W.M.; Nybakken, G.E.; Crawford, E.D.; Schwab, A.D.; Smith, W.F.; Fairman, R. Toward the development of peptide nanofilaments and nanoropes as smart materials. Proc. Natl. Acad. Sci. USA 2005, 102, 12656–12661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutevelis, E.; Woolfson, D.N. A Periodic Table of Coiled-Coil Protein Structures. J. Mol. Biol. 2009, 385, 726–732. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug. Deliv. Rev. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Li, S.; Zhang, X.; Guo, C.; Peng, Y.; Liu, X.; Wang, B.; Zhuang, R.; Chang, M.; Wang, R. Hydrocarbon Staple Constructing Highly Efficient α-helix Cell-Penetrating Peptides for Intracellular Cargo Delivery. Chem. Commun. 2020, 56, 15655–15658. [Google Scholar] [CrossRef]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2. [Google Scholar] [CrossRef]
- Silva, E.A.; Mooney, D.J. Spatiotemporal control of vascular endothelial growth factor delivery from injectable hydrogels enhances angiogenesis. J. Thromb. Haemost. 2007, 5, 590–598. [Google Scholar] [CrossRef]
- Hiemstra, C.; Zhong, Z.; Van Tomme, S.R.; van Steenbergen, M.J.; Jacobs, J.J.L.; Otter, W.D.; Hennink, W.E.; Feijen, J. In vitro and in vivo protein delivery from in situ forming poly(ethylene glycol)-poly(lactide) hydrogels. J. Control. Release 2007, 119, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Teixeira, L.M.; Krouwels, A.; Dijkstra, P.; van Blitterswijk, C.; Karperien, M.; Feijen, J. Synthesis and characterization of hyaluronic acid-poly(ethylene glycol) hydrogels via Michael addition: An injectable biomaterial for cartilage repair. Acta Biomater. 2010, 6, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Gu, Y.; Qin, L.; Chen, L.; Chen, X.; Cui, W.; Li, F.; Xiang, N.; He, X. Injectable thermosensitive hydrogel-based drug delivery system for local cancer therapy. Colloids Surfaces B Biointerfaces 2021, 200, 111581. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Tang, J.; Li, H.; Huang, Y.; Yin, C.; Li, D.; Tang, F. Antitumor effects of self-assembling peptide-emodin in situ hydrogels in vitro and in vivo. Int. J. Nanomed. 2021, 16, 47–60. [Google Scholar] [CrossRef]
- Guvendiren, M.; Lu, H.D.; Burdick, J.A. Shear-thinning hydrogels for biomedical applications. Soft Matter 2012, 8, 260–272. [Google Scholar] [CrossRef]
- Simonovsky, E.; Miller, Y. Controlling the properties and self-assembly of helical nanofibrils by engineering zinc-binding β-hairpin peptides. J. Mater. Chem. B 2020, 8, 7352–7355. [Google Scholar] [CrossRef]
- Ren, P.; Li, J.; Zhao, L.; Wang, A.; Wang, M.; Li, J.; Jian, H.; Li, X.; Yan, X.; Bai, S. Dipeptide Self-assembled Hydrogels with Shear-Thinning and Instantaneous Self-healing Properties Determined by Peptide Sequences. ACS Appl. Mater. Interfaces 2020, 12, 21433–21440. [Google Scholar] [CrossRef]
- Bencherif, S.A.; Sands, R.W.; Bhatta, D.; Arany, P.; Verbeke, C.S.; Edwards, D.A.; Mooney, D.J. Injectable preformed scaffolds with shape-memory properties. Proc. Natl. Acad. Sci. USA 2012, 109, 19590–19595. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, M.H.; Shea, L.D.; Peters, M.C.; Mooney, D.J. Bioabsorbable polymer scaffolds for tissue engineering capable of sustained growth factor delivery. J. Control. Release 2000, 64, 91–102. [Google Scholar] [CrossRef]
- Paquin, F.; Rivnay, J.; Salleo, A.; Stingelin, N.; Silva, C. Multi-phase semicrystalline microstructures drive exciton dissociation in neat plastic semiconductors. J. Mater. Chem. C 2015, 3, 10715–10722. [Google Scholar] [CrossRef] [Green Version]
- Hassan, C.M.; Peppas, N.A. Structure and morphology of freeze/thawed PVA hydrogels. Macromolecules 2000, 33, 2472–2479. [Google Scholar] [CrossRef]
- Berillo, D.; Mattiasson, B.; Galaev, I.Y.; Kirsebom, H. Formation of macroporous self-assembled hydrogels through cryogelation of Fmoc-Phe-Phe. J. Colloid Interface Sci. 2012, 368, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.R.S.; Rao, K.S.V.K.; Rao, K.M.; Reddy, N.S.; Eswaramma, S. pH sensitive poly(methyl methacrylate-co-acryloyl phenylalanine) nanogels and their silver nanocomposites for biomedical applications. J. Drug. Deliv. Sci. Technol. 2015, 29, 181–188. [Google Scholar] [CrossRef]
- Lyu, L.; Liu, F.; Wang, X.; Hu, M.; Mu, J.; Cheong, H.; Liu, G.; Xing, B. Stimulus-Responsive Short Peptide Nanogels for Controlled Intracellular Drug Release and for Overcoming Tumor Resistance. Chem. Asian J. 2017, 12, 744–752. [Google Scholar] [CrossRef]
- Tang, C.; Miller, A.F.; Saiani, A. Peptide hydrogels as mucoadhesives for local drug delivery. Int. J. Pharm. 2014, 465, 427–435. [Google Scholar] [CrossRef]
- Liu, H.; Bi, X.; Wu, Y.; Pan, M.; Ma, X.; Mo, L.; Wang, J.; Li, X. Cationic Self-Assembled Peptide-Based Molecular Hydrogels for Extended Ocular Drug Delivery. Acta Biomater. 2021, 131, 163–171. [Google Scholar] [CrossRef]
- Taka, E.; Karavasili, C.; Bouropoulos, N.; Moschakis, T.; Andreadis, D.D.D.; Zacharis, C.K.K.; Fatouros, D.G.G. Ocular co-delivery of timolol and brimonidine from a self-assembling peptide hydrogel for the treatment of glaucoma: In vitro and ex vivo evaluation. Pharmaceuticals 2020, 13, 126. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Wang, T.; Xia, L.; Wang, L.; Qin, M.; Li, Y.; Wang, W.; Cao, Y. Single-molecule study of the synergistic effects of positive charges and Dopa for wet adhesion. J. Mater. Chem. B 2017, 5, 4416–4420. [Google Scholar] [CrossRef]
- Li, Y.; Liu, H.; Wang, T.; Qin, M.; Cao, Y.; Wang, W. Single-Molecule Force Spectroscopy Reveals Multiple Binding Modes between DOPA and Different Rutile Surfaces. ChemPhysChem 2017, 18, 1466–1469. [Google Scholar] [CrossRef]
- Li, Y.; Qin, M.; Li, Y.; Cao, Y.; Wang, W. Single molecule evidence for the adaptive binding of DOPA to different wet surfaces. Langmuir 2014, 30, 4358–4366. [Google Scholar] [CrossRef]
- Wang, T.; Li, Y.; Wang, J.; Xu, Y.; Chen, Y.; Lu, Z.; Wang, W.; Xue, B.; Li, Y.; Cao, Y. Smart Adhesive Peptide Nanofibers for Cell Capture and Release. ACS Biomater. Sci. Eng. 2020, 6, 6800–9807. [Google Scholar] [CrossRef] [PubMed]
- Geckil, H.; Xu, F.; Zhang, X.; Moon, S.; Demirci, U. Engineering hydrogels as extracellular matrix mimics. Nanomedicine 2010, 5, 469–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, X.; Avci-Adali, M.; Alarçin, E.; Cheng, H.; Kashaf, S.S.; Li, Y.; Chawla, A.; Jang, H.L.; Khademhosseini, A. Development of hydrogels for regenerative engineering. Biotechnol. J. 2017, 12, 1600394. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, H.; Mihara, H. Soft materials based on designed self-assembling peptides: From design to application. Mol. Biosyst. 2013, 9, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xing, R.; Bai, S.; Yan, X. Recent advances of self-assembling peptide-based hydrogels for biomedical applications. Soft Matter 2019, 15, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, X.; Dong, X.; Zhang, L.; Sun, Y. Biomimetic design of affinity peptide ligand for capsomere of virus-like particle. Langmuir 2014, 30, 8500–8508. [Google Scholar] [CrossRef]
- Gray, B.P.; Brown, K.C. Combinatorial Peptide Libraries: Mining for Cell-Binding Peptides. Chem. Rev. 2014, 114, 1020–1081. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Lewis, P.L.; Shah, R.N. Tailoring nanostructure and bioactivity of 3D-printable hydrogels with self-assemble peptides amphiphile (PA) for promoting bile duct formation. Biofabrication 2018, 10, 035010. [Google Scholar] [CrossRef]
- Horii, A.; Wang, X.; Gelain, F.; Zhang, S. Biological designer self-assembling peptide nanofiber scaffolds significantly enhance osteoblast proliferation, differentiation and 3-D migration. PLoS ONE 2007, 2, 190. [Google Scholar] [CrossRef]
- Danesin, R.; Brun, P.; Roso, M.; Delaunay, F.; Samouillan, V.; Brunelli, K.; Iucci, G.; Ghezzo, F.; Modesti, M.; Castagliuolo, I.; et al. Self-assembling peptide-enriched electrospun polycaprolactone scaffolds promote the h-osteoblast adhesion and modulate differentiation-associated gene expression. Bone 2012, 51, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, H.; Matsubara, D.; Mihara, H. Functionalization of self-assembling peptide materials using molecular recognition of supramolecular peptide nanofibers. Polym. J. 2020, 52, 913–922. [Google Scholar] [CrossRef]
- Yang, S.; Wang, C.; Zhu, J.; Lu, C.; Li, H.; Chen, F.; Lu, J.; Zhang, Z.; Yan, X.; Zhao, H.; et al. Self-assembling peptide hydrogels functionalized with LN- And BDNF- mimicking epitopes synergistically enhance peripheral nerve regeneration. Theranostics 2020, 10, 8227–8249. [Google Scholar] [CrossRef] [PubMed]
- Adak, A.; Das, G.; Khan, J.; Mukherjee, N.; Gupta, V.; Mallesh, R.; Ghosh, S. Extracellular Matrix (ECM)-Mimicking Neuroprotective Injectable Sulfo-Functionalized Peptide Hydrogel for Repairing Brain Injury. ACS Biomater. Sci. Eng. 2020, 6, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Hellmund, K.S.; von Lospichl, B.; Böttcher, C.; Ludwig, K.; Keiderling, U.; Noirez, L.; Weiß, A.; Mikolajczak, D.J.; Gradzielski, M.; Koksch, B. Functionalized peptide hydrogels as tunable extracellular matrix mimics for biological applications. Pept. Sci. 2020, 113, e24201. [Google Scholar] [CrossRef]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert Opin. Drug Deliv. 2017, 4, 429–444. [Google Scholar] [CrossRef]
- Mircioiu, C.; Voicu, V.; Anuta, V.; Tudose, A.; Celia, C.; Paolino, D.; Fresta, M.; Sandulovici, R.; Mircioiu, I. Mathematical modeling of release kinetics from supramolecular drug delivery systems. Pharmaceutics 2019, 11, 140. [Google Scholar] [CrossRef] [Green Version]
- Brandl, F.; Kastner, F.; Gschwind, R.M.; Blunk, T.; Teßmar, J.; Göpferich, A. Hydrogel-based drug delivery systems: Comparison of drug diffusivity and release kinetics. J. Control. Release 2010, 142, 221–228. [Google Scholar] [CrossRef]
- Lamprecht, A.; Yamamoto, H.; Takeuchi, H.; Kawashima, Y. Microsphere design for the colonic delivery of 5-fluorouracil. J. Control. Release 2003, 90, 313–322. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Liu, S.; Chen, X.; Zhang, Q.; Wu, W.; Xin, J.; Li, J. Multifunctional hydrogels based on β-cyclodextrin with both biomineralization and anti-inflammatory properties. Carbohydr. Polym. 2014, 102, 869–876. [Google Scholar] [CrossRef]
- AAmorim, C.; Veloso, S.; Castanheira, E.; Hilliou, L.; Pereira, R.; Pereira, D.; Martins, J.; Jervis, P.; Ferreira, P. Bolaamphiphilic Bis-Dehydropeptide Hydrogels as Potential Drug Release Systems. Gels 2021, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.B.P.O.; Veloso, S.R.S.; Castanheira, E.M.S.; Figueiredo, P.R.; Carvalho, A.T.P.; Hilliou, L.; Pereira, R.B.; Pereira, D.M.; Martins, J.A.; Ferreira, P.M.T.; et al. An injectable, naproxen-conjugated, supramolecular hydrogel with ultra-low critical gelation concentration—prepared from a known folate receptor ligand. Soft Matter 2022, 18, 3955–3966. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Metters, A.T. Hydrogels in controlled release formulations: Network design and mathematical modeling. Adv. Drug Deliv. Rev. 2006, 58, 1379–1408. [Google Scholar] [CrossRef] [PubMed]
- Ghannam, M.; Abu-Jdayil, B.; Esmail, N. Flow behaviours comparison of crude oil–polymer emulsions. Int. J. Ambient Energy 2018, 39, 581–593. [Google Scholar] [CrossRef]
- Chakroun, R.W.; Wang, F.; Lin, R.; Wang, Y.; Su, H.; Pompa, D.; Cui, H. Fine-Tuning the Linear Release Rate of Paclitaxel-Bearing Supramolecular Filament Hydrogels through Molecular Engineering. ACS Nano 2019, 13, 7780–7790. [Google Scholar] [CrossRef]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. J. Control. Release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Heuser, T.; Weyandt, E.; Walther, A. Biocatalytic Feedback-Driven Temporal Programming of Self-Regulating Peptide Hydrogels. Angew. Chem. Int. Ed. 2015, 54, 13258–13262. [Google Scholar] [CrossRef]
- Cui, T.; Li, X.; He, S.; Xu, D.; Yin, L.; Huang, X.; Deng, S.; Yue, W.; Zhong, W. Instant Self-Assembly Peptide Hydrogel Encapsulation with Fibrous Alginate by Microfluidics for Infected Wound Healing. ACS Biomater. Sci. Eng. 2020, 6, 5001–5011. [Google Scholar] [CrossRef]
- Choe, R.; Yun, S.I. Fmoc-diphenylalanine-based hydrogels as a potential carrier for drug delivery. E-Polymers 2020, 20, 458–468. [Google Scholar] [CrossRef]
- World Health Organization: Cancer. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 10 February 2021).
- Sun, Z.; Song, C.; Wang, C.; Hu, Y.; Wu, J. Hydrogel-Based Controlled Drug Delivery for Cancer Treatment: A Review. Mol. Pharm. 2020, 17, 373–391. [Google Scholar] [CrossRef]
- Norouzi, M.; Nazari, B.; Miller, D.W. Injectable hydrogel-based drug delivery systems for local cancer therapy. Drug Discov. Today 2016, 21, 1835–1849. [Google Scholar] [CrossRef]
- Vishnubhakthula, S.; Elupula, R.; Durán-Lara, E.F. Recent Advances in Hydrogel-Based Drug Delivery for Melanoma Cancer Therapy: A Mini Review. J. Drug Deliv. 2017, 2017, 7275985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Zheng, C.; Xiong, F.; Ran, W.; Zhai, Y.; Zhu, H.H.; Wang, H.; Li, Y.; Zhang, P. Recent Progress in the Design and Application of Supramolecular Peptide Hydrogels in Cancer Therapy. Adv. Healthc. Mater. 2021, 10, 2001239. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Arabi, L.; Alibolandi, M. Doxorubicin-loaded composite nanogels for cancer treatment. J. Control. Release 2020, 328, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Vyas, M.; Simbo, D.A.; Mursalin, M.; Mishra, V.; Bashary, R.; Khatik, G.L. Drug Delivery Approaches for Doxorubicin in the Management of Cancers. Curr. Cancer Ther. Rev. 2019, 16, 320–331. [Google Scholar] [CrossRef]
- Roychoudhury, S.; Kumar, A.; Bhatkar, D.; Sharma, N.K. Molecular avenues in targeted doxorubicin cancer therapy. Futur. Oncol. 2020, 16, 687–700. [Google Scholar] [CrossRef]
- van der Zanden, S.Y.; Qiao, X.; Neefjes, J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. FEBS J. 2020, 288, 6095–6111. [Google Scholar] [CrossRef]
- Karavasili, C.; Andreadis, D.A.; Katsamenis, O.L.; Panteris, E.; Anastasiadou, P.; Kakazanis, Z.; Zoumpourlis, V.; Markopoulou, C.K.; Koutsopoulos, S.; Vizirianakis, I.S.; et al. Synergistic Antitumor Potency of a Self-Assembling Peptide Hydrogel for the Local Co-delivery of Doxorubicin and Curcumin in the Treatment of Head and Neck Cancer. Mol. Pharm. 2019, 16, 2326–2341. [Google Scholar] [CrossRef]
- Mei, L.; He, S.; Liu, Z.; Xu, K.; Zhong, W. Co-assembled supramolecular hydrogels of doxorubicin and indomethacin-derived peptide conjugates for synergistic inhibition of cancer cell growth. Chem. Commun. 2019, 55, 4411–4414. [Google Scholar] [CrossRef]
- Mei, L.; Xu, K.; Zhai, Z.; He, S.; Zhu, T.; Zhong, W. Doxorubicin-reinforced supramolecular hydrogels of RGD-derived peptide conjugates for pH-responsive drug delivery. Org. Biomol. Chem. 2019, 17, 3853–3860. [Google Scholar] [CrossRef]
- Martin, A.D.; Thordarson, P. Beyond Fmoc: A review of aromatic peptide capping groups. J. Mater. Chem. B 2020, 8, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yu, T.; Peng, L.; Sun, Q.; Wei, Y.; Han, B. Advancements in Hydrogel-Based Drug Sustained Release Systems for Bone Tissue Engineering. Front. Pharmacol. 2020, 11, 622. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, X.; Chen, H.; Peng, J.; Yang, X. Small Peptide-Doxorubicin Co-Assembly for Synergistic Cancer Therapy. Molecules 2020, 25, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, E.; Diaferia, C.; Gallo, E.; Morelli, G.; Accardo, A. Stable formulations of peptide-based nanogels. Molecules 2020, 25, 3455. [Google Scholar] [CrossRef] [PubMed]
- Mauri, E.; Papa, S.; Masi, M.; Veglianese, P.; Rossi, F. Novel functionalization strategies to improve drug delivery from polymers. Expert Opin. Drug Deliv. 2017, 14, 1305–1313. [Google Scholar] [CrossRef]
- Chan, K.H.; Lee, W.H.; Ni, M.; Loo, Y.; Hauser, C.A.E. C-Terminal Residue of Ultrashort Peptides Impacts on Molecular Self-Assembly, Hydrogelation, and Interaction with Small-Molecule Drugs. Sci. Rep. 2018, 8, 17127. [Google Scholar] [CrossRef] [Green Version]
- Milcovich, G.; Antunes, F.E.; Farra, R.; Grassi, G.; Grassi, M.; Asaro, F. Modulating carbohydrate-based hydrogels as viscoelastic lubricant substitute for articular cartilages. Int. J. Biol. Macromol. 2017, 102, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yang, C.; Lu, C.; Zhang, F.; Yuan, Z.; Zhuang, X. Two-Dimensional Porous Polymers: From Sandwich-like Structure to Layered Skeleton. Acc. Chem. Res. 2018, 51, 3191–3202. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| k | n | Drug Release Mechanism | Reference | |||
|---|---|---|---|---|---|---|
| ac–(RADA)4–CONH2 | pH 7.4 | 0–≈450 h | – | 0.141 | Case-I transport | [165] |
| IDM-1 | pH 5.5 | 0–24 h | 0.0219 | 0.6282 | Non-Fickian difusivo | [166] |
| pH 6.5 | 0.0068 | 0.7975 | Non-Fickian diffusion | |||
| pH 7.4 | 0.0046 | 0.7857 | Non-Fickian diffusion | |||
| 1-RGDH | pH 5.5 | 0–24 h | 0.0342 | 0.8084 | Non-Fickian diffusion | [167] |
| 24–168 h | 0.1374 | 0.2265 | Case-I transport | |||
| pH 6.5 | 0–24 h | 0.0185 | 0.7946 | Non-Fickian diffusion | ||
| 24–168 h | 0.0766 | 0.2039 | Case-I transport | |||
| pH 7.4 | 0–24 h | 0.0127 | 0.7768 | Non-Fickian diffusion | ||
| 24–168 h | 0.0471 | 0.2189 | Case-I transport | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, C.B.P.; Gomes, V.; Ferreira, P.M.T.; Martins, J.A.; Jervis, P.J. Peptide-Based Supramolecular Hydrogels as Drug Delivery Agents: Recent Advances. Gels 2022, 8, 706. https://doi.org/10.3390/gels8110706
Oliveira CBP, Gomes V, Ferreira PMT, Martins JA, Jervis PJ. Peptide-Based Supramolecular Hydrogels as Drug Delivery Agents: Recent Advances. Gels. 2022; 8(11):706. https://doi.org/10.3390/gels8110706
Chicago/Turabian StyleOliveira, Carlos B. P., Valéria Gomes, Paula M. T. Ferreira, José A. Martins, and Peter J. Jervis. 2022. "Peptide-Based Supramolecular Hydrogels as Drug Delivery Agents: Recent Advances" Gels 8, no. 11: 706. https://doi.org/10.3390/gels8110706
APA StyleOliveira, C. B. P., Gomes, V., Ferreira, P. M. T., Martins, J. A., & Jervis, P. J. (2022). Peptide-Based Supramolecular Hydrogels as Drug Delivery Agents: Recent Advances. Gels, 8(11), 706. https://doi.org/10.3390/gels8110706