Excitons in Carbonic Nanostructures


1
Palladin Institute of Biochemistry, Leontovicha st. 9, 01030 Kyiv, Ukraine
2
Yuriy Fedkovych National University, 58012 Chernivtsy, Ukraine
C 2019, 5(4), 71; https://doi.org/10.3390/c5040071
Submission received: 26 September 2019
/
Revised: 29 October 2019
/
Accepted: 2 November 2019
/
Published: 12 November 2019
(This article belongs to the Special Issue Optical and Electronic Properties of Carbon-Based Nanomaterials and Composites)
Abstract
:Unexpectedly bright photoluminescence emission can be observed in materials incorporating inorganic carbon when their size is reduced from macro–micro to nano. At present, there is no consensus in its understanding, and many suggested explanations are not consistent with the broad range of experimental data. In this Review, I discuss the possible role of collective excitations (excitons) generated by resonance electronic interactions among the chromophore elements within these nanoparticles. The Förster-type resonance energy transfer (FRET) mechanism of energy migration within nanoparticles operates when the composing fluorophores are the localized electronic systems interacting at a distance. Meanwhile, the resonance interactions among closely located fluorophores may lead to delocalization of the excited states over many molecules resulting in Frenkel excitons. The H-aggregate-type quantum coherence originating from strong coupling among the transition dipoles of adjacent chromophores in a co-facial stacking arrangement and exciton transport to emissive traps are the basis of the presented model. It can explain most of the hitherto known experimental observations and must stimulate the progress towards their versatile applications.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Serendipitous discovery of the photoluminescent properties of carbon nanoparticles [1,2] took researchers by great surprise. It is known that graphite is an ideal light absorber with an extremely broad continuum of electronic transitions, from UV to infrared, and with a similarly strong ability to trap the absorbed light [3]. Being a “totally black” material, it is simply incapable of emitting visible light. Regarding other structures formed by inorganic carbon, the diamonds demonstrate ideal light transparency and only by creating the point defects in their sp3 hybridized carbon network [4] can they be made light absorbing and emitting [5]. Graphene is also transparent and non-emissive due to the fact of its zero band gap [6], but when its sp2 hybridized monolayer structure is rolled into a carbon nanotube, a weak absorption and emission is observed [7] in the near-IR region [8]. Graphite, graphene, carbon nanotubes, and also fullerenes are known as very potent electron-transfer quenchers for other fluorophores [9]. Yet, why are very small particles made of carbon strong absorbers and emitters of light in the visible range of spectrum?
An even greater surprise was waiting for scientists when they started production of nanoscale fluorescent materials from virtually any organic or inorganic source of carbon using a broad variety of carbonization or fragmentation methods and isolating them from reaction products in fractions of several nanometers in size [10]—they started to become very efficient. Despite strong variability in size, composition, structural order, solubility, and other reported parameters, the photophysical and spectroscopic properties of these new emitters demonstrated some general features [11,12]. These features turned out to be quite different from those observed in organic dye molecules [13] or nanoparticles of inorganic origin such as semiconductor quantum dots [14]. This fact raised many difficulties in fitting them into a well-established set of knowledge about light absorbing and emitting materials. Resolving them is not easy since, here, it is not possible to start from monomeric dyes and observe the effects of their assembly with respect to the monomers, such as the shifts of the excitation energies and redistribution of the oscillator strength. Commonly, synthetic chemists deal with reactions in which the starting components and reaction conditions are well determined; pathways can be predicted based on previous knowledge and the properties of products are expected. This refers also to organic molecular nanomaterials that can be obtained by self-assembly of molecular components, so that their fluorescence-related electronic transition energies are maintained or slightly perturbed [15]. Here, we have a different situation. The fluorescent nanoparticles are obtained from nonfluorescent starting materials in empirically chosen conditions and it remains to be discussed why they are fluorescent and how to make their fluorescence optimal for particular application.
The discovery of fluorescent carbonic nanomaterials has attracted the attention of many researchers coming from different fields of physics, chemistry, and material science who bring with them their concepts, ideas, and even terminology. As it was noted References [11,16,17], the presently suggested explanations for the photoluminescence of carbon nanomaterials, often called dots, are very divergent. If we accept that individual nanoparticles contain multi-chromophoric units, we have to think if these chromophores interact and, if yes, how theses interactions happen. These ideas can be classified into two broad categories. One is based on the assumption that each luminescent particle represents an assembly of molecules emitting independently, forming an integrated fluorescent ensemble [18]. They could be the aromatic islands within the particle core [19] or their charge–transfer complexes [20] formed with polar groups on the particle surface [21]. The other explanations consider the presence of collective effects but very different in nature. They could arise due to the interaction among fluorophores that are confined in nanoparticles at close distances. Then, the multiple steps of the excited-state energy transfer could be realized according to the well-known Förster-type resonance energy transfer (FRET) mechanism [22]. The FRET mechanism describes the energy migration among localized electronic systems. In contrast, the collective excitations (excitons) may propagate within the whole particle volume [21,23] or through the surface [24,25] in a collective manner. In this case the emission should be observed at exciton-hole recombination sites [26,27] that could be the localized surface traps [28] or specific edge states [29].
One may consider that all these mechanisms operate in parallel demonstrating the specificity of a particular type of carbon nanoparticles. Meanwhile, some features of their spectroscopic performance are very general. Among them are the broad-range spectral tunability, the unusually large Stokes shifts observed even in cryogenic conditions [30], and the collective photophysical behavior in single-molecular experiments [31]. Moreover, on a single-molecular level, these structures do not show spectroscopic or lifetime heterogeneity. It can be observed that hundreds and even thousands of publications with their exponential growth in recent years still have not provided a consistent view of the origin of emission of carbon nanomaterials. This hinders significantly the development of new materials with desirable characteristics and their efficient use.
The goal of this Review was to provide a broad view of the physical mechanisms responsible for the fluorescence of carbon nanoparticles based on collective excitonic effects. This Review focused on the possibility of the formation and propagation of Frenkel excitons, i.e., the quantum coherent excitations that spread over the whole nanoparticle or its significant part performing as a system of organized interacting fluorophores. In my view, the exciton theory can explain the key features of the photophysical behavior of fluorescent nanocarbon structures. I believe that such a platform will allow for the creation of new steps for a better understanding of the unique features of these nanoscale systems.
2. The Structures and Properties of Fluorescent Nanocarbon Materials
2.1. The Broad Family of Fluorescent Nanocarbons
In order to comprehend the generality of the observed spectroscopic effects, it would be reasonable to draw the reader’s attention to the whole range of diverse objects that display them. Fluorescent carbon nanoparticles [32] are represented by graphene (G-dots) [33,34,35] and graphene oxide (GO-dots) [12] derivatives and, more recently, by carbon dots (C-dots) [36,37,38,39,40]. Presently, the latter are the most popular in research and applications [17,40,41,42,43]. The simple reason for this is that they are quite easily available. They can be made by simple burning [44] or microwave heating [45] of different organic matter [46] and even of natural products such as coffee grounds [47] and soy milk [48]. From these predecessors, the obtained particles may incorporate different reactive groups forming heterocyclic structures with the participation of O, N, and S atoms [49]. Such heteroatoms forming the polar groups can also be introduced into G-dots and GO-dots which allows easy solubility of these nanostructures in water. High surface-to-volume ratios [50] and the presence of polar groups on their surface [51,52] may be strong determinants of their properties. Regarding the sizes of these particles, they vary from several to tens of nanometers and may exhibit broad variation in shapes. The structures of spherical (or almost spherical) shapes are usually considered to have zero dimensionality (0-D) which gave them the name “dots” [53] even if they are the fragments of two-dimensional structures of graphene (G-dots) [33,54] or graphene oxide (GO-dots) [12].
Commonly, fluorescent carbon nanoparticles are characterized by a high amount of carbon–carbon bonding, predominantly in sp2 hybridization of the graphene type [55], but also by the features of disordered or diamond-type sp3 hybridized structures [1,56]. This immediately suggests that the π–electronic conjugation must exist along their graphene sheets, but it may be irregular and disrupted into small fragments [57]. Graphene oxide dots differ from G-dots by the presence of oxygen-containing groups (epoxy, hydroxy, carboxy) not only at the sheet edges but also within the sheet planes. If these groups are located on the basal plane, they disrupt the extended regular conjugation, which leads to the presence of isolated sp2 domains [34]. These basic structural elements form the carbon core. The polar groups formed by organic heterocycles during or after the synthesis of nanoparticles are exposed to the surface.
Structural variability determines the diversity of spectroscopic properties of carbon nanoparticles that may be very significant. In the family of G-dots, one may observe very broad absorption bands in the UV region, approximately 230–280 nm, and a long tail extending into the visible range [58]. Their broad UV absorbance band in the region 230–320 nm can be attributed to π–π* electronic transitions in the aromatic carbon rings, and its long-wavelength tail to n–π* transitions of carbonyls or other connected groups. Surprisingly, the excitation at this very strong UV absorption band does not lead to fluorescence emission at all, which was indicated by many researchers. Several authors could only find “blue” emission corresponding to the resolved UV excitation band [59]. In contrast, the major emission band of G-dots in the visible range is commonly excited at the wavelengths 320–370 nm. It is the far edge of the major absorption band where the absorption at their excitation band maximum is low. By exciting at these wavelengths, a variety of fluorescence colors can be observed in different preparations. Nitrogen doping and oxygen enrichment of the periphery of G-dot structures does not change the character of their light absorption and emission [60].
The spectroscopic properties described for GO-dots are quite similar to that of G-dots [12]. It was reported that their light emission can be blue [61], green [62,63] or red [64] when the excitation band is shifted from its typical position at 320–370 nm to longer wavelengths. These properties can be changed by dopants and other factors. The introduction of nitrogen atoms into graphene oxide structures resulted in the change of initially green luminescence [65] into the bright blue emission [66]. The switching between these colors can be achieved just by the variation of temperature on their synthesis [67]. Also, the starting material in their synthesis is a strong determinant of emission [68]. Their spectrally heterogeneous emission can be revealed by variation of pH [58].
Fluorescent carbon dots (C-dots) are the most versatile but less defined and less characterized members in the family of nanocarbon materials. Meanwhile, they share similar properties with G-dots and GO-dots [69] which are discussed above. It is true that they can be made in one step by solvothermal treatment of any organic matter [10]. But their synthesis based on well-defined molecular precursors is preferable, since it is the way to achieve functional carbon materials with controlled surface chemistry, mesoscopic morphology, and microstructure [70]. Commonly studied C-dots are formed by carbon–carbon bonding predominantly in the graphene-type sp2 hybridization [55], but they reveal a relatively high amount of the diamond-type sp3 hybridization (or disorder) of carbon atoms [1,56]. This suggests that the π–electronic conjugation must exist along the graphene sheets, but this conjugation may be irregular, especially in view of the presence of disrupting hydroxyl and carbonyl groups [57]. Based on these data, the core of a C-dot can be viewed as a highly defected composition of coexisting aromatic and aliphatic regions, the elementary constituents of which are graphene, graphene oxide, and diamond [71]. They are assembled in proportions and with variations of surface groups that depend on the starting material and the conditions of their synthesis (Figure 1).
2.2. Going Deeper into Nanocarbon Structures and Their Formation
Being fused into nanoparticles, aromatic fluorophores should be located at distances of the order of one nanometer or even closer and possess, at least partially, regular arrangement. This arrangement may be variable within the nanoparticle and also among the nanoparticles in the ensemble. Therefore, we have to consider two types of structural heterogeneity—within a particle and among the particles. Addressing this issue is complicated, since the exact packing arrangements in carbon nanoparticles on the atomic level remain unresolved and, moreover, a great diversity is expected between different structures in their ensemble. Important information can be obtained by combining spectroscopy and microscopy that could allow for study on a single particle level.
It is known that graphene sheets, even distorted, possesses a high level of in-plane regularity resulting in coupling of π–electrons into the “electron clouds” [72]. However, in graphite-like structures, the interactions among graphene layers are weak, being represented by dispersion forces only. The latter can be easily disrupted [73], but there exist other factors contributing to the stability of nanoparticles. When aromatic heterocycles are attached to graphene planes, their binding is also governed by dispersion forces with only a minor contribution of electrostatic interactions [74]. Therefore, the major factor stabilizing the whole “dots” is not the interactions among graphene planes but rather the hydrophobic force arising from screening the core structure of nanoparticles from water or from other polar solvents and exposing the relatively polar heterocycles on the surface.
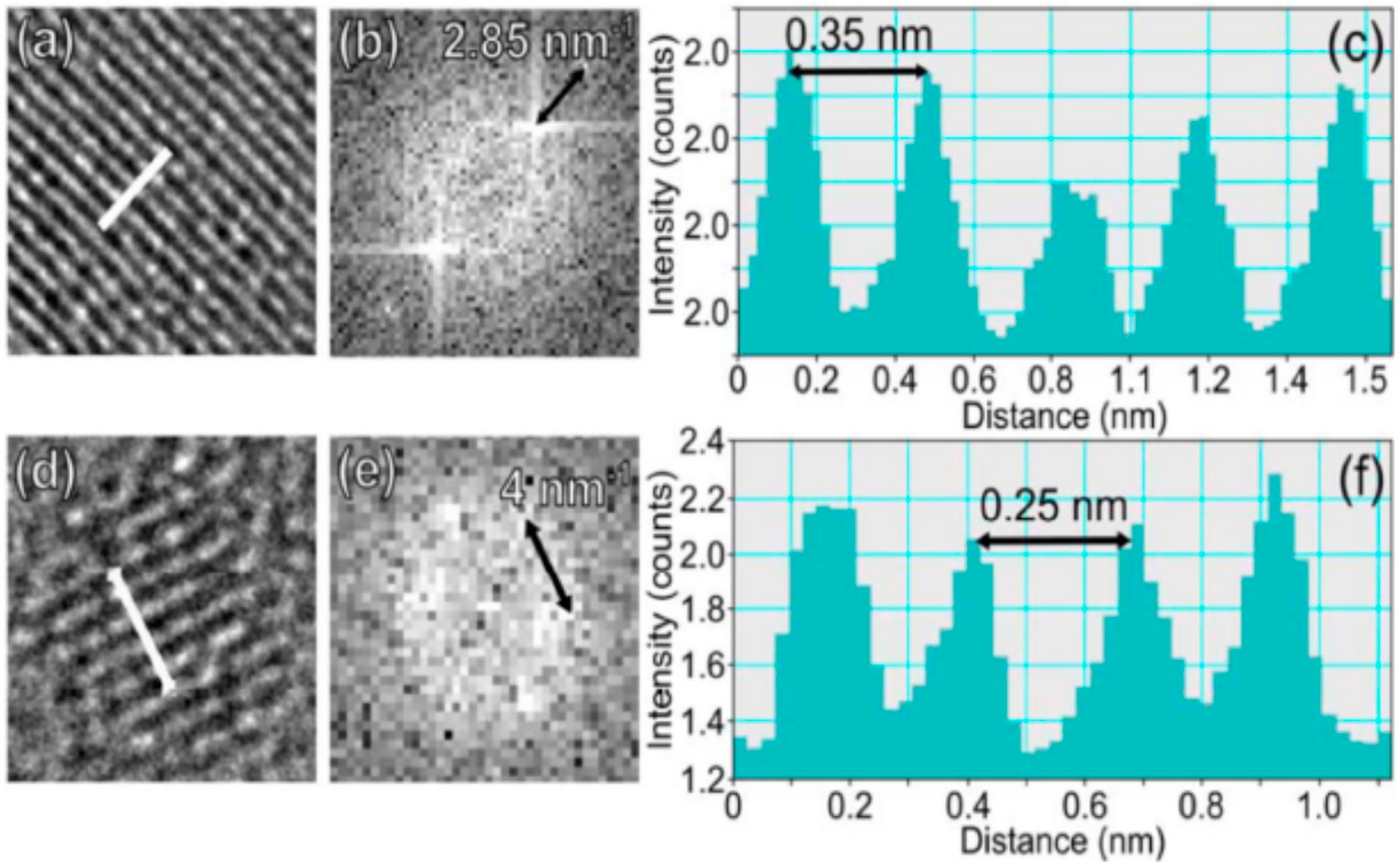
Figure 2 demonstrates some of the examples of C-dots structures obtained by high-resolution electron microscopy and which are identified as bright spots in the fluorescence images [31]. They were are “green” C-dots obtained by microwave treatment of sucrose solutions with added polyethylene glycol and possessing, in aqueous media, the excitation and emission band maxima at 425 and 530 nm, correspondingly [68]. The observed nanostructures have different diameters varying from nearly 7 nm to 20 nm, different aspect ratios from nearly 1.5 (Figure 2c,d) to 1 (Figure 2e,f), and some possess a characteristic onion-like shape structure such as reported previously [75]. Such polydispersity is typical for C-dots. Their size-dependent separation in other studies [76,77] showed fluorescence emission in all obtained fractions.
Regarding fine intramolecular structures, two types of particles were resolved in the HRTEM images. One of them, shown in Figure 2c–f, demonstrates a lattice spacing of around 0.35–0.36 nm, which agrees well with the <002> plane of graphitic carbon [40] (we call it the crystal structure I). The second type of C-dot is characterized by a fully crystalline structure, 2–5 nm in diameter (Figure 2g,h). A lattice spacing of nearly 0.24 nm was also observed in different studies [78] and corresponds to the <100> crystal plane of graphite [40] (crystal structure II). All the observed C-dots were composed of multiple layers of graphene as determined by the analysis of electron diffraction patterns (see Figure 3). Similar carbonic nanostructures have been found in other studies using alternative top-down or bottom-up synthetic methods (see References [40,79] and citations therein). Some of the C-dots, which possessed an onion-like shape, exhibited an amorphous carbon shell.
Thus, considering the photophysical properties of C-dots, one must account for the versatility of these carbon core structures and their intra-particle ordering. Such observations are important in view that the function of carbon core in generating fluorescence emissive states is highly disputable. Some authors suggest its major role [80]. In contrast, the C-dots possessing an sp3 diamond core were found not to differ in fluorescence from those with a graphitic one [25]. Recently, a new version of optically active C-dots was obtained, possessing a nanocrystalline beta carbon nitride core and a disordered surface shell that hosted a variety of polar functional groups [81]. Being quite different from common C-dots as sp2-carbon materials, they still possess similar properties, such as strong and wavelength-tunable fluorescence in the visible region [82]. These data present arguments for only a passive role of the carbon core in photophysical behavior of carbon nanostructures.
Considering the in-particle ordering and stability, these features are determined by the mode of particle formation. Persistence in ordering of graphene layers for C-dots formed by a “top-down” technique is expected, and spontaneous arrangement of them in the case of nanoparticles formed in “bottom-up” synthesis is possible. But in the latter case, the π–π stacking forces among aromatic hydrocarbons cannot be strong enough for forming the stable structure [83], since they are known to be inherently weak (being an interaction among molecular quadrupole moments) [84]. Therefore, much stronger interactions are expected to play the major role in stabilizing the structures. This may result in ordering of their terminal polar groups extended to the surface that will be organized in stacks in the same planar mode and additionally stabilized by electrostatic interactions among them.
In the course of nanoparticle formation, the following scenario can be also realized. The synthesis of dipolar aromatic fluorophores and their assembly may be the primary factors guiding the ordering within the nanoparticle by their own π–stacking interactions with the contribution of electrostatic attraction. The known self-assembled structures of organic dyes are formed in this way [85,86]. In such multi-chromophore systems, a relative orientation of the dipolar dyes, pointed preferably in opposing directions, compensates for their individual dipole moments resulting in strong attraction forces. In C-dots, those may be the aromatic fragments with carboxylic and hydroxy groups [34] that, with the inclusion of nitrogen, may be complemented by pyrrole and pyridine derivatives [87] and with the participation of sulfur—by thiophenes [87]. All of them possess a strongly asymmetric electronic charge distribution and, thus, the tendency to aggregate in a stacked manner.
If the C-dots are formed in this way of stacking of aromatic dipoles as a primary process, then the carbonization with the formation of a particle core may proceed later. In a number of brilliant studies, it was shown that, in C-dots formed by thermal treatment of citric acid + amine, the aromatic fluorophores and their associates are observed well before the carbonization occurs [18,88,89,90,91]. Moreover, the fluorescent C-dots may totally consist of organic dyes [92,93,94]. As we seen below, these facts may suggest the principle of formation of ordered H-aggregated structures. Such formation and ordering of fluorophores may drive the assembly of the whole particle with the carbonization occurring or not occurring at the latter steps.
2.3. Variability in Quantum Yield, Emission Color, and the Strong Stokes Shifts
Carbon nanoparticles can be fluorescent if they contain carbon and oxygen only, particularly when they are made from carbohydrates or organic acids. However, their properties can be modified dramatically if different heteroatoms are included into their structures as the components of precursors during their synthesis (doping) or by binding after the synthesis (passivation). Doping with nitrogen [87,95], sulfur [96], and, less frequently, by phosphorous [97] and boron [98,99] atoms is shown to increase dramatically the fluorescence quantum yield. The most popular nitrogen-doped structures are formed of citric acid as a carbon source with the addition of ethylene diamine, urea, and amino acids as the source of nitrogen.
Manipulation with the emission color is important not only for various applications but also for understanding the electronic states of C-dots, since it is the means to modulate the energy gaps between the excited and ground states and to observe the excitation–emission correlations. Bright fluorescence of C-dots is usually observed in the blue or green spectral range when excited at 330–450 nm. Orange-red emission was also reported with the shift of excitation to longer wavelengths together with the strong decrease in intensity. This fact can be attributed to the heterogeneity of emitters [100] that appear on inter-particle [31] or intra-particle [101] levels. However, quite recently, scientists achieved strong emission in this long-wavelength region by variation of precursors and experimental conditions. This extended the application areas of C-dots including their use in light harvesting devices [102]. Here, we focused on cases when the most emissive fluorescent band shifted together with the shift of the correspondent excitation band maximum. These shifts were achieved by doping (variation of precursors), change of synthesis conditions or passivation (after-treatment).
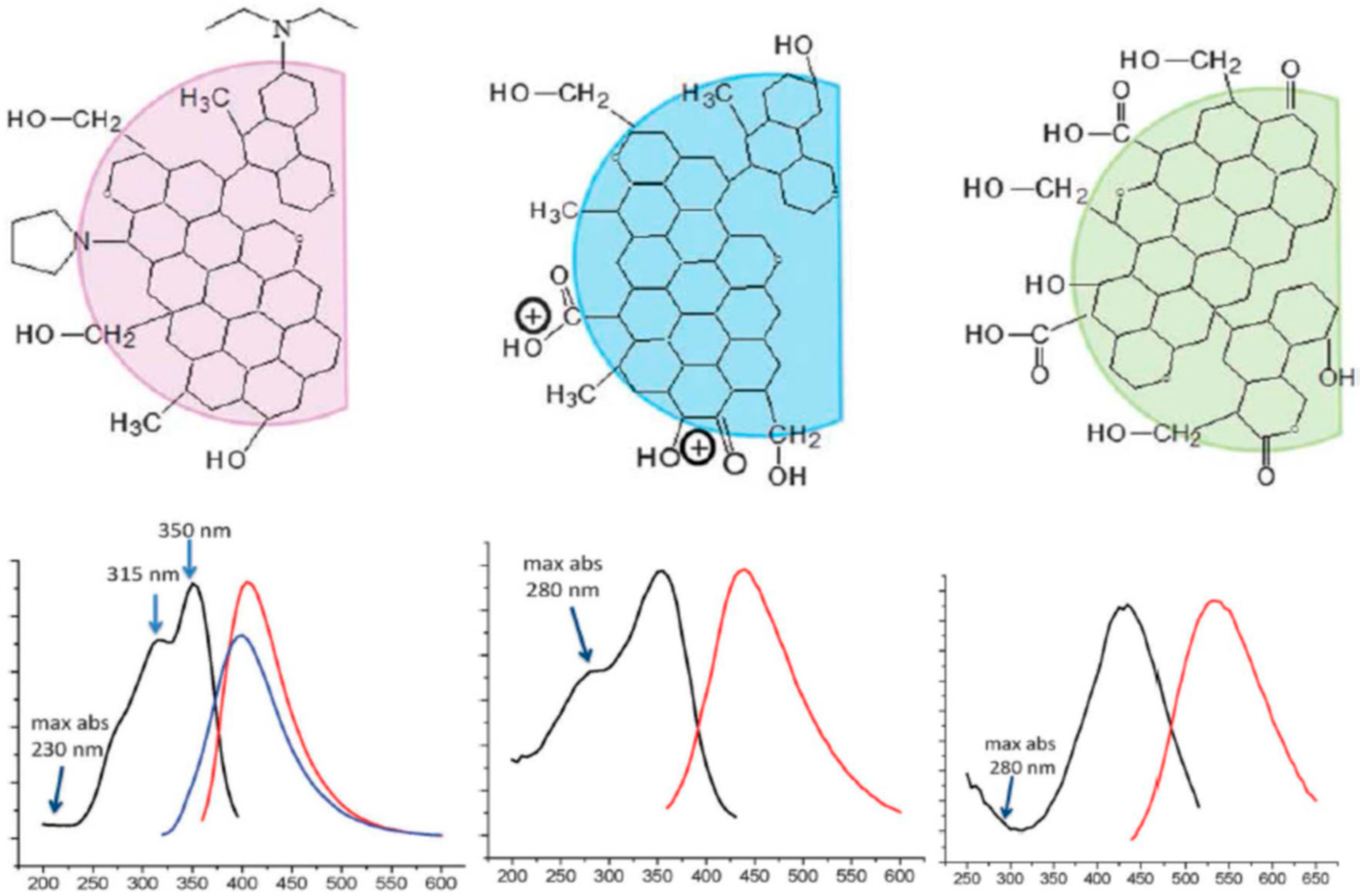
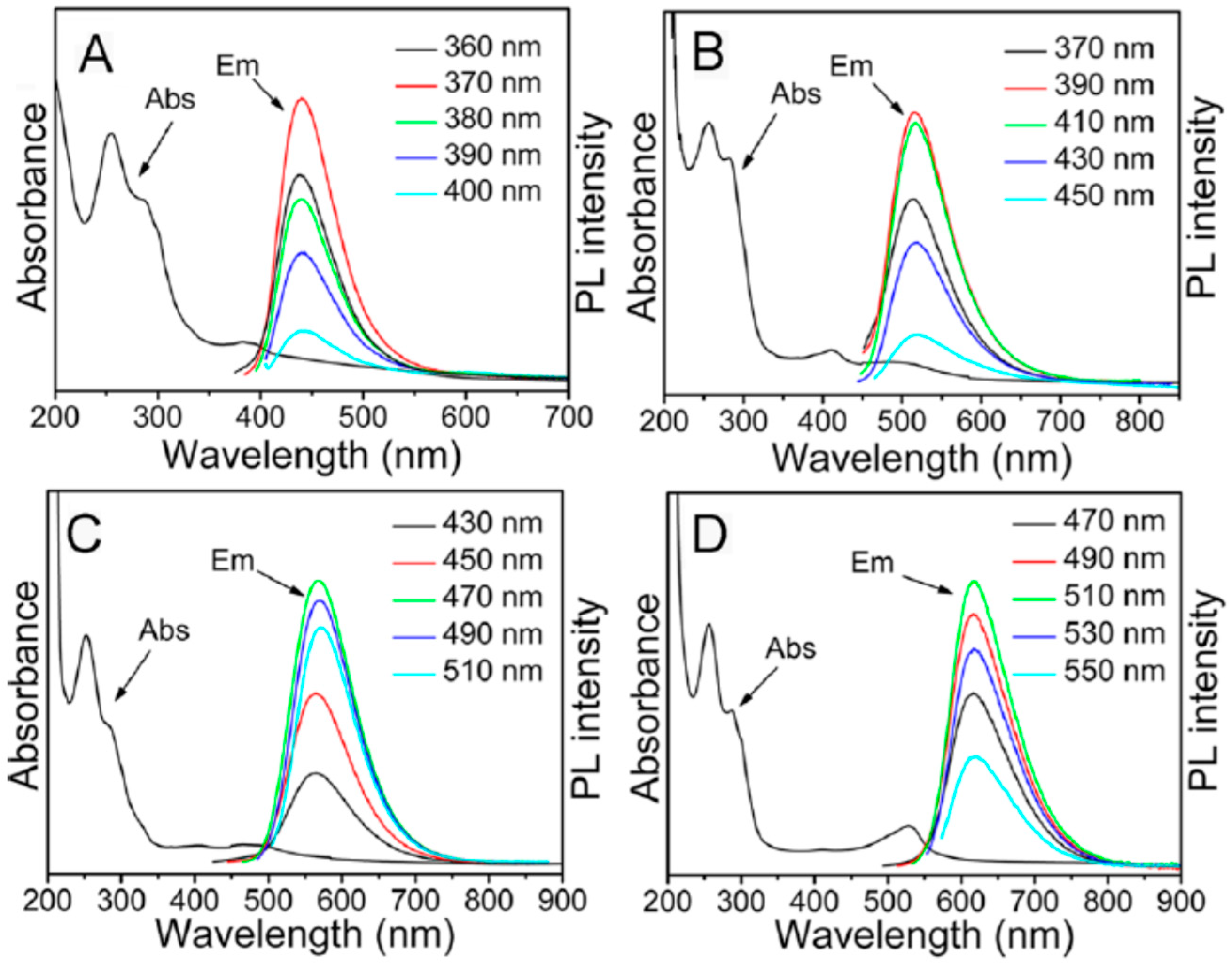
Multicolor C-dots could be obtained by different oxidative or reductive treatments of citric acid or ethylene glycol [104]. Hydrothermal synthesis of C-dots in one pot from urea and p-phenylene diamine resulted in fractions displaying different colors in their emission (with the band maxima ranging from 440 to 625 nm) [103]. These fractions were separated by column chromatography. Each of them demonstrated high optical uniformity, so that every sample showed only one peak in the excitation spectrum and only one peak in the excitation-independent emission spectrum (Figure 4). All of these fractions were characterized by high quantum yields (up to 35%) in water and quite similar mono-exponential fluorescence lifetimes. They did not differ in particle size or in the UV absorption spectra representing the carbon core. The only essential difference among them was the degree of oxidation at the particle surface. Therefore, the shifts in excitation and emission spectra to red were associated with the increase in the incorporation of oxygen species into their surface structures. In a recent publication by these authors [105], the bright red emitting C-dots were obtained from citric acid and ethane diamine in formamide. Essentially, being excited and emitting light of quite different energies, these preparations demonstrated quite similar Stokes shifts, suggesting the same photophysical mechanism of their emission.
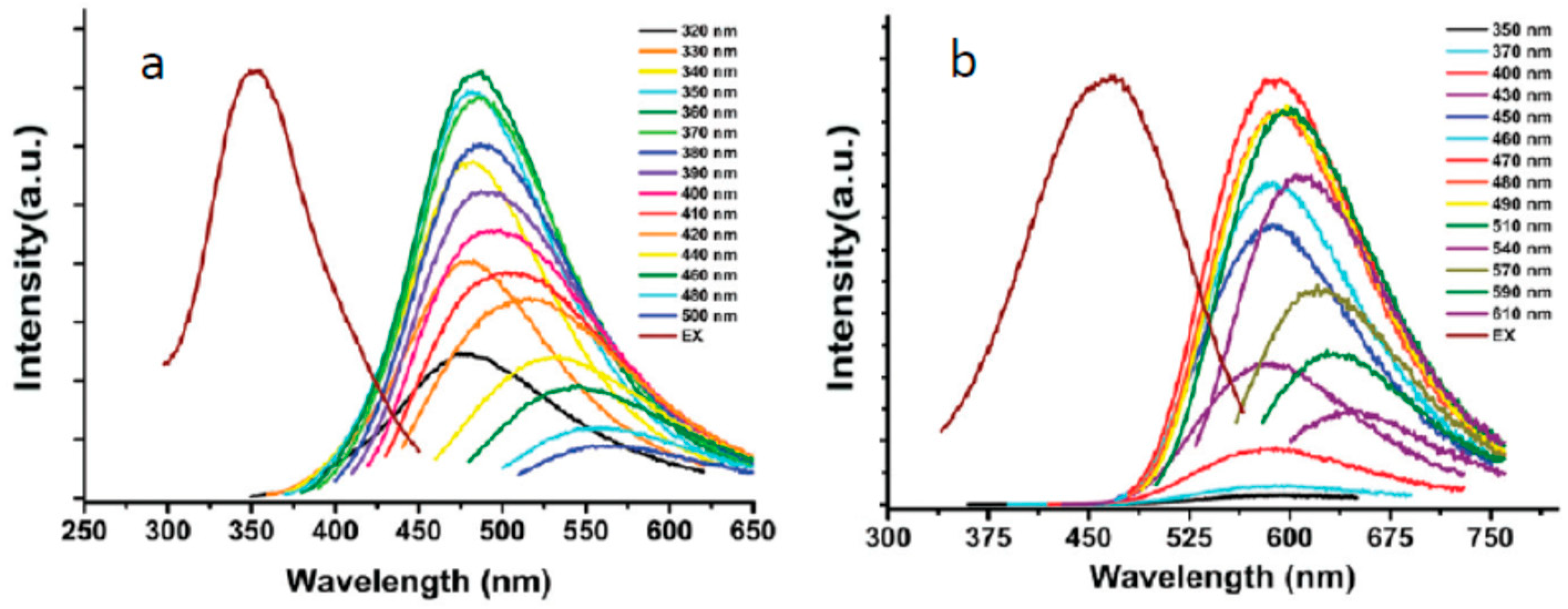
Adsorption of cations on C-dots surface [106] also favors the strong red shifts of fluorescence spectra as well as an increase of nitrogen content in starting materials [107]. Phenylenediamine isomers were used as starting materials for producing the C-dots of different colors [108]. Synthesis of blue, green, and red C-dots was reported by changing the solvent in the solvothermal reaction of citric acid and urea from water to ethanol and then to DMF [109]. Also, the sequential passivation with polyethylene glycol and 2,2′- (ethylenedioxy)bis(ethylamine) (EDA) resulted in the synthesis of the green, blue, and orange emissive C-dots [110]. The passivation changed the state of oxygen and nitrogen atoms. In these cases, the spectra were found to be excitation wavelength dependent (Figure 5). Essentially, the regularity of almost equal Stokes shifts can be observed not only for the main excitation and emission bands but also for photoselected wavelength-shifted emission.
Thus, we observed a strong Stokes shifts in the C-dots emission that displayed the general regularity independent of their structure, precursors, conditions of synthesis and spectroscopic studies, and excitation and emission wavelengths. They are quite similar to the particles emitting light over the whole range of the spectrum, from blue to red [111], so that the excitation spectra shift according to the shift of the fluorescence bands demonstrates Stokes shifts similar in magnitude. Therefore, clear separation among these relatively broad bands is commonly observed [11,43,112]. Such spectroscopic behavior is peculiar for C-dots of different composition with crystalline or amorphous carbon cores or with the inclusion of nitrogen-, sulfur- or phosphorous-containing groups at their surface. Moreover, the nanoparticles derived from graphene [59,69] and graphene oxide [12,113] also possess this feature. As we will see below, it is hard to explain such regularity based on the molecular type of fluorescence emitters, and, thus, the involvement of excitonic states must be suggested.
The strong Stokes shifts resulting in separation of absorption and emission bands together with high quantum yields and long fluorescence lifetimes are the key factors in achieving amplified spontaneous emission and lasing. Laser based on carbon nanoparticles in organic solvents was demonstrated in References [114,115].
3. The Key Questions to be Resolved
3.1. Why Are Carbon Nanoparticles so Emissive?
One of the mysteries of carbon nanomaterials is their bright unquenched fluorescence emission in the visible range. Due to the high amount and density of sp2 hybridized islands rich in π-electrons, such particles are the strongest light absorbers in the UV spectral range. However, they are also known as the strongest fluorescence quenchers acting as electron acceptors [9]. Graphene [116,117] or graphene oxide [118,119] were found to be very efficient in quenching all the emission of the dyes attached to them. Meanwhile, it is known that the fluorescent moiety of C-dots demonstrates strong electron–donor ability when interacting with external acceptors [55,120]. Moreover, their emission demonstrates the electron transfer quenching on interaction with other carbon materials such as graphene oxide and carbon nanotubes [121]. Why, then, is the visible emission not quenched by the intrinsic aromatic (sp2) carbon clusters?
This is probably because the two types of excited states, those generated by absorbing UV light (that are potential quenchers) and the emitters generated by visible light are uncoupled. Such uncoupling is seen in observational experiments of UV emission (at 303 nm) with deep UV excitation [122]. It may be due to the existing great difference in the energies of these states making negligible the overlap integral in FRET [71]. Experimental data show no indication of FRET from the UV light absorbers of the first type to emissive species of the second type which is evidenced by the absence of additional maxima in excitation spectra in the UV range [68]. In some cases, the excitation band in the UV was still found, but, surprisingly, the UV excitation may lead to emission in the near-UV that is quite separated from that excited and emitted in the visible region [123]. Back transfer resulting in quenching is even less probable. The other explanation that does not disregard the first one is that the structural separation between fluorophores at the particle periphery and the carbon core is so large that the overlap of their electronic wave functions is small, and this does not allow the electron-transfer quenching.
The other question is related to the visual light emission itself. Why is there no self-quenching of fluorescence within the carbon nanoparticle excited in the visible region? It should be noted that upon formation of π–π-stacked dye ensembles, many new relaxation pathways beyond those given for dye monomers may become active. It is well known that in aggregated organic dyes and even in concentrated dye solutions, concentration quenching is a very general phenomenon [124], whereas the aggregation-induced emission (AIE) [125,126] can be observed only in special cases. Commonly, AIE is achieved by elimination of direct π–π interactions among the dyes in aggregate with the aim to retain their molecular-type photophysics and, in this way, avoid concentration-dependent quenching [127]. A variety of excitation energy traps could appear with the formation of carbon nanostructures, similar to that of associations of organic dyes [128]. The generally known mechanism of quenching is the formation of excimers and exciplexes. The term exciplex (excited complex) is used for general description of an electronically excited molecular complex of definite stoichiometry. More detailed classification includes homo-excimers (excited dimers) (electronically excited complexes formed between identical atoms or molecules) and hetero-excimers (electronically excited complexes formed between two non-identical atoms or molecules). Traditionally, the excimer formation was considered as the reason for fluorescence quenching [129] and, indeed, with few exceptions (e.g., pyrene derivatives [130]), such complexes are non-emissive, and avoiding them is the problem when constructing nanoscale fluorescence emitters made of organic dyes [131]. Thus, the nanoparticles and nanocomposites made of fluorescent dyes are often designed to avoid the excimer formation.
In addition, the non-fluorescent species may be formed already in the ground state as the charge–transfer complexes and H-dimers [132]. They originate due to the close proximity of the dyes in their ensemble and their concomitant electronic coupling. Moreover, in networks of organic molecules, the loss of the excitation energy that competes with the energy transfer among the fluorophores can follow several mechanisms, and the transfer process may be directed along multiple routes terminating in quenching. The question is, why does this not happen in carbonic nanostructures? A possible explanation is the formation of excitonic states, in which the mechanisms of energy propagation and quenching are quite different. A closer look at the cases of aggregation-induced emission reveals that, in addition to the high rigidity of molecules in aggregate (suppression of intramolecular loss of excitation energy), the structural requirements should be satisfied for making impossible the formation of non-fluorescent associations among constituting dyes [131]. It is hard to presume that all known cases of fluorescent nanocarbon particles conform to such conditions. Thus, the question as to how the concentration quenching is avoided in carbonic nanostructures remains without a convincing answer.
3.2. What Determines the Excitation and Emission Peak Positions?
Carbon nanomaterials demonstrate an incredibly broad range of fluorescence emitting colors. The first reported carbon dots were excited in the violet range and emitted blue light [1], and in most of early studies the emission was reported in the blue-green range of the spectrum [95]. Nowadays, the emissive carbon dots covering the whole visible spectral range down to the near infrared have been reported [133,134]. They emit bright fluorescence with band maxima observed in yellow [135,136], orange [137,138], and red [78,139,140] colors. The near-IR absorbing and emitting nanoparticles were recently obtained [141,142]. Thus, the spectroscopic coverage by these emitters is very broad, similarly to the well-known visible fluorescent proteins [143]. The important difference is that in fluorescent proteins, the structure is well known down to the atomic level and all the steps of its formation are well understood, whereas, in our case, the origins of all spectroscopic properties are debated.
The key point of this discussion is the relative role of conjugated sp2 domains within the carbon core that can be light emitters and energy transfer donors and of the “surface states” that can be molecular-like emitters [144] or the sites of emissive recombination of excitonic electrons and holes [27]. Some authors consider the role of the core as central [80,113], whereas others suggest as central the coupling between the surface and the core [145], their dual involvement in emission [146], or complete absence of core contribution [82,103,147]. In single-particle experiments (see Figure 2) the species with a crystalline and amorphous core were studied in the same sample, and they demonstrated rather similar characteristics with respect to their fluorescence [31]. Assuming the concept of the central role of surface-exposed fluorophores, it is not easy to predict and modulate the emission color based on their structures and chemical modifications. Moreover, there is evidence that the emissive states are highly delocalized on the disordered surface shell thus involving simultaneously a large number of functional groups [82].
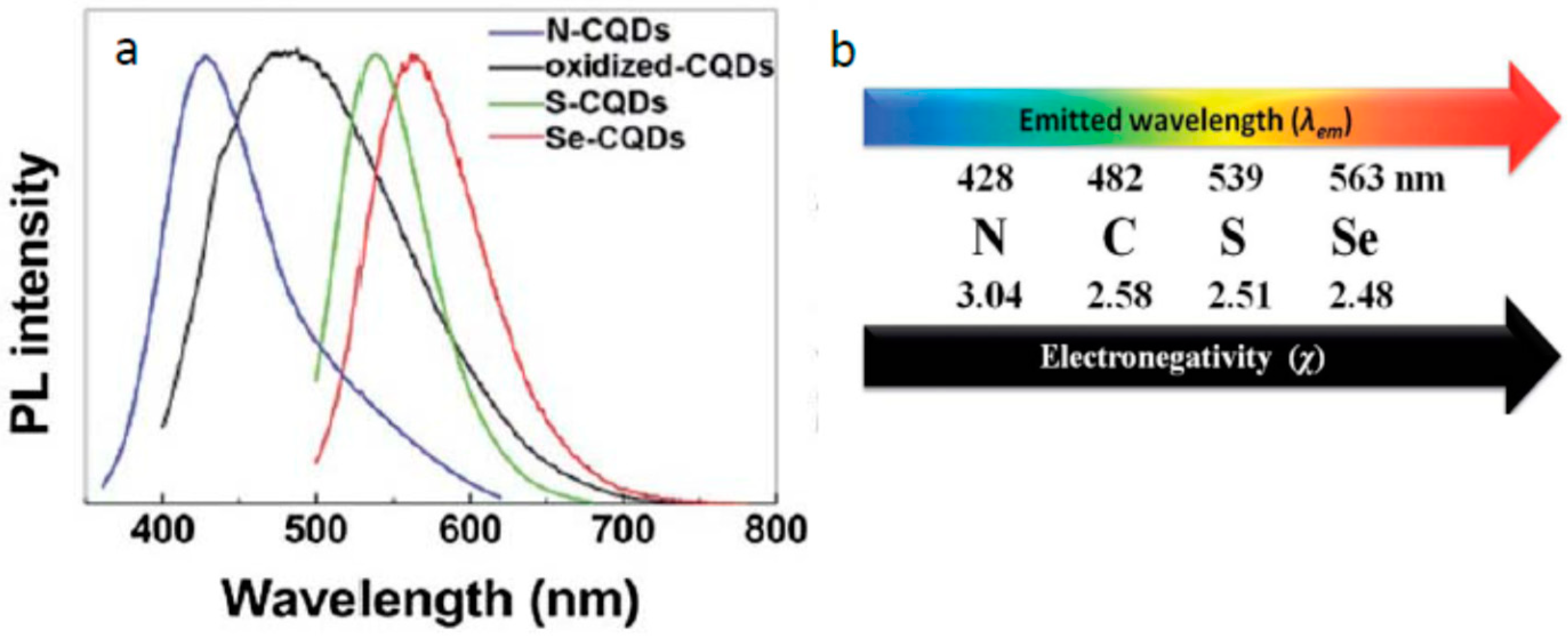
It is frequently assumed that all different proposed mechanisms are possible, they can operate in parallel, and it is the particle design that may provide a preference for one of them. Indeed, the particle design is very important but, if we stand on this position, our efforts for optimizing the optical properties will remain empirical. An extended discussion of these issues can be found in a number of recently published reviews [11,17,43,148,149] from which one can derive the key role of surface-exposed groups, their passivation, modification, oxidation or reduction in generation and modulation of fluorescence emission. Presenting many examples, these works show that the surface modifications play an important role in controlling the emissive states without altering the electronic states of the core. Thus, it was found that a strongly red-shifted fluorescence emission could be achieved by doping the electron-withdrawing fluorine atoms [139]. One of the correlations was found in heavily doped species of C-dots between spectral positions and the electronegativity of doping atoms [150]. Doping with the heteroatoms in a sequence of N, S, and Se resulted in strong shifts of fluorescence spectra to the red (Figure 6). In another study performed on separated carbon dots fractions, it was found that the fluorescence increased with the increasing degree of oxidation [103]. The dependence of emission color on the extent of exposed carbonyl groups was also reported [151]. The role of this and many other factors that determine the spectroscopic behavior is the major point of discussion in the current literature. At present, it has not led to a definite answer that can be used as a guideline for creating materials with optimal and well-predictable properties.
Of particular interest is the concentration-dependent tuning of fluorescence emission in carbonic nanoparticles which must be the result of shifts in their association–dissociation equilibrium. The unexpectedly strong spectral shifts to longer wavelengths with the concentration increase were observed by several authors [152,153]. Thus, these shifts were reported from 514 to 585 nm [154], 404 to 583 nm [155], and even 400 to 630 nm [156]. This means that carbonic nanoparticles may interact in solutions, and this interaction substantially decreases the energy of emitted quanta.
The other interesting phenomenon is related to the “bottom-up” synthesis of C-dots. When they are synthesized in the same reaction pot, the nanoparticle formation may proceed along two or more parallel routes in well-resolved discrete forms with recognizable spectroscopic properties, instead of the expected broadly heterogeneous species [134,157,158]. The observed dual emissions can be excitation wavelength-independent, and these properties can be retained in the solid state [158]. It was reported that the two blue and orange excitation-emission channels can be disconnected and excited at two different wavelengths [158]. Reporting on this, some authors believe that the different emissive sites are formed within the same nanoparticles, e.g., belonging to intrinsic states and also to solvent molecules connected on the surface [159]. However, in some cases, these forms can be separated by chromatographic techniques [157,160]. The possibility of selectively bleaching one of these emissive forms due to the fact of its much lower photostability was shown [161], as well as its quenching of inter-particle interactions [162]. Single-particle studies [101] have demonstrated that different resolved spectral forms can be attributed to individual particles, suggesting the possibility of integrating different types of electronic transitions within the nanoscale composition. Transition from one emissive form to another may also proceed in the course of synthesis and as a function of reaction temperature and, thus, it can be thought that carbon particles initially form an association of molecular fluorophores and then they attain the carbonic core with a dramatic change of the emission [88,91,163]. All this suggests that thermodynamic and kinetic drivers exist for ordering, resulting in quite discrete supramolecular forms possessing discrete electronic states.
Summarizing, we state that our present lack of knowledge does not allow for establishing clear relations between the structure and spectra in a carbon nano-world. What is surprising is the fact that the carbonic nanostructures—despite their varied history of formation from different sources, their chemical differences, and the resulting tremendous variability of structures—demonstrate a broad range of spectral differences and possess quite similar photophysical behaviors.
3.3. Why Are the Strong Stokes Shifts Systematically Observed?
Now we refer to unusually very strong and regular Stokes shifts in carbonic nanoparticles. As it was demonstrated by numerous authors, the substantial shifts of visible emission with respect to the excitation band, reaching or even exceeding the level of 3000–6000 cm−1, are quite common for all fluorescent carbon nanomaterials. It was reported that such strong Stokes shifts are almost polarity independent when studied in solutions [164] and are retained in solid states [165]. Meanwhile, it is known that such strong Stokes shifts cannot be generated by molecular forms of aromatic hydrocarbons if they do not create strong molecular dipoles interacting with polar solvents in the excited states or participate in some excited-state reaction such as electron or proton transfer.
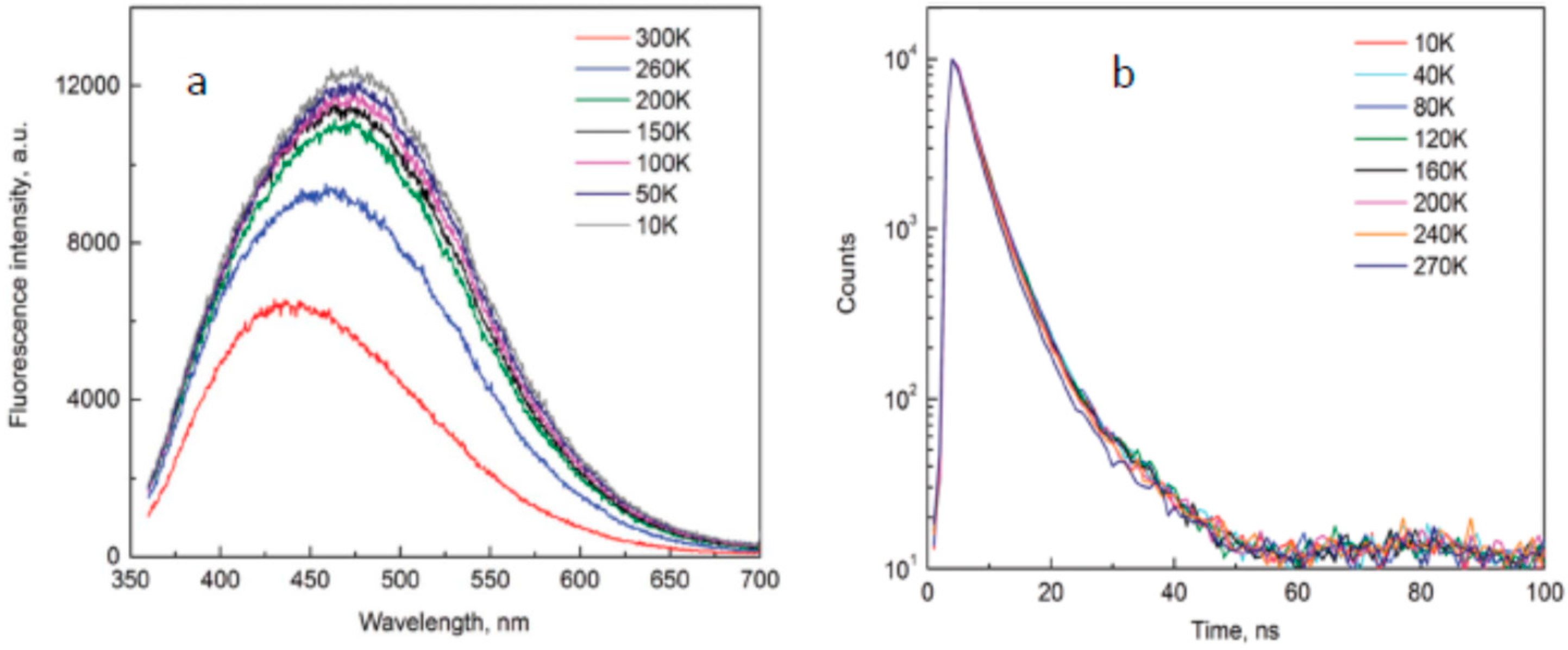
A molecular view suggests several explanations for such essential losses of excitation energy, and the involvement of dielectric relaxations in excited states of the constituting fluorophores interacting dynamically with their environment is one of them. The requirements for achieving such properties are the strongly distributed charge resulting in a strong dipole moment in the excited state and its location in a highly polar mobile environment. In this case, the decrease in the excitation energy should proceed before or during the emission. Indeed, in some studies, the authors detected the solvent-dependent variations of spectra [78,166,167,168] and the relaxation-related shifts in time-resolved emission bands when solution was used [168,169]. However, the magnitude of the observed time-resolved effects was not sufficient to explain the whole phenomenon. Moreover, the Stokes-shifted emission was found to be retained in solid state [111] and in cryogenic conditions [26,30] in which the solvent environment is removed or immobilized as well as in single-molecular studies of isolated nanoparticles immobilized on support [31].
The other possibility that potentially exists is the formation of fluorescent excimers and exciplexes due to the pairwise interaction among the fluorophores if they are located closely [170]. When one of them is excited, it forms a complex with the other, resulting in a strong loss of energy leading not only to quenching but, potentially, to the red-shifted fluorescence emission. Such cases are known and can be seen in some arrangements in molecular crystals [171] and conjugated polymers [172]. The most emissive excimers were described for pyrene. On formation of its excimers in solutions, the fluorescence spectrum shifts from ~400 to ~485 nm. Inspired by these analogs, the researchers made an attempt to explain the Stokes shifts in C-dots by constructing the system of multiple excimers [80]. However, as noted above, in most of the known cases, such interactions result in emission quenching. Therefore, it is hard to imagine that this mechanism can be of general value operating for very different fluorophores that emit brightly at different wavelengths. The intramolecular excited-state reactions, such as isomerizations, charge transfer, and proton transfer, may generate strong shifts in the fluorescence spectra [173,174]. These reactions require a specific arrangement within a fluorophore of specific groups of atoms and, in view of the diversity of studied structures, it is hard to imagine them being of general value.
Thus, we cannot explain the observed effects by considering the carbon nanoparticle as a box packed with aromatic fluorophores emitting individually. However, there remains a possibility that the fluorophores interact weakly, which allows an energy flow from the short-wavelength excited to long-wavelength emitting fluorophores within the aggregate. According to FRET theory, such dipole–dipole resonant interaction among fluorophores does not require their exact ordering and can propagate on a nanometer scale [174]. The requirement for dipole–dipole resonance is the overlap of the fluorescence spectrum of the donor and absorption spectrum of the acceptor. Following this concept, in studies of carbon nanoparticles and considering them as a system of fluorophores, we need to consider multiple FRET steps not only because their expectant close location but also because of the poor overlap between the averaged excitation and emission spectra that is always seen in experiments. Meanwhile, the experiment does not show evidence for intra-particle energy flow that could result in strong temporal Stokes shifts [68].
3.4. What Is the Origin of Spectral Heterogeneity and Tunability?
The spectroscopic heterogeneity of most nanoscale carbon samples is clearly evidenced by the broad unstructured emission bands, their dependence on excitation wavelength, and non-exponential kinetic traces in fluorescence decay [175]. Continuous shifts of fluorescence emission as a function of excitation wavelength (see Figure 5) and, correspondingly, of excitation spectra as a function of recording wavelength are the indications of emission tunability. Such shifts are usually accompanied with a great loss of intensity [176]. Its origin is strongly debated in the literature and a clear explanation is presently lacking so that many authors connect it with structural heterogeneity [17,175] and with the presence of impurities and by-products [177]. In carbonic nanomaterials, this feature is present to a different extent, from being very strong to its complete absence [99,178,179]. Its relation to the red edge effects [175,180] seems to be incorrect. Since the spectral selection can be observed also on the “blue edge” of the excitation band (see Figure 5) and the excitation spectra also shift as a function of emission wavelength, the most probable is the interpretation in terms of the ground-state heterogeneity [100,181].
Regarding the origin of such spectral heterogeneity, the first question to be asked is whether it appears within or among the nanoparticles. It may appear due to the structural variety of fluorescence emitters and to their in-particle interactions leading to selection of their differently shifted excitation spectra form’s inhomogeneously broadened contour [182]. Shifting the excitation wavelength, one can produce photoselection of species whose fluorescence is the brightest at this wavelength, and such an emission spectrum becomes wavelength shifted. Existing methods of preparing carbonic nanostructures do not allow for excluding this type of heterogeneity. Therefore, it is suggested that each luminescent particle represents an assembly of different emitters [18] such as the aromatic islands within the particle core [19] or their charge–transfer complexes [20] formed with polar groups on the particle surface [21]. Understanding of such possibilities is poor due to the vague knowledge of the principle of formation of emissive states. The emission may come from several independently formed fluorophores within the same structure being photoselected by excitation energy. There may also be the assemblies of fluorophores attaining excitonic properties. Confined within the nanostructure, molecular or excitonic emitters may transfer their excited-state energy to electron–hole recombination sites of terminal acceptors that may be variable. With our present knowledge, there is no clarity regarding these issues.
In many cases, however, the spectral heterogeneity was not observed, particularly for blue [183] and green [62] emitting carbon dots. Moreover, as it was shown by several authors, the spectroscopic features of heterogeneity can be removed in purification procedures. Such purified nanoparticles demonstrate excitation wavelength-independent emission and single-exponential lifetimes [160,184]. In principle, the excitation wavelength independence of fluorescence spectra can be observed in two cases: when the light emitters in their ensemble are truly identical in spectroscopic properties and also when the differences exist but they can be averaged on a scale faster than the emission [100,174]. Therefore, it was important to provide single-particle studies on those carbon dots that demonstrated significant spectroscopic heterogeneity in solutions. It was shown in Reference [31] that single carbonic nanostructures can exist without in-particle spectroscopic heterogeneity and display single-exponential decays, though in a population they may differ strongly in these parameters. The features characterizing heterogeneity in ensembles should be the result of the summation of the individual contributions. In another study [101], it was shown that intra-particle heterogeneity is still possible. However, it may be the result of forming associations among nanoparticles since they are able to aggregate (see Section 3.1). In general, all these data do not support the ideas advocated by different authors: that the excitation may travel within the nanoparticles more or less randomly to randomly distributed terminal acceptors resulting in the in-particle heterogeneity of emission. Direct proof of this mechanism is lacking.
Does the interaction with a solvent influence the spectroscopic properties of carbonic nanoparticles? The experiments show that their spectral positions may not be significantly affected by the solvent change [185] or by drying the sample and its studying in solid state [165,186,187]. The study of single particles applied on a support also show the spectra close to that measured in solutions [31]. However, in solutions, if the surface-exposed groups are involved in emission, they should interact with the solvent and we should see some evidence for that. Indeed, solvent effects were noticed modulating the positions of emission spectra that can be plotted as a function of polarity [165,167,188]. With the increase in solvent polarity, the red shifts of these spectra were observed, and the shifts reported by some authors were very dramatic, e.g., from green (530 nm) in toluene to red (620 nm) in water, and similar changes in color were achieved through their incorporation to polymer matrices of different polarities [165]. Such effects are similar or even more substantial than that in charge–transfer organic dyes chosen as polarity indicators [189]. Dielectric polarizability and hydrogen bonding with protic solvent molecules [166,190] may contribute to this spectral change [167]. Regarding hydrogen bonding, its effect is local and should involve only small proton accepting/donating surface groups. In contrast, the dielectric polarization is a collective effect; it requires the dynamic generation of a reactive field in a fluorophore environment in response to the appearance of its high dipole moment due to the strongly increased charge separation in the electronic excited state [191]. How, then, is such a dipole moment formed? In organic dye molecules this happens by redistribution of π–electron density, but it is hard to comprehend how the electron density is shifted in the case of carbon dots. Does that occur over the whole nanoparticle or only at its local emissive site?
It is interesting to see if there is a connection between the solvent–dependent shifts and spectral tunability discussed above. The shifts induced by polar solvation are possible if the fluorophore environment is highly dynamic on the timescale of emission. If it is, then the effects of inhomogeneous broadening should not be seen (the dynamics will average the sub-states with different excitation energies). In contrast, in the case of ground-state heterogeneity, variable species in ensemble will respond to solvent in a more or less similar way, generating the total solvent effect [181]. In reported cases, the solvent–dependent fluorescence spectra were not affected by modulation of excitation wavelength [164,188], indicating that the polarity effects and wavelength tunability were uncoupled. This suggests considering the nature of carbonic nanoparticles to be highly dynamic. Is this a general trend? The situation is not so simple, since the solvent–dependent shifts may show spectroscopic heterogeneity and the emission from two or more poorly resolved emission sites can be involved [192]. Therefore, the absence of easily visible regularity in solvent effects and in their site-selectivity raises many suggestions regarding the presence of different emissive states that are stabilized to different extents by the surrounding solvent environment.
3.5. What Is the Photophysical Mechanism Governing the Emission?
In every fluorescent carbonic nanoparticle, we can find atomic structures arranged into aromatic heterocycles rich in π-electrons that can be located either in the inner core or in outer shell. They may be attached to the particle surface, and they can be coupled in different ways and may allow the transfer (and exchange) of electrons, protons, and excitation energy. This transfer can attain collective characters and propagate as excitons terminating with the emissive electron–hole recombination. Based on the similarity in spectroscopic behavior with other well-studied emitters, different mechanisms of the emissive process were suggested and are actively being discussed. Even the question of where the emissive sites are located [193] do not have a definite answer, since the excitation can migrate among different sites, and the sites of exciton–hole recombination cannot be easily identified.
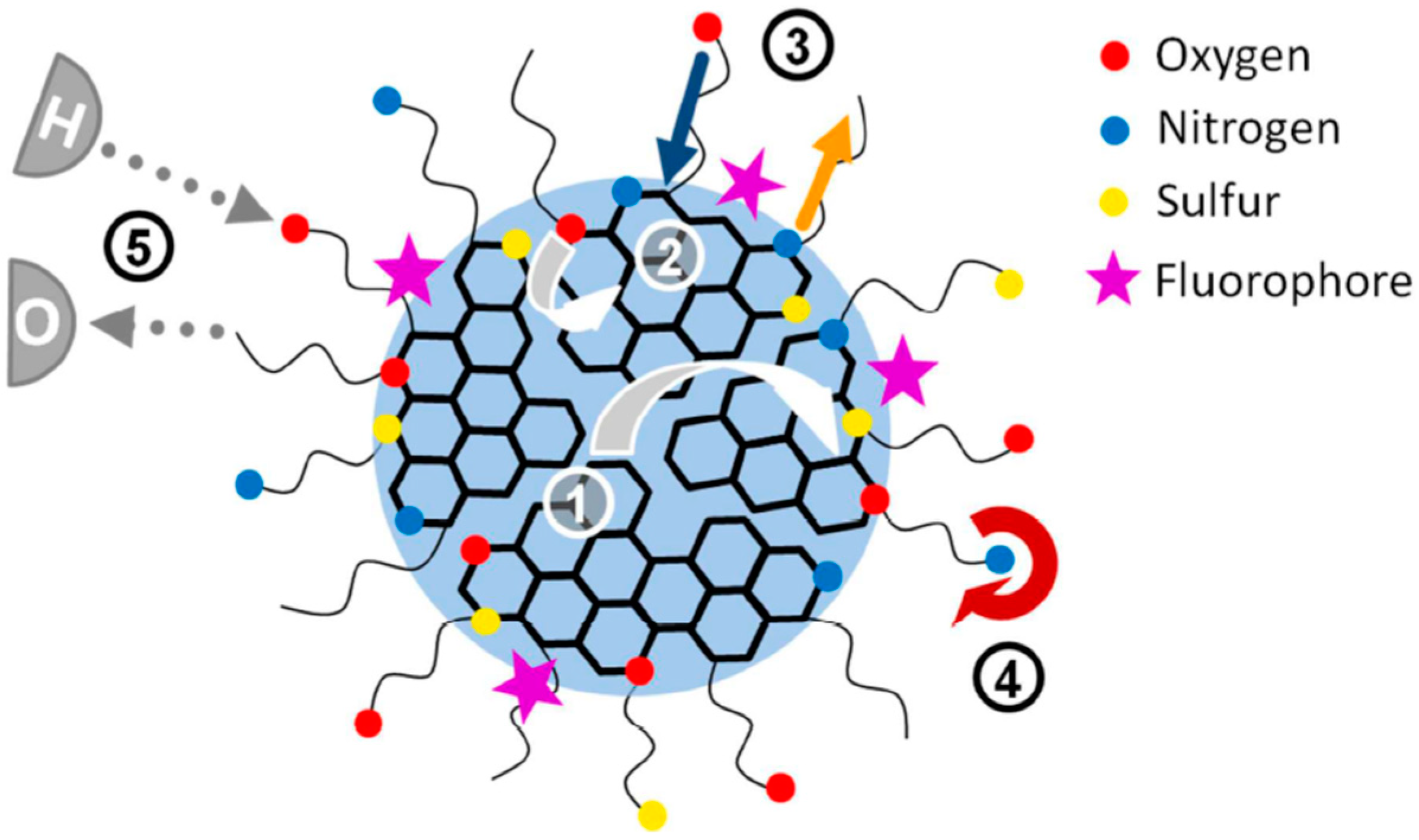
The situation becomes even more complicated when observing several emitting sites excited and emitting at different wavelengths and acquiring the possibility of achieving white color emissive carbon dots [159,194]. Since these initial excited states subsequently decay through a variety of pathways, different mechanisms may be involved here [166,192,195]. Figure 7 illustrates this complicated situation, in which different frequently discussed interactions and transition pathways in N- and N,S-doped nanoparticles are illustrated.
It is surprising that, with all the diversity of structures, interactions, and excited-state reactions, all types of fluorescent carbon nanomaterials tend to show several recurrent properties, such as a bright and broad (60–100 nm in width) fluorescence emission, which can usually be tuned across the visible range by changing the excitation energy, demonstrating unusually strong Stokes shifts that are never observed in unmodified aromatic hydrocarbons. The similarity among the different types of nanocarbons extends to fluorescence lifetimes that are always in the nanosecond time range, usually longer than that of typical monomeric dyes [69]. The fluorescence quantum yields were already reported to be relatively high, 30–40% [45,49], and some recent data report a much higher level [196,197]. On the other hand, random formation of particle surface groups, their modification and heteroatom doping, and the interaction of these heterogeneously formed surfaces with solvent must result in microscopic heterogeneity, the issue raised by many researchers. Variation of starting material composition and of the conditions of synthesis (e.g., temperature, exposition time, pressure) generates an innumerable number of structural variants. Nowadays, these factors are selected by empirical trials with the aim to achieve the optimal parameters for various applications.
4. Chromophore Behavior in Nanoscale Ensembles
Since multiple fluorophores are potentially present in carbon nanostructures, the knowledge of the collective behavior of organic dyes in their crystalline and aggregated forms may help us to comprehend the origin of their fluorescence. The theory of molecular excitons is central in this description. By definition, excitons are the mobile neutral quasi-particles in the form of electron–hole pairs that appear in solid bodies and molecular associations upon electronic excitation [198]. Excitons can be considered as the propagating collective excitations in a system of interacting molecules, in which the participating fluorophores lose their individuality and the whole system responds (emits light or is quenched) as one unit. The hole is the positively charged site, from which the electron is abstracted. The electron and the hole remain bound by Coulomb forces. Their propagation is coupled, so that they carry energy but not the charge.
Depending on the strength of electron–hole bonding, the excitons can be of two major types [199]. There are Wannier–Mott loosely bound excitons such as those observed in semiconductor nanocrystals—quantum dots (QDs). They are often called large radius excitons because, in these cases, the electrons absorbing the light quantum are promoted to conduction band and behave as free charge carriers. They can propagate to a large distance that is limited only by a crystal or particle size, and their recombination with the holes results in fluorescence. The other type of excitons that we will consider is the strongly bound Frenkel excitons. Possessing high electron–hole affinity, they are typical for the complexes of organic molecules and conjugated polymers [200]. If these excitons are formed in dye aggregates, the electrons remain localized on molecular sites, while the excitation is coherently delocalized over many fluorophores [201].
4.1. On the Involvement of Wannier–Mott Loosely Bound Excitons
In the case of loosely bound excitons, the light absorption results in transition of an electron from the valence band to the conduction band. This generates the motive bound pairs of conduction-band electrons and valence-band holes, so that the emission is coupled with the loss of energy released on electron–hole recombination. Electrons and holes are attracted by Coulomb interactions, but because of dielectric screening, this attraction is weak. The exciton wave function is delocalized, and the exciton is allowed to move freely. In QDs, the quantized energy levels appear because the wave function of the exciton becomes confined in all three spatial dimensions. Such a quantum confinement effect determines the optical properties [202] so that the positions of absorption and emission bands become determined by the nanoparticle sizes. The smaller they are, the fewer energy levels can be occupied by the electrons and the greater the distance among their individual levels. Therefore, the decrease in particle size results in blue shifts of their emission [203,204]. It is because of small bandgaps (the energy gaps to an electron conductance state) and high dielectric constants that the excitons with such properties are typically found in QDs. It is a question if these conditions can be realized in carbonic nanostructures. However, theoretical studies have shown the possibility of generating the loosely bound excitons in graphene nanoscale monolayers with the inclusion of polar groups at the sheet edges [205,206]. The evidence for their presence was obtained in experiments on graphene oxide sheets [207].
In line with this concept, the bandgap in carbonic nanostructures could be generated by the quantum confinement of the delocalized π–electrons in the conjugated sp2 carbon networks [11,77]. There were many suggestions to attribute the origin of fluorescence emission in the visible spectral range to the exciton–hole recombination sites [26,27] that could be located on particle surfaces [28] or on sheet edges [29]. In this case, the exciton propagation length may involve the whole particle volume [21,23] or the whole particle surface [24,25].
If this concept is correct, then, like in semiconductor nanocrystals, the emission wavelength should depend strongly on the particle size. Such dependence must be seen when the particles made of the same material are separated according to their sizes—the smallest should emit blue and the largest red. Though such dependence was reported by some authors [76,77], its confirmation was not obtained in later research [208]. In some publications [145,209], the size effects were considered to be important but screened by more efficient effects of surface states.
Another critical issue of this concept is the difficulty in explaining the large Stokes shifts which in typical QDs are only ~15 nm from the absorption onset [203]. The exciton free motions and their recombination do not require significant energy relaxations. High local dielectric screening of charges needed for formation of loosely bound excitons is the characteristic of low bandgap inorganic semiconductors [210]. However, such type of electron delocalization may exist in ensembles of aromatic hydrocarbons when their surface is exposed to a polar environment, e.g., in short carbon nanotubes that can be fluorescent [211]. It is not clear if this effect may operate in three-dimensional carbon nanostructures. Also, the nanoscale systems with loosely bound excitons confined in all three directions display optically isotropic fluorescence response [212] and because of such isotropic three-dimensional behavior, the QDs are considered as “dots”. However, in some rare cases the excitation transition dipole moment was observed (e.g., in single silicon nanocrystals) and it is attributed to local defect centers [212]. Still, the analogy of carbon nanostructures with such systems is unclear and the numerous experimental data on C-dots show high level of their optical anisotropy on excitation by polarized light (see Section 5.2).
These facts witness that the theory of Wannier–Mott loosely bound excitons explaining the optical properties of inorganic semiconductors may not operate in carbon nanostructures or operate only in a limited number of cases.
4.2. The Excitonic States in Aggregates of Organic Dyes
The aggregates of organic dyes demonstrate quite different behavior from that of semiconductor crystals. The collective effects that appear on their association are well described by the theory of molecular excitons that are often called the Frenkel excitons. Here, due to the absence of essential dielectric screening, the electron–hole pairs demonstrate stronger Coulomb attraction. Therefore, they are called the strongly bound or small-radius excitons. As the neutral quasi-particles, they may propagate through the aggregate while at each instant of time, the electron and the hole occupy the same molecular site. This process, by which excitons spread in a wave-like manner over spatially separated molecular dyes is called coherent exciton delocalization [201].
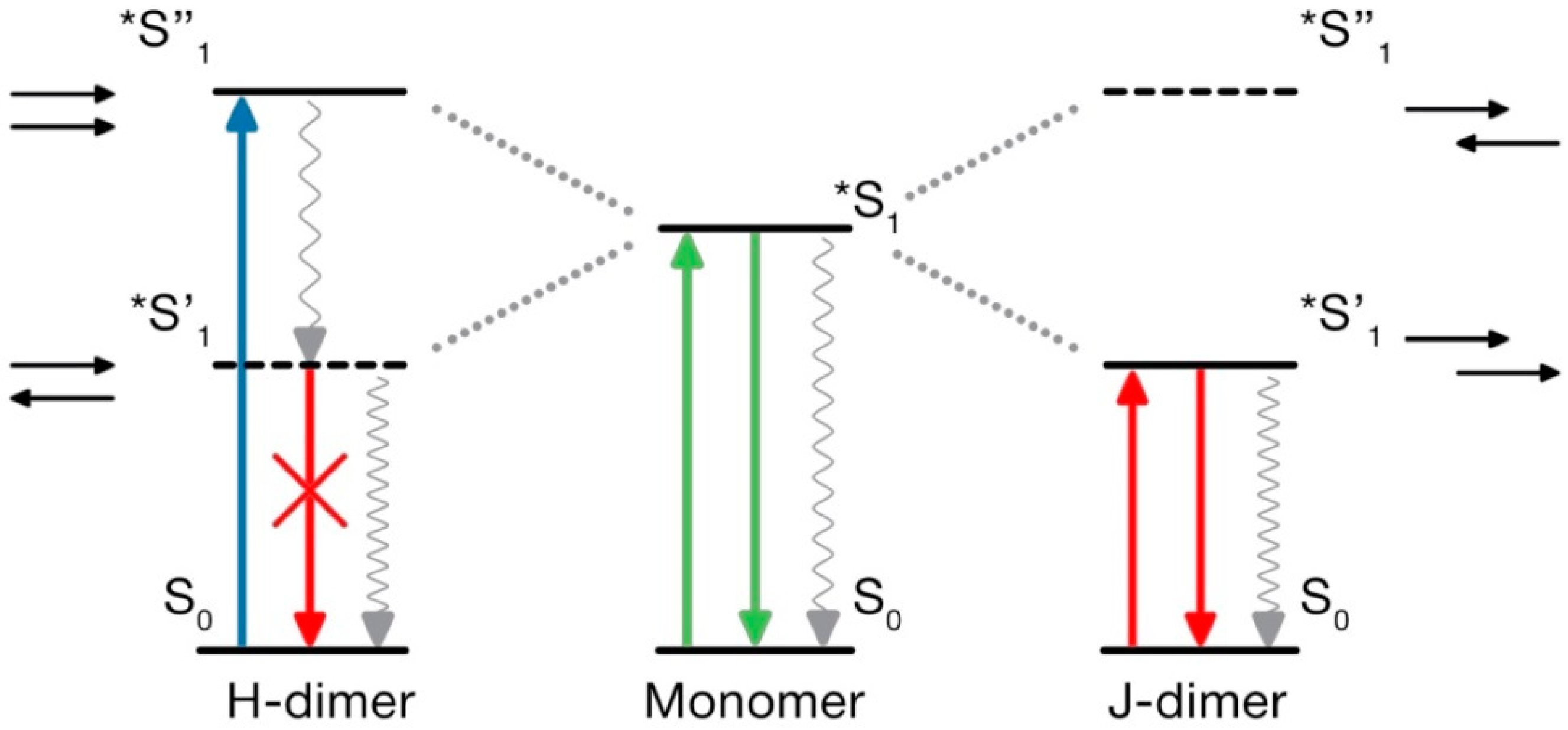
The theory of molecular excitons was developed by Alexander Davydov [213], and it has brilliant confirmation in studies of molecular crystals formed by organic dyes [214]. In this description, the aggregates are presented by ordered structures of constituting chromophores with their purely electrostatic intermolecular coupling but without significant overlap of electronic density. The electronic excitation is delocalized over many monomers in the form of excitation waves [215], leading to new spectroscopic phenomena [213]. In order to display them in organic dyes, the high crystallinity in the aggregates may not be needed, but they should assemble into well-ordered intermolecular arrangements with a definite packing geometry. The simple model linking this geometry and photophysical properties is based on a dimer of aromatic molecules that is represented by point dipoles. Suggested by Michael Kasha [216,217,218], it was very productive in explaining major spectroscopic effects (Figure 8). This model was extended to molecular lattices [219] and became the basis of many new developments.
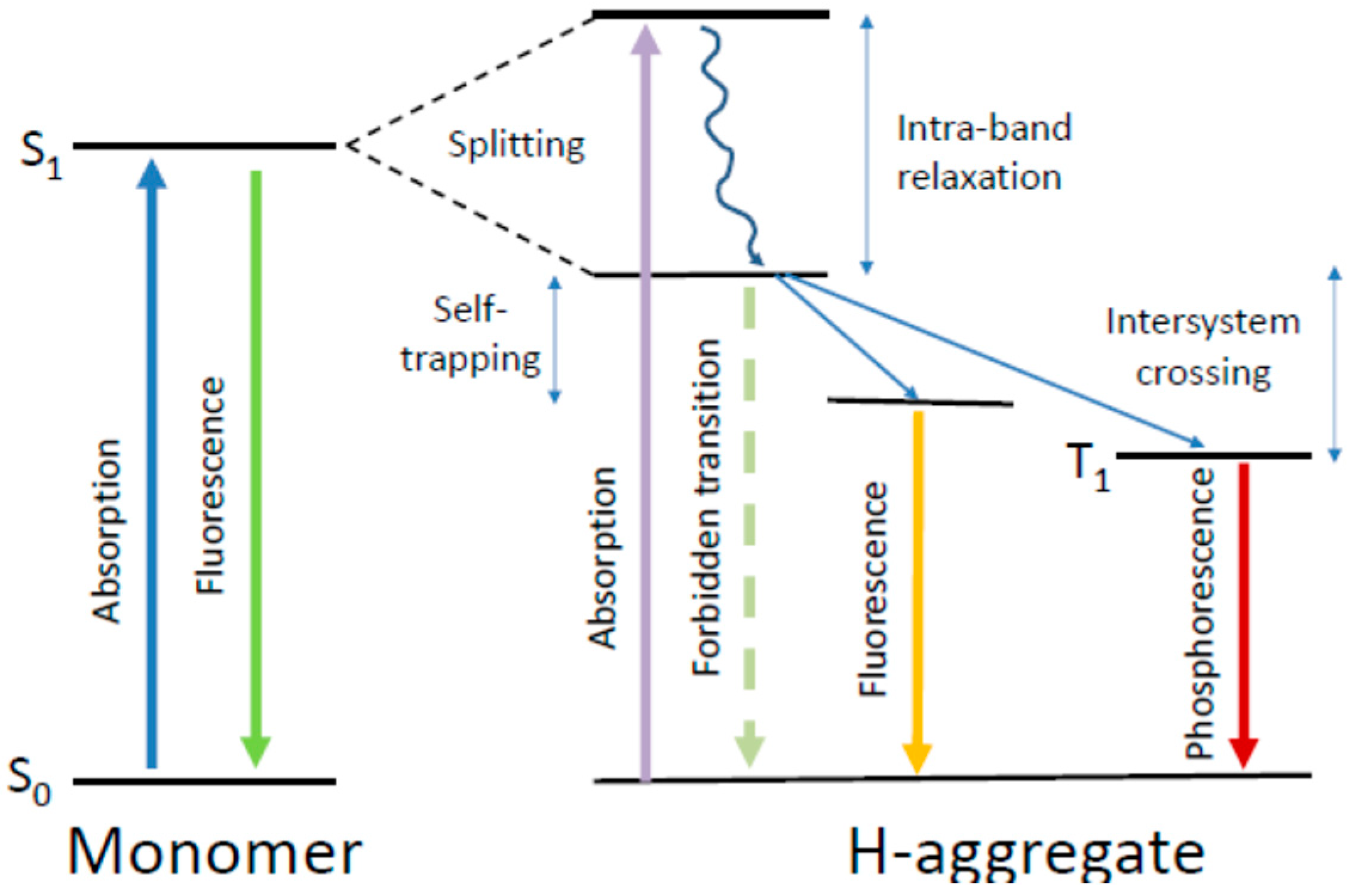
In the primary Kasha model, the point dipoles in the dimer-approximating interacting molecules are arranged so that the transition dipoles are oriented along their long axes. The electronic transition energies RE calculated as the functions of their orientation in the dimer. The results demonstrated the splitting of the excited state energy into two levels occurring due to the electronic state degeneracy. The model shows that the angle among the transition dipoles determines whether a transition is allowed to the higher or lower levels. Essentially, if the dipoles have opposing orientations and if the vector sum of their transition dipole moments is zero, then the electronic transition to the lower level is forbidden and cannot be accessed by electronic excitation. But the transition to the upper level is allowed, which provides the possibility of absorbing the light quantum at higher energies (shorter wavelengths) in comparison to that of a monomer. The emissive level is located on a much lower energy scale, and the transition from this state to the ground state with fluorescence emission is forbidden. This is the case of H-dimers.
The other limiting case is of J-dimers where the transition dipoles are in line with the molecular axis of the dimer. In this case, the transition to the higher excited-state level is forbidden but allowed at the lower level. Consequently, the maximum absorption of the dimer becomes red-shifted relative to the absorption of the monomer. Since, in J-dimers, transitions to a low energy level on excitation are only allowed, and the fluorescence emission proceeds from the same level without participation of vibrational modes, and the negligible Stokes shift is observed. This case approximates the behavior of J-aggregates, the most typical of which are the aggregates of cyanine dyes [220]. In addition to very small Stokes shifts between extremely narrow absorption and emission bands, they demonstrate very short fluorescence lifetimes. This is definitely not the case of carbon nanoparticles in view of their broad and strongly Stokes-shifted emission bands and rather long nanosecond lifetimes. Therefore, we will focus our discussion on the properties of H-aggregates only.
The forbidden character of emissive transitions in H-dimers is one of the consequences of the Kasha model [217]. Meanwhile, in different experiments on H-aggregates, these structures formed of assembled organic dyes demonstrate fluorescence emission with rather high quantum yields [221,222]. Several explanations were suggested for these discrepancies. One is based on considering the vibronic contributions to electronic transitions. It was predicted that the effect of exciton–vibrational coupling, being negligible for J-aggregates, should be strong for H-aggregates [223]. In J-aggregates, the 0–0 transitions among the lowest vibronic levels are the most active, which makes these spectral bands very narrow and the Stokes shifts very small. In contrast, in H-aggregates, even if the 0–0 transition is forbidden, the higher excited-state vibronic bands may be optically active. This may result not only in bright emission but also in large widths of both absorption and emission spectral bands. Thus, the broad short-wavelength-shifted absorption spectra and the long-wavelength-shifted emission bands are considered to be characteristic for π–π-stacked H-aggregates [224,225].
The other effect that can make the H-aggregates emissive is the absence of ideal co-planar alignment of dyes in aggregate, so that each distortion makes the electronic transition partially allowed [226]. The finite emissive transition probability can be achieved even by slight rotation of the coupled dyes in the closely π–π-stacked sandwich aggregates [222,225] so that a small level of structural disorder may lead to highly emissive H-aggregates. In further development of the Kasha model, it was considered that, at small intermolecular distances, a significant wavefunction overlap among neighboring molecular orbitals becomes essential and that the charge–transfer interactions may increase the stability of H-aggregates [227]. The H-aggregates may provide effective short-range exciton coupling due to the wavefunction overlap, and this should facilitate their charge–transfer behavior. Finally, the collective dynamics and interactions in aggregate composed of a number of stacked chromophores can be different from that predicted by the Kasha dimer model [223,228].
These new experimental facts and developments in the theory required reconsidering the popular notion that the H-dimers or H-aggregates, if produced by dye association, formeing nanoparticles or thin films, are always nonfluorescent serving only as the excitation energy traps. The quite common bright emission of H-aggregates [225,229,230] is recognized by their most easily detected characteristic feature, which is the strong short-wavelength shift of absorption spectra relative to that of monomers and the strong long-wavelength-shifted emission.
Interesting was the experimental observation that the H-dimers of cyanine dyes that are nonfluorescent in solutions can become brightly fluorescent in thin films due to the fact of their somewhat twisted configuration [224]. Imperfect packing is also expected in polymeric dye structures and indeed, in oligofluorenes, the emissive H-aggregates are formed by stacking interactions in the main chain [231]. Moreover, in single crystals of para-distyrylbenzene, the quantum yield of H-aggregate emission can reach 78% [224]. Rigid dye environment in aggregate can contribute to suppressing the non-emissive relaxation channels. It was reported that such rigidity and screening from quenching by water can be achieved by complexation of H-aggregates with calix[4]arenes [232] or cyclodextrins [127]. Fluorescent H-aggregates allow rather broad variations of intermolecular configurations [221] including formations of fiber-like structures [18]. They allow broad variability in the positions and widths of absorption spectra as a function of the number of assembled fluorophores and of their relative configurations [233]. In all these cases, the fluorescence quenching due to the energy migration to non-emissive energy traps can be minimal.
The experiments show that the fluorescence spectra of H-aggregates, in contrast to J-aggregates, are characterized by substantial widths. They can be even broader than in the spectra of constituting dye molecules. In organic dyes, the broad spectra are due to the presence of pronounced vibronic progressions forming the spectral contour that are usually hidden under an envelope of a broad electronic band. In H-aggregate also, different electronic–vibrational transitions are allowed to populate. Moreover, some transformations of spectra may occur due to the forbidden character of 0–0 transition, resulting in further apparent bathochromic shifts [226,228]. The presence of strong electron–vibrational coupling may also increase the fluorescence quantum yield of H-aggregates [234].
4.3. Coherent and Non-Coherent Exciton Transfer
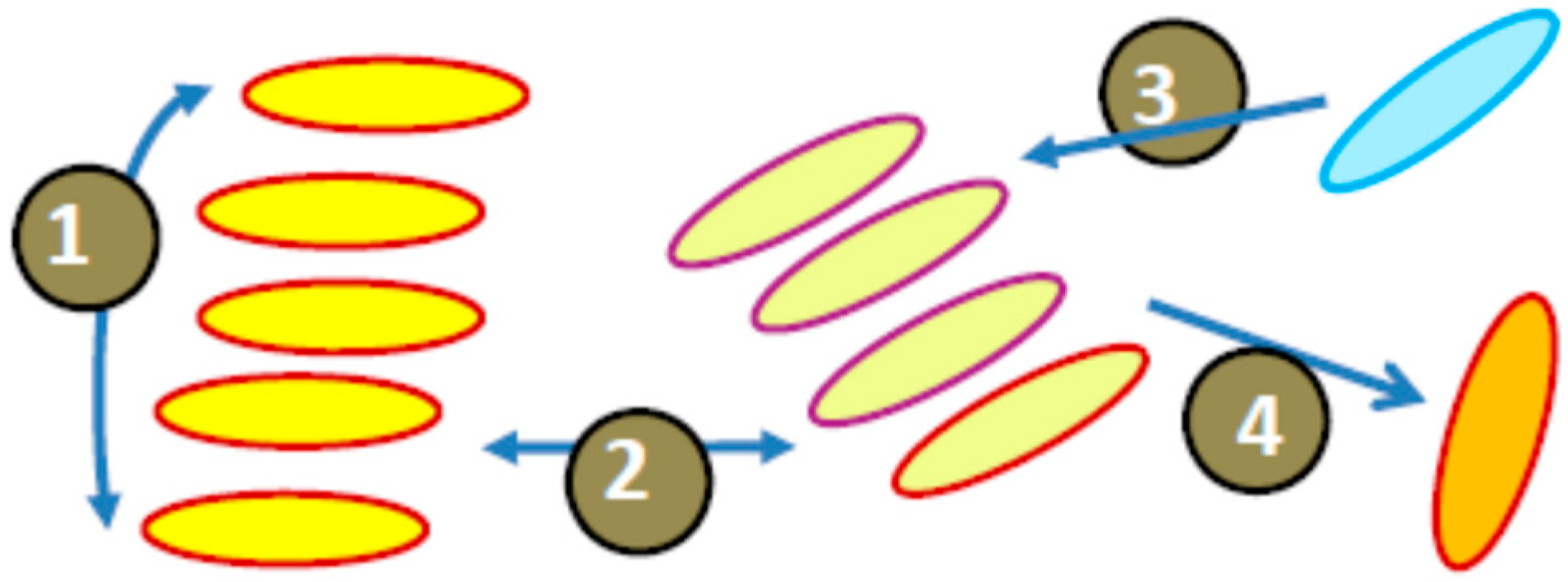
In all cases when more than one chromophore contributes to the light absorption and emission of each carbonic nanoparticle, we have to consider their interactions within short distances. The possibilities can exist for the transfer (or exchange) of excitation energy among them. The excitation energy transfer is the process where the energy absorbed at one site (the donor) is transferred to another nearby site (the acceptor). Usually, in mechanistic descriptions of these effects in multi-chromophore systems, the two limiting cases are discussed [235]. First is the incoherent hopping, in which the excitation energy jumps randomly as a diffusive energy transport among molecular-type forms. Here, the Förster theory of electronic energy transfer (FRET) [22] is applicable. In these cases, the assumption of weak donor–acceptor electronic coupling arising from the dipole–dipole interactions (Förster limit) is satisfied. In contrast, the Frenkel excitons (i.e., electron–hole pairs) spread in a wave-like manner over spatially separated molecules resulting in the states that behave as new effective light absorbers that span multiple molecules [236]. Coherent exciton delocalization is the process that creates a regime, in which the stronger electronic coupling delocalizes the excitation to produce excitonic states. In this case, the energy donors and/or acceptors are already the excitons shared among strongly interacting fluorophores (Frenkel limit). Many examples from chemical and biological worlds show that such coherence effects exist and that they are robust to the presence of disorder and noise [214,237].
In molecular aggregates, both Frenkel-type coherent and Förster-type incoherent energy transfers can be continuously connected to each other. The collective transition dipoles in molecular aggregates can be much larger than the molecular transition dipoles; therefore, the excitonic states may dramatically accelerate the energy transfer [228]. Some authors call such acceleration a “super-transfer” [238]. In a system of identical fluorophores or excitons formed by them, multiple steps of excited-state energy transfer can be realized according to the FRET mechanism. This process is often considered as exciton diffusion, in which the coherence is lost [230]. A modified version of Förster theory, accounting for the role of excitonic donors and acceptors promoting energy transfer, was developed [239]. Essentially, fluorophores (excitons) participating in this Förster-type resonance excited-state process do not lose their individuality. Before transferring the excitation, the donor energy becomes thermally relaxed, the phase relationship in dipole–dipole resonance is lost, and the energy migration dynamics lose the electronic coherence. Different types of energy transfer in molecular systems are depicted in Figure 9.
There are many examples of systems in which the coherence in energy transfer is preserved. They include some conjugated polymers [240] and pigment clusters found in systems of photosynthesis [241,242]. Excitons in these aggregates can be delocalized across many fluorophores and may show both coherent and incoherent energy transfer. When the dimension of the structure is reduced to a size comparable to the critical distance of energy transfer, its modulation can be possible by variation of nanoparticle size (but not to such an extent as in the case of loosely bound excitons).
The other important factor is the interaction of excitons with the lattice phonons (the collective vibrations of the lattice) that include the vibrations coupled to optical transitions [243]. Such interaction causes the loss of coherence in excitonic coupling. But it is favorable for FRET, since it increases the matching in energy between donor and acceptor and, improving the resonance conditions, increases the transfer probability. Therefore, due to the increased phonon population, the FRET rate rises with the increase in temperature. Such dependence is lost in cryogenic conditions.
Excitons can be electronically coupled among themselves. Such interactions can lead to two exciton bound states, called biexcitons. Experimental evidences for biexciton states were found in molecular aggregates [244]. Interestingly, no signature of biexcitons is found in the J-aggregate. The co-facial stacking of the molecular components in the H-aggregate system, resulting in strong exciton–exciton interaction, is the origin of this effect. The biexciton states are characterized by gigantic two-photon absorbance [245], which may have relation to still unclear two-photonic properties of carbonic nanoparticles (see Section 5.5).
A remarkable change in color can be observed on some C-dots aggregating in solutions (see Section 3.2) and, when comparing them in liquid and aggregated states [246], one can find its explanation. It can be suggested that the coherent exciton transfer operating within the particle is complemented by incoherent hopping on the particle aggregation.
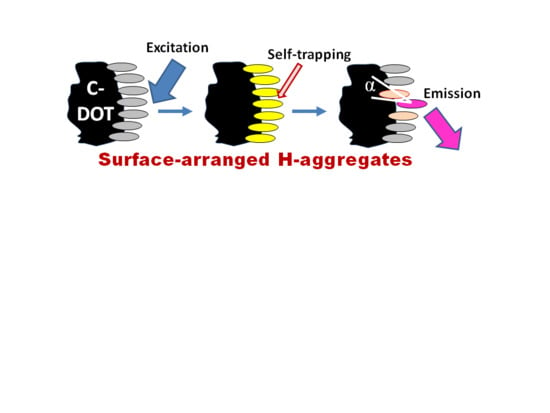
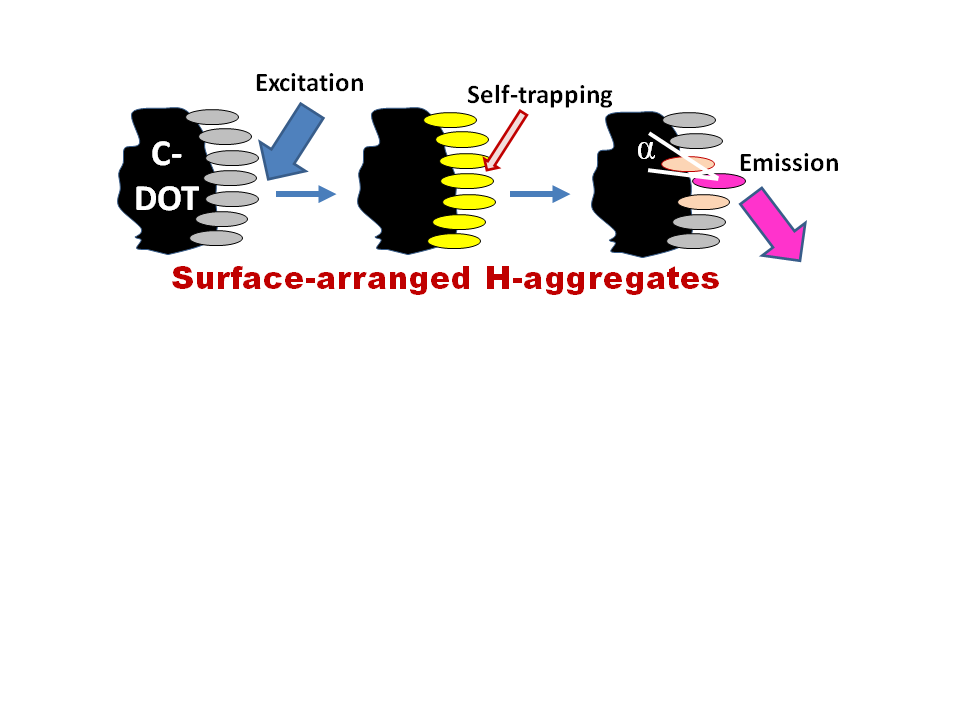
4.4. Exciton Self-Trapping
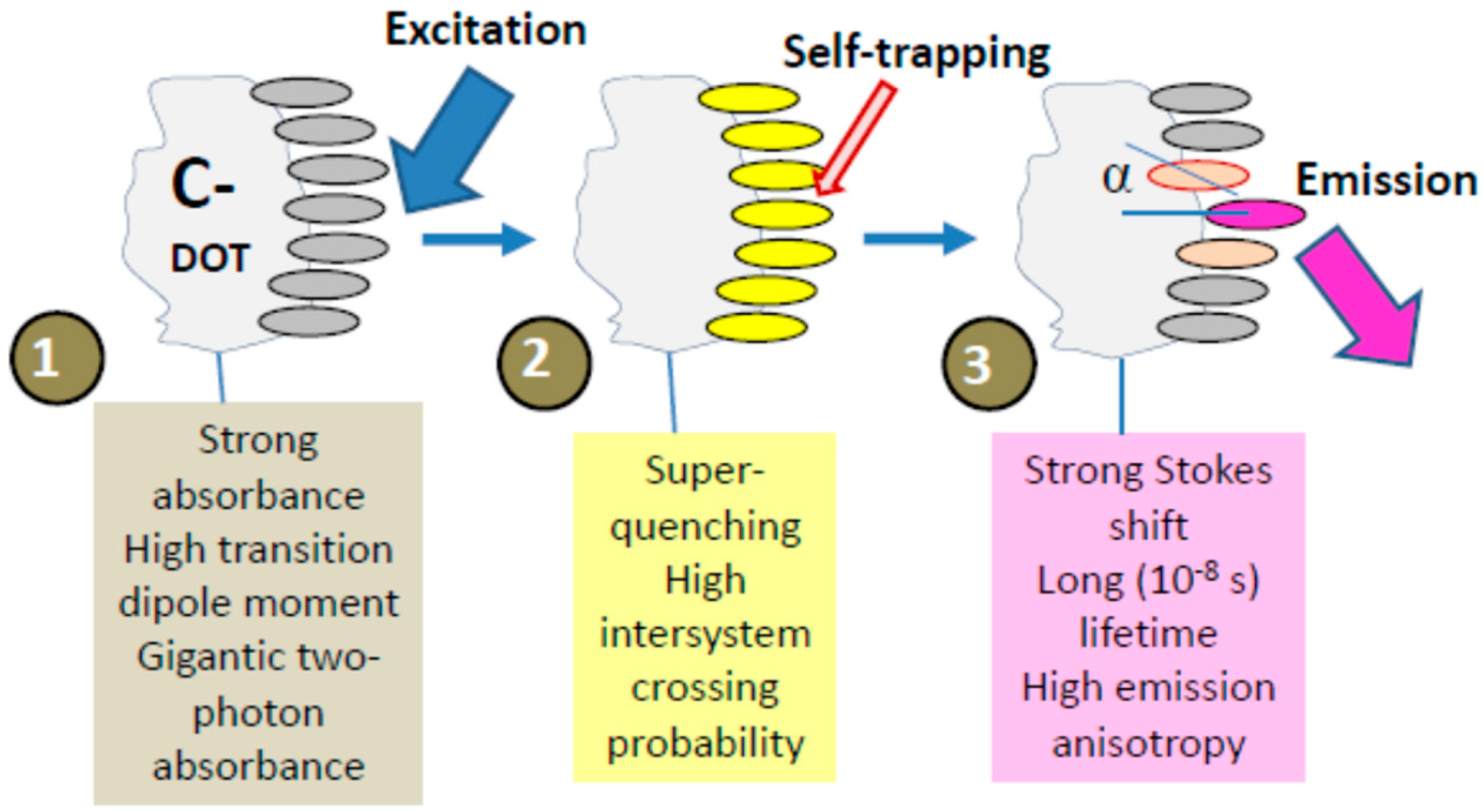
An important factor that has to be accounted for in the photophysics of molecular aggregates is the localizing influence of static and dynamic disorder in a molecular ensemble. It limits the coherence range of the exciton by establishing a balance with the delocalizing influence of resonant intermolecular coupling. Coherent exciton delocalization is not ideal also, because it can be modulated and even disrupted by molecular vibrations, particularly by the collective lattice vibrations (phonons). In organic materials, the excitons interacting with its surroundings can couple to these vibrations resulting in a significant nuclear relaxation in the participating molecules that accompanies the electronic excitations. Such dynamic localization, produced by coupling to internal and external vibrational modes, imperfections of aggregates, and additional disorder induced by the environment restricts the size of the exciton wavefunction up to its localization at a particular site [247]. The suppression of a phonon-coupled exciton motion that occurs in such a deformable lattice and which prevents its further propagation is called exciton self-trapping.
This self-trapping not only stops the exciton mobility but also decreases its energy, and, thus, a large reduction in energy by several hundred millielectronvolts can be achieved. Hence, it modifies not only the transport but also the optical properties of excitons. The magnitude of this change is connected directly with the strength of the exciton–phonon coupling [248]. The local geometrical distortion of the trapping site that needs the input of energy is a direct consequence of self-trapping. The interplay between energy loss and geometrical deformation determines the site of exciton immobilization. Since, at this site, the ground state becomes destabilized, the excitation energy decreases, resulting in the red shift of the measured emission spectrum. Because of this, in the stacked molecules of organic dyes, the exciton self-trapping may result in the appearance of huge Stokes shifts. The self-trapping strength increases if the excitons are created along molecular stacks of participating dipolar molecules that induce strong polarization of the surroundings [249]. Since the excitation energy transfer from the self-trapped excitons is suppressed, this results in high polarization anisotropy of emission.
It should be emphasized that the scenario of the exciton self-trapping is very different for the Frenkel and Wannier–Mott excitons [250,251]. It was shown rigorously that the self-trapping in one-dimensional Frenkel excitons is realized without barrier [252], while for the Wannier–Mott excitons it proceeds by overcoming the self-trapping barrier [253]. The presence or absence of a self-trapping barrier is a factor that allows for distinguishing the two mechanisms. It can sharply affect the temperature dependence of the exciton dynamics.
A first attempt to attract researchers to molecular excitons as the means to explain the properties of carbon dots was made [80]. The authors suggested that the model system should comprise three types of polycyclic aromatic hydrocarbon molecules (i.e., pyrene, perylene, and anthracene) embedded in a polymer matrix. In some sense, this model simulates the spectroscopic behavior of C-dots by explaining the Stokes shifts. These shifts are attributed to the exciton self-trapping in its hydrocarbon core. Still, the self-trapping of excitons was not proved and, since then, it remained a good idea. In other research [27], the propagation and self-trapping of the hot (thermally unrelaxed) carriers within the nanoparticle were considered within the Wannier–Mott exciton model. The observed high level of polarization of C-dots emission seemed to be in support of this idea. Meanwhile, the absence of a change in their emission intensity at the variation of the amplitude of the applied electric field [27] together with the arguments presented above calls into question the applicability of the Wannier–Mott [199] exciton model.
Thus, from the basic photophysics we derive that the exciton self-trapping owing to the strong exciton–phonon interaction must dramatically change the photophysical and spectroscopic properties. However, a consistent atomistic understanding of the underlying processes is still lacking, which impedes the application of this powerful theory to carbon nanosystems.
4.5. What Is Special in H-Aggregates of Organic Dyes?
Focusing on excitonic H-aggregates of organic dyes, several prejudices have been revised in recent years that allow for a broader view of the appearance and properties of excitonic H-aggregates. Let me count them:
(1) The H-aggregates are nonfluorescent. This is not true. Such statement comes from the Kasha dimer model (Figure 8) showing that, with an ideal collinear arrangement of interacting dipoles, the emissive electronic transition is forbidden. However, as it is discussed in Section 4.2, their bright fluorescence emission is observed in many cases, and the mechanisms that remove these restrictions are many. They include non-ideal relative location and orientation of coupled chromophores, intramolecular vibrations and their coupling, and the coupling with lattice vibrations (phonons) [227,234].
(2) Molecular disorder destroys the exciton coherence in H-aggregates. This may be true but only when this disorder is static and if it exceeds some (not well established) limiting value [223]. In the case of its existence, the excitons are delocalized only over some part of the system [254]. The decreased delocalization (coherent length) is determined by the competition between intermolecular coupling strength and structural disorder, so that, with the increase of disorder, a shift to a more localized emission mode is expected [255]. Dynamic disorder is different. It is produced by fluctuations of the monomer transition frequencies or by intermolecular couplings that are faster compared to the exciton transfer [227]. Operating on a very fast time scale, it may even be favorable for the display of H-aggregate fluorescence.
(3) The chromophores forming fluorescent H-aggregates should be identical in chemical structure. No, this is not true. The heterogenic H-aggregates can be formed, and their excitonic coupling can be efficient. The necessary condition for efficient hetero-aggregates is their resonant coupling (that includes electron–vibrational coupling) that can be also achieved among dissimilar monomers [256]. This fact was proved in the studies of different systems, including the dye assemblies in solutions coupled by their dipole–dipole interactions [257], the mixed films incorporating different dyes [258], and also the dyes assembled on a DNA scaffold [259].
Figure 10 allows for comparing of electronic state energy diagrams for isolated fluorophore molecules and for the H-aggregates of organic dyes. In all cases, the light absorption was an ultrafast process. Molecules, if they do not participate in coupled reactions, after their electronic and nuclear relaxations, emit light or decay in a non-emissive way to the ground state S0 from the same electronic energy level S1. The fluorescence emits from the same vibrationally relaxed excited molecule. In contrast, in the π-conjugated H-aggregates, as in dimers (Figure 8), due to the fact of their electronic conjugation, the light absorption drives the system to the higher level of the split excited energy state S1. The energy is distributed over a number of molecules, and the relaxations that precede the emission are more dramatic, resulting in a strong decrease in excitation energy. Here, the propagating exciton is coupled with the phonon modes. The motions of the nuclei coupled with phonons result in the exciton self-trapping occurring on a very fast (sub-picosecond) timescale with a change of nuclear geometry [233]. Thus, both electronic and nuclear sub-systems are reorganized in time on an ultra-fast scale, generating the self-trapped excitons that become the emissive species. The fate of such exciton is diverse. It can emit light, propagate in a diffusive manner (by FRET), be quenched or exhibit the intersystem crossing into the triplet state, T1, with the emission of phosphorescence. All that can further lower the energy of the excited state.
In this way, the excitons demonstrate very interesting properties. Their absorption is dominated by completely delocalized excitations, while fluorescence comes from the recombination of self-trapped excitons at the localized sites [260]. The exciton self-trapping, the effect of torsional disorder, and the localization of excitations in aggregates are all consequences of the strong nonlinear coupling between electronic and vibrational degrees of freedom leading to the concentration of excitation energy in these localized states.
Summarizing this section, I presented the two versions of exciton theory that are applied in order to explain the optical properties of carbonic nanostructures. The concept of Wannier–Mott excitons is borrowed from the theory and experiments in semiconductor physics. Regarding Frenkel excitons, their origin comes from the crystals of organic dyes. The latter are the small-radius excitons that are characterized by propagating electronic excitation but without the free electron motions in the conduction zone, since they are bound to correspondent holes residing in nuclear structures. They are easily recognized in spontaneously formed dye aggregates of the J and H type. Due to electronic band splitting, the H-aggregates display strong Stokes shifts and, because of involvement of electron–vibrational and electron–phonon interactions, their electronic spectra are broad. The energetic advantage of forming the H-aggregates in multi-chromophore assemblies is their arrangement in sandwich-type stacks stabilized by attractive dipole–dipole interactions in which the transition dipoles can be parallel and inversely oriented to each other. They can collectively act as the donors or acceptors for other groups of molecules to which they are weakly coupled. They can form localized (self-trapped) states that can produce highly polarized emission.
5. The Novel Excitonic Concept
Whereas the “molecular view” is dominant in the discussions of fluorescence mechanisms of carbon nanoparticles [18,93,144], suggesting molecular emitters located in the particle core [19], being extended to the surface [20] or forming the whole particle [94], there are spectroscopic properties that do not fit. In addition to the discussed above (Section 3.3) systematically observed very strong Stokes shifts, carbonic nanoparticles may possess the fluorescence lifetimes, τF, that are unexpectedly much longer than that of typical organic dyes (e.g., fluorescein and rhodamines [261]). Their reported τF values can be found on the level of 10–11 ns [262,263] and, in some reported cases, up to 15 ns [264], being at similarly high levels in solutions and solid state [265]. In addition, their appreciable room-temperature phosphorescence was found, even in those carbon dots which were made of only light atoms and could basically display only weak spin-orbit coupling [266,267,268]. These nanoparticles demonstrate very high two-photon absorption (TPA) cross-sections (on the level of 40,000 GM) combined with high luminescence quantum yields [24,269], whereas similar properties of molecular fluorophores can be achieved only with a very special design and still at a low levels of emission intensities. This unique behavior of nanocarbon particles is not always observed and reported, but there are many relevant documented cases in support. They need to be explained.
Our focused interest on molecular excitons was inspired by the success in understanding photosynthetic light harvesting systems [235,270,271]. Excitonic states of natural pigments in photosynthesis increase substantially the absorption cross-section for incident light without the cost of dissipative processes that might result from diffusive trapping of excitation energy absorbed by a weakly coupled aggregate of molecules. The described mechanisms underlying the ultrafast dynamics of excitons, electronic couplings among the excitonic states, rapid trapping of exciton in the lowest state being captured by the exciton manifold, and other interesting events may be relevant to the systems that we overview here. Our primary aim was to establish this relevance.
5.1. Carbonic Nanoparticles as Collective Emitters
Exciton in molecular aggregates is considered as a collective quantum emitter in a sense that constituting chromophores lose their individuality and respond to electronic excitation as one unit. The observation of a “superquenching” effect is a simple test for that. When any quencher regardless of the mechanism of quenching interacts with any unit of the excitonic system, not only this unit but the entire system becomes quenched [272,273]. This effect explains the very efficient quenching ability of all types of carbon nanostructures and is explored in different chemical sensor applications. Thus, the detection limit for sensing mercury cation [274,275] as well as of many other metal ions [276,277] reached several nanomoles.
Both electron donors and acceptors are able to efficiently quench the fluorescence emission of carbon nanoparticles [120] which suggests the participation of these quenchers in the scavenging of the surface-confined holes and electrons. The photo-generated electrons can be used for the reduction of different compounds including CO2 conversion and obtaining H2 from water [278]. The disruption of the electron–hole radiative recombination resulting in quenching occurs locally, but the whole nanoparticle may provide the mechanistic framework in harvesting of the photo-generated electrons for productive purposes [279]. The observed superquenching effect is likely a result of an amplified energy transfer occurring in the C-dot due to the exciton propagation, analogous to another system exhibiting Frenkel excitons, the conjugated polymer nanoparticles [280,281] and J-aggregates of cyanine dyes [282,283].
The methods of single-molecular spectroscopy allow for obtaining information about the photophysical behavior of individual nanoparticles. Thus, the superposition and averaging of the characteristics of many particles leading to static disorder can be avoided. Modern techniques allow for obtaining the fluorescence spectra and decay rates of a selected nanoparticle and tracing its emission in time until it is bleached. A temporal development among different states can be followed within a broad time range, and not only can the short-range populated intermediate states be identified [284] but the long-range behavior as well. The particles can lose their fluorescence reversibly (blink) and irreversibly (bleach); thus, a focused search could respond to questions if they do that as one unit or in multiple steps demonstrating the presence of a number of fluorescent sites within a single nanoparticle. This allows for distinguishing the fluorescence response between the assembly of molecular-type emitters and collective emitters, the excitons. The emission of several exciton traps in parallel is not excluded [285].
Moderate blinking of C-dots have been reported [286], and it was also observed that they can be photobleached when studied in solutions [287]. The experimental data obtained with single C-dots deposited on cover glass plates [31] demonstrate the characteristic single-step transient blinking and photobleaching (Figure 11). They suggest the presence of single excited-state levels in each particle that can be reversibly or irreversibly inactivated in one step.
Figure 11 represents data from different authors. It was reported that oxidized C-dots display the single-step on–off behavior [286]. Regarding the reduced C-dots, the signatures of multiple steps were also observed, which may be the result of formation of additional defect-related emissive traps but also of particle aggregation. The two-state blinking was reported by one more group of researchers, and the ability of its tuning by an electron acceptor serving as molecular quencher was demonstrated [288]. As mentioned above, such a superquenching effect is characteristic of excitonic states [289]. A correlation was found between the brightness and bleaching rates [290]. Due to the fact of these properties, the C-dots were successfully applied in different versions of super-resolution microscopy (nanoscopy) [31,291,292].
Interesting is the blinking statistics derived from experiments on blue-green [31] and yellow-red emitting [293] single C-dots with amorphous core structures studied on a solid support. In both cases, the statistic of the off-state durations extending from 0.05 s to tens of seconds could be well fitted with a power law function. Such temporal behavior was observed both at room and at cryogenic temperatures [293]. This regularity was also observed for semiconductor nanocrystals, where the fluorescence intermittency originates from the statistic of trapped charges within a particle or on its surface [294]. It seems to be universal for different nanoscale emitters [295]. In contrast, for dye molecules, the blinking statistic is typically exponential. Thus, it may be suggested that the stochastic dynamics of intermittency is governed by photo-induced energy fluctuations generating the exciton traps.
Thus, we come to an important conclusion. The carbon dots represent a class of integral quantum emitters, the observed intermittency of which demonstrates the stochastic two-state (on–off) switching. Such all-or-none bleaching and blinking behavior [31] provides strong support for the idea of considering the C-dots as the excitonic H-type aggregate emitters [16]. In all aspects, they behave as collective emitters with its particular energy gaps, so that their exciton length may extend to the whole nanoparticle.
5.2. Optical Anisotropy and Macrodipoles
One of the mysteries of carbonic nanomaterials is the fact that they generate highly polarized fluorescence emission. When studied in ensemble-averaged conditions, in the solid state, the emission anisotropy reaches its limiting value [27] and, in viscous solutions, it decays on the timescale of the emission (ns) starting from a high initial level [193]. It demonstrates rather homogeneous spectral behavior, being almost independent of excitation and emission wavelengths [27,68]. The loss of anisotropy in time, when studied in solutions, can be typically explained by rotational diffusion of nanoparticles [296]. Indeed, anisotropy was observed to be strongly viscosity dependent. However, with the account of the size of particles and solvent viscosity, their rotational correlation time should be much longer, of the order of tens-hundreds nanoseconds, so that on a time scale of fluorescence decays, the whole-particle rotational mobility should not provide an important effect [68]. Therefore, some other depolarizing factors must be involved.
Putting together the results of numerous observations, we note a paradox. On one side, some strong spectroscopic relaxations are manifested leading to dramatic Stokes shifts. On the other side, they still occur with the retention of the initial orientation of the excitation and emission dipoles that must be witnessed for the rather simple dynamics of the relaxation process.
Demonstrating the presence of anisotropic absorption and stimulated emission in different solvents, a much faster decay of anisotropy was found in water [168]. The authors connected this observation with the excited-state proton transfer reaction, in which the exposed polar groups of C-dots serve as proton acceptors. Indeed, such reactions can proceed in picoseconds, and they may result in polarization loss and substantial Stokes shifts [173,297]. However, this proposed mechanism cannot be universal, since it requires the involvement of specific groups on the particle surface.
The ideas based on the exciton concept have started to be applied to resolve these issues. The large values of anisotropy across the spectra were attributed to localization of excitons. Considering the Wannier–Mott loosely bound exciton model (see its critics above), it was suggested that the observation of linearly polarized florescence can be due to the recombination of hot carriers [27]. According to this interpretation, the excitonic states being excited to the conduction band generate hot carriers where the momentum alignment and anisotropy excited by the linear polarized light are conserved during the emission. In this case, the carriers should not be thermalized; otherwise their emission should lose polarization. However, the arguments presented for that are not convincing and conflict with a commonly accepted view that, in molecular systems, the vibrational relaxation is one of the fastest events [298]. In semiconductor QDs, such cooling rates, which are due to the coupling with lattice vibrations, are observed in early picoseconds, which is faster than the emission rates by approximately three orders of magnitude. Meanwhile, for special cases of QDs [299] and halide, perovskites [300] were described for which the electron carrier cooling was greatly retarded. In these cases, a slower carrier cooling was achieved when the spacing among their discrete electronic levels was much larger than the phonon energy. Slowing the electron cooling was also described for nano-sized graphene sheets [300], but it may not be realized in C-dots lacking the well-ordered graphene core.
Important is another aspect that can be derived from the measurements of optical anisotropy. Since the whole particle is seen as a collective quantum emitter, this emitter can be excited directly possessing a fixed linear excitation transition dipole, and this dipole should not change its original orientation in the excited state. If so, then the well-documented effects of solvation [167] should be attributed to the dynamic interaction with the solvent of this dipole. Experiments provided on a single particle level have shed more light on this issue [31,293].
The single-molecular studies suggest a straightforward way for measuring the orientation and dimensionality of the excitation transition dipole moment (TDM) by scanning an emitter through the focal region of an azimuthally polarized laser beam [212]. Figure 12a–e shows the excitation patterns of individual fluorescent C-dots, for which the TDM is oriented along the plane of support [31]. The image of every nanoparticle is represented by two nearby bright spots of elliptical shape, which correspond to a fixed linear excitation TDM. Fixed orientation of the pattern indicates that the TDM did not undergo reorientation during the acquisition time. The orientation and dimensionality of the emission TDM can be determined using defocused imaging [301]. Observed images, as seen in Figure 12f–j, can be very well fitted with a model that assumes that the emission originates from a single fixed dipole [31]. Comparison of the excitation patterns and defocused images of the same individual C-dots showed that both excitation and emission transition dipoles of the particles were oriented parallel to each other within the error of measurement. This finding implies that each nanoparticle has only one optically active fluorescent site which is directly excited by the laser light without any energy transfer from other optically active groups [31,168]. No change in orientation of the dipole moment was observed on blinking which demonstrates that the structural anisotropy is the inherent property of these nanoparticles and it is responsible for the existence and orientation of dipoles. Therefore, these nanoparticles cannot be considered zero-dimensional “dots” [16].
As a possibility to describe the optical anisotropy, one may also take into account the involvement of charge–transfer (CT) excitons [247,302]. They were introduced as an intermediate case between Frenkel and Wannier–Mott excitons and, as a small electron–hole separation may exist, they may be considered as a version of small-radius excitons. This theoretical concept was applied in ionic crystals to analyze the cases when the electron and the hole occupy adjacent molecules and are coupled by electrostatic force. Their interesting features are the formation of a large static dipole moment and high efficiency in the generation of self-trapped states. They can also couple with Frenkel excitons to attain new features [303]. When the energy difference between the lowest Frenkel exciton and the nearest-neighbor charge–transfer excitons becomes small, then their strong mixing determines the nature of the lowest energy states. The high polarization of electronic spectra is characteristic for these states. Regarding carbon nanostructures, this issue was not elaborated.
5.3. Strong Two-Photon Absorbance
The requests for very bright fluorophores for two-photon biological imaging have brought the attention of researchers towards carbonic nanostructures [304]. Compared with traditional one-photon fluorescence microscopy, two-photon imaging has the advantage of generating high-energy visible fluorescence from low-energy excitation in the near-IR region. The nonlinear effect of the dependence of light absorption on illuminating light intensity allows for forming very sharp images in the microscope focal plane. In this respect, the properties of carbonic structures were found to be outstanding. Already, in the initial steps of studying the C-dots, their two-photon cross-section σ was estimated to be of the order of 40,000 GM (Goeppert–Mayer units, 1 GM = 1 × 10−50 cm4 s/photon) [305]. Similar or even higher σ values were reported later [24,306] and the high two-photon absorption efficiency of G-dots and GO-dots was also demonstrated. These values have made the carbonic nanoparticles competitive to the best-performing semiconductor QDs [269]. They showed to be considerably better than the best performing molecular organic dyes [307]. Thus, regarding the dyes commonly used in microscopy [308], this difference is by about three orders of magnitude (for fluorescein and rhodamines σ ~30–50 GM).
Such superior performance cannot be explained just by assembling individually emitting molecular fluorophores (two quanta arriving simultaneously excite single fluorophore according to its σ value). On the molecular level, the general trends for improving the two-photonic absorbance are known. They are focused on providing the planarity and high π–electronic conjugation within the molecule and on inducing its dipolar, quadrupolar, and higher-order asymmetry. How are these rules realized in the best performing G-dots and C-dots? This issue is obscure, and the only clearly seen fact is the strong modulation of the two-photon absorbance by doping with nitrogen [24] or boron [98] atoms.
The solution to this issue can be found if we draw an analogy with the more deeply studied associates of organic dyes. We can find that their theoretically [309] and experimentally [310] analyzed cooperative enhancement of two photon absorption is due to the generation of excitons. In this regard, also, their performance is dramatically different from the behavior of organic dyes assembled in detergent micelles [311] or polymer conjugates [312], in which high performance is achieved due to only to the local concentration effects. Within the frame of exciton theory, the intensive nonlinear optical response could be explained by the occurrence of the large oscillator strength generated by one or few delocalized excitonic states. Such states can be generated already in dimers. Thus, the excitonic effects arising in butadiene-linked porphyrin dimers show a large σ value, ~104 GM, which is 500 fold higher than that for the parent monomer [313]. A gigantic increase of σ was also reported for dendrimers formed of pyrene core and fluorine dendrons [314]. A connection was found between the two-photon absorbance and the extent of π-conjugation [315]. Despite the absence of full understanding [307], the cooperative effects that appear in molecular aggregates are well documented and have started to be explored for dramatic enhancement of the two-photon absorption efficiency [310]. Thus, the analogy is straightforward, but its detailed analysis is lacking.
5.4. Molecular Disorder and Relaxations
Now we refer to the mechanisms of the broadening of the fluorescence spectra in carbonic nanostructures. Their fluorescence spectra usually have a full width at half-maximum (FWHM) of 80–100 nm and more, which is of similar value and sometimes larger than that of common organic dyes. If compared to semiconductor quantum dots with their typical widths of 25–30 nm, such difference is dramatic. This fact is an additional witness for the fundamental differences in excitonic properties among these systems.
There is a common way to distinguish the homogeneous and inhomogeneous broadening components in electronic spectra. The homogeneous line width is determined by electron–vibrational and electron–phonon interactions that result in the fluctuations of transition frequency [316]. These fluctuations are very fast processes; therefore, by variation of excitation energy (wavelength) within the absorption band, no photoselection among different sub-sites can be achieved on a timescale of fluorescence emission. In H-aggregates, the transitions from the ground state to different excited-state electron-vibrational levels plus the interactions with lattice phonons form the homogeneous contour of the spectrum.
An indication of strong coupling between electronic transitions and lattice phonons was obtained in single-particle studies [31,168]. It was noted that all the individual fluorescence spectra of C-dots contained tails at the side of lower energies. Deconvolution of spectra into two Lorentzian components allowed assigning the lower energy satellite peak to the excitation of optical phonons coupled to the radiative recombination of the charge carriers. According to these data, the energy band separation that corresponds to a coupling between electronic transitions and phonons lies in the range of 100–140 mEv. In view that the interactions between electrons and lattice vibrations result in a bound state with a decreased energy, as compared to a system without such interaction (stepwise localization of electron and hole [243]), the phonon energies could be estimated. The processes of excitonic relaxation from upper to lowest excitonic states inside the excitonic zone generating the Stokes shift should be almost independent of the temperature. Such temperature-independent fluctuations within a manifold of these levels result in broad and structureless spectral bands.
Inhomogeneous broadening is different. It is caused by static disorder in the population of fluorophores due to the fact of their distribution on interaction energies with the environment [181]. It can be both static and dynamic, and the transition from dynamic to static mode can be seen in cryogenic studies as a dramatic reduction in the line width and Stokes shifts, the appearance of zero-phonon lines, etc. [317]. Resolving these cases by applying cryogenic experiments could contribute to the progress in understanding the photophysics of carbonic nanostructures. If the C-dots are the ensembles of emitting molecules and the molecular motions and the electronic relaxations coupled with them exist in ambient conditions, these motions should essentially be frozen in cryogenic experiments.
Two low-temperature regions were distinguished in these studies [30]. With a temperature decrease to 80 K, the fluorescence band steadily increased in intensity together with a correspondent increase in lifetime, but underwent a rather small shift or narrowing [26,30]. In other studies provided in this range, a stronger shift to shorter wavelengths was observed together with the band narrowing and increased variation of its position as a function of excitation wavelength [318]. The suppression of thermally activated processes may be observed in this range; they may include the relaxations in interactions with a dipolar solvent that freeze on cooling. According to our results (Figure 13), the decrease of fluorescence quenching on cooling in the entire measured temperature range, from 300 K to 10 K, was not associated with the change in the rate of fluorescence emission. This result is very unusual, since in molecular fluorophores in condensed media, thermal quenching is always dynamic and results in the decrease of lifetime [319]. In contrast, in excitonic systems, this can happen due to the generation of exciton traps that must be temperature dependent. In this case, it can be observed as static quenching.
The other region from 80 K down to 10 K demonstrates a quite different behavior. Here, no changes in spectra are observed. The emission intensities and the lifetimes remained temperature independent. Thus, no additional signatures of suppressed molecular relaxations were detected in the experiment [30]. Essentially, as it is seen in Figure 13, the emission band remained very broad and strongly Stokes-shifted from the excitation band. This behavior does not depend on the excitation wavelength which suggests the absence of inhomogeneous broadening contributions to the spectra [181]. One may notice that, on deep cooling, the spectra remaining with temperature-independent intensity are even more highly shifted to longer wavelengths compared to the room-temperature spectra. This is a result of the appearance of phosphorescence contribution that is quenched at higher temperatures (see below).
Such a remarkable difference in the C-dots’ behavior in two temperature regions fits to the Frenkel exciton concept explaining the presence of a strong Stokes shift and the absence of its variation in deep cryogenic conditions. In this case, in contrast to Wannier–Mott excitons, the relaxation from the upper to the lowest excitonic states inside the excitonic zone occurs without activation energy and, therefore, must be (almost) temperature independent [250,252]. Based on these data, it was suggested that the most general feature of carbon nanoparticles, the very large Stokes shifts and the considerable differences between the absorption and excitation spectra, are the result of formation of the dynamic defect which is the self-trapped Frenkel exciton. The exciton–lattice interaction and the local overheating that is caused by the exciton relaxation at the trapping site generate the dynamic formation of the distorted domain in the H-aggregate.
5.5. Triplet State Generation
Usually, carbonic nanoparticles are formed of only light atoms which results in a small dielectric constant and weak spin-orbit coupling. Therefore, their ability to provide the singlet-triplet conversion and generate the phosphorescence emission may be expected as weak. In the world of organic molecules, only the dyes that include heavy atoms (such as tetra-bromo derivative eosin and tetra-iodo derivative erythrosine) are brightly phosphorescent in aqueous solutions. Such emission can be easily distinguished from fluorescence by strong wavelength-shifted emission bands and the longer lifetimes by many orders of magnitude. It is known also that the surrounding rigidity of the dye favors the phosphorescence and, thus, increases the range of phosphorescent dyes [320]. Aggregation of dye molecules [218], their insertion into solid matrices [321], and removing oxygen from the medium [322] or deep freezing of solutions [323] favor the appearance of phosphorescent emission. In carbon nanostructures, heavy atoms can be present as the components of starting materials in synthesis, as the result of their after-treatment or as impurities. The conditions of particle rigidity and protection from diffusion of oxygen can also be realized. Accordingly, the phosphorescence of C-dots was detected when they were incorporated into solids, such as polymer films [324]. In H-aggregated molecular assemblies of organic dyes, a bright room-temperature phosphorescence was achieved in solid powders [325] and even in water solutions [326] due to the fact of their special design [327]. Meanwhile, the basic mechanism of their triplet exciton generation [321,328] remains unclear as well as in nanocarbon systems.
In our cryogenic studies of C-dots, an unexpected broadening of the emission band extending to longer wavelengths was observed (see Figure 13a), suggesting the contribution of a new luminescence band strongly shifted to longer wavelengths. For a more detailed analysis, the emission spectrum at 10 K was deconvolved into two Gaussian functions. The long-wavelength component was characterized by very long lifetime (0.64 s) and, therefore, it was attributed to phosphorescence [30]. Its intensity and lifetime were independent of temperature in the 10–80 K region, but, above 80 K, these parameters started to demonstrate a strong dependence with the characteristic activation energy derived from Arrhenius plots of approximately 25 meV. This suggests that the quenching of phosphorescence is due to the fact of a single activational dynamic process.
Such behavior of ultra-long phosphorescence emission may be connected with the generation of triplet excitons. It was found recently that these excitons can be generated through intersystem crossing in associates of simple organic molecules [329] and their crystals [330], even if they do not contain heavy atoms. Since the planar organic molecules can be coupled in H-aggregates, this provides an effective means of stabilizing and protecting the triplet excitons. Their generation is favored in H-aggregates formed by the charge–transfer dyes [39]. It was observed that the duration of phosphorescence in H-aggregates may be much longer than that of constituting molecules, by several orders of magnitude, reaching the level of second [327,329].
6. The Basic Model of Excitonic States
As we have seen from the discussion in previous sections, the ability of forming the Frenkel-type excitonic states must be determined by molecular structures that allow realizing the electronic nature of the π-conjugated systems. The electronic coupling within these systems determines the pathways of formation and the thermodynamic stability of the H-aggregates and of their photophysical properties. Meanwhile, it is very difficult to employ the theoretical background elaborated on the models of molecular crystals, conjugated polymers or dye dimers and aggregates to carbonic nanostructures. The greatest problem is in versatility of these structures. It is hard to single-out the “basic” structure, for which the fundamental mechanism generating the emissive state(s) should be clearly seen, and to consider other possible mechanisms as “modulating”. Still, I will try to do that based on experimental data obtained for the most typical carbonic nanostructures and the knowledge of exciton behavior in molecular aggregates.
6.1. The Role of Particle Core
In elucidating the mechanisms of emission of carbon nanoparticles it is quite natural to study separately the photophysical transformations leading to emission in the nonpolar particle core and polar particle shell. The role of the carbon core is one of the key issues discussed in the current literature. In G-dots, the sheets forming the core are the necessary element of the structure [162]. In other carbonic nanostructures, these core elements may be present or not, and, if present, they can be seen in crystalline or amorphous forms (see, e.g., Figure 2). Therefore, it can be expected that the primary role of the core is in stabilizing the structure by constituting the planar sheets interacting among themselves by van der Waals interactions. The magnitude of these weak interactions among the planes should provide a stabilizing effect. These sheets, being analogous to nanographenes [331], may extend to the particle surface and terminate by the charge–transfer fluorophores that combine nonpolar and polar fragments. In this way, the planar layered core structure may guide the planar arrangement of the latter.
In cases when the core structures are represented by clusters of aromatic π-electronic groups of atoms, they can perform their own optical activity [80]. However, their ability to be organized into the excitonic H-aggregates is expected to be low. Probably, dependent on their design they can serve as the independent emitters or as the FRET donors [146]. But their more general contribution to generated emission is expected to be in providing the characteristic pattern of exciton–phonon interactions to emitting excitonic structures [31,293] as discussed in Section 2.2. In this role, the core, whatever it is, can mediate a complex interplay between electronic and vibrational processes and participate in the self-trapping of excitons and also in enhancing their emission.
6.2. The Role of Particle Shell
The key role in generating and modulating the optical effects should be attributed to the particle shell that is the target of many modifications and solvent-induced perturbations dramatically changing the spectroscopic behavior [17,103,148,151]. In the course of synthesis and structural modifications, the shell can be formed of structurally asymmetric amphiphilic charge–transfer fluorophores [20,332]. Recently, the arguments have been presented for attributing this role to the coupled π–electron system with carbonyl groups [151]. For being at the interface between the nonpolar core and polar solvent, their most energetically favorable configuration can be achieved if they are planar and extend the polar groups to the surface with or without the structural guidance by the particle core. These surface-exposed fluorophores should be arranged in stacks, which is typical for the self-assembly of organic dyes [85,86].
The dense stacking arrangement of fluorophores may be stabilized by stronger dipole–dipole interactions [85,333] due to the charge transfer properties of the partners [334] and, particularly, by the electrostatic complementarities between the electron-rich and electron-deficient regions of the stacked aromatic groups. Increased stabilization can be provided by electrostatic interactions between complementary charges of the neighbors [335]. This is the most energetically favorable configuration for the formation of the H-type excitons.
Here, the analogy with the so-called quasi-one-dimensional (1-D) molecular crystals can be seen. They are formed by planar aromatic molecules arranging in 1-D face-to-face stacks with the appearance of their overlapping electronic π-orbitals [336]. In the case of “top-down” synthetic procedure, such preferential arrangement of surface-exposed fluorophores must be realized by after-treatment of an already formed core that can operate as a template for surface modification by polar groups [337]. Regarding the “bottom-up” (e.g., solvothermal) synthesis, the stacks of aromatic heterocyclic fluorophores may appear on the first steps of synthesis [144,264], ahead of carbonization and core formation. This core–shell (or shell-only) configuration of fluorophores stabilized by complementary positive and negative charges [85] are the sites of binding-release of protons and ions of different signs. The presence of both electron donating and electron accepting groups of various strength may explain the remarkable electrochemical properties of carbon nanoparticles [338] and their high efficiency in different redox reactions that involve both donation and acceptance of electrons [120,339,340].
6.3. The Self-Trapped Excitons and Their Emission
The essential feature of excitons is not only their ability to absorb light collectively as one unit formed by a π-conjugated network but also to emit light locally at the electron–hole recombination site. Our model, schematically presented in Figure 14, depicts the most general sequence of events occurring with light absorption and emission in H-aggregate forming carbonic nanostructures.
These events could be the following. Light absorption by an H-aggregate located within the particle shell possesses all the features of collective excitation that propagates over the whole excitonic unit and demonstrates its characteristic features, such as the strong one-photon absorbance and gigantic absorbance of multiple photons, short-wavelength position of the absorption band, and high transition dipole moment. The propagating exciton has many possibilities to decrease its high energy. If it is quenched, it can exhibit the superquenching effect as well as blinking and bleaching (see Section 5.1). It can also demonstrate the intersystem crossing that results in conversion to triplets with the appearance of phosphorescence emission (see Section 5.5). However, the fastest and the most efficient will be the intra-band relaxation provided by the exciton–phonon interaction as discussed in Section 4.4. Such relaxation proceeding without an activation barrier allows for achieving a high efficiency of energy conversion and its release in the form of emission. It allows for avoiding the dissipative processes that might result from diffusive trapping of excitation energy that could be unavoidable in weakly coupled aggregated fluorophores.
The ultrafast attainment of the self-trapping exciton localization site occurs with the aid of collective vibrations (phonons) creating a dynamic defect. As a result, the exciton appears in the state of the lowest excited-state energy, and some part of this energy is released in the form of ultrafast local heating. This brings a local distortion of structure that breaks the H-aggregate stacking configuration with the neighbors and results in a change of angle α between their dipoles (see Figure 14). As it was suggested [30], the distorted local fluorophore arrangement attains the properties of J-aggregate. (Such H- to J-transformation is known in molecular aggregates [341].) This opens an allowed transition for the emissive decay from this localized site, and, therefore, the fluorescence can be highly polarized and with a high quantum yield. Since the emission of J-aggregates is always shifted to longer wavelengths, this explains the strong Stokes shift.
Thus, this model suggests mechanistic realization of proposed earlier surface energy traps, the existence of which was noted in the pioneering work on fluorescent C-dots [1] and postulated in many subsequent studies [17]. The essential features of this mechanism are elaborated and illustrated here. They include the “antenna effect” in light absorption, the excitation energy transformation involving conformational variables coupled with collective vibrations, and the emission from the trap site. We present the view that, mechanistically, the exciton transport in carbonic nanostructures is similar to that in molecular aggregates of organic dyes and features strong resemblance to that in photosynthetic antenna systems [236,271]. A similar level of coherent effects is expected to play an important role in carbonic nanostructures.
7. Concluding Thoughts and Prospects
In conclusion, we have to recognize that, despite tremendous efforts and an innumerable number of publications, the research community still has not achieved a clear understanding of the origin of fluorescence emission of carbonic nanoparticles and the means to optimize their desired properties. The broad structural variability, unavoidable in conditions of synthesis from multiple predecessors and along multiple commonly hidden routes, creates a diversity of interpretations. Such a situation obviously generates statements, such as “the unique interplay between electronic states of different nature” or “the different surface state and groups govern the emission”, which explain nothing. These difficulties are expected, since we are addressing materials that are always complex, possessing heterogeneous compositions and high structural disorder, and variable in size or under the influence of surface effects. Meanwhile, as it was often noted, these nanoscale emitters demonstrate surprising general regularities. The similarities of spectroscopic behavior between G-dots, GO-dots, and C-dots [11,12,69] allow us to believe that they have a common origin. We observe that passivation and doping by different heteroatoms change dramatically the spectroscopic properties of different carbon nanostructures shifting their emission strongly to the red [133,134]. It becomes the actual task to understand how this changes the of energy of excitonic states. The common origin between C-dots and GO-dots demonstrating green emission was attributed to surface states with the participation of carboxyl and carbonyl groups [69]. Attachment of heteroatoms may dramatically change the configuration and energetics at these sites, as they serve as exciton traps. Many routes can be followed to optimize their properties if we start to understand the photophysical mechanisms in the background of these effects.
With an attempt to comprehend these general features, the researchers try to explore their preceding theoretical and experimental knowledge by finding the analogs with previously studied and better characterized systems. A number of such analogs was suggested (e.g., short sheets of aromatic islands, clusters of organic dyes or semiconductor-like nanocrystals), but they are still not accepted broadly due to the presence of opposing arguments. Thus, are these carbon nanosystems that we study unique? Should we develop new physics to explain their properties? In my opinion, no. The closest systems that could be suggested are the molecular aggregates demonstrating the excitonic effects. Therefore, the present work was motivated by the necessity to provide critical analysis of the photophysical behavior of carbonic nanoparticles based on the well-developed physical theory of molecular excitons.
Being suggested for explaining the properties of crystals and self-assembled aggregates of organic dyes, the concept of molecular excitons has developed into a powerful theory applied for the analysis of different phenomena as diverse as the conjugated polymers and biological systems of photosynthesis. Excitonic states are considered a powerful mechanism to substantially increase the absorption cross-section for incident light and display a high efficiency of energy conversion and release. In molecular systems, their formation provides the means to reduce dissipative processes that might result from diffusive trapping of excitation energy in a weakly coupled aggregate. Thus, molecular excitons demonstrate the features of which many scientists working in research and application of carbonic nanostructures are not well familiar. Moreover, these features are quite surprising, since it is hard to imagine a single quantum system that is able to absorb the light quanta collectively and emit them locally. Based on this property, different aspects of nanocarbon behavior can be explained, as it was described above.
With our present knowledge, we can formulate only a very general framework for considering the carbonic nanostructures as the excitonic H-aggregate systems. We have to complement the obtained results with our understanding of the precise mechanism of exciton self-trapping and photon emission and to connect them with the properties of real structures. We have to find the ways to optimize these properties in design and construction, carrying on the idea of the presence of a hidden reserve that has yet to be revealed and explored.
I assume that many experimental findings in our field may not fit the above presented integrated view based on excitonic concept or may request its further development, but its ability to serve a platform for discussion and analysis may appear fruitful. Standing on this platform, we can address different questions of fundamental value. Many of them could be directed to the H-aggregates that are able to generate the excitons serving as the structurally determined antennas for absorbing and transforming the light quanta and to the coupling of these processes with molecular and lattice vibrations. In fact, there are the vibrations that may be considered as noise, but there are those that allow the H-aggregates to demonstrate bright fluorescence emission. All that happens in the presence of static disorder, and the understanding of the extent of this disorder that could allow the carbonic nanoparticles to be emissive is very important.
Other questions have to be addressed to the structure, location, and properties of exciton self-localization (trapping) sites. Are they arranged randomly or, in contrast, according to some hidden deterministic rule? The dynamics of excitons are thought to be dictated by competition between the delocalizing effect of electronic coupling among the H-aggregate elements and the localizing influence of electron–phonon coupling. Then, the exciton self-trapping is induced by the amplitude of fluctuations occurring on their ultra-short (fs-ps) characteristic timescales. Accordingly, the self-trapping is accompanied by re-arrangement of the structure by local heating within this time range. To what extent? Should it be coupled with a very small change in the equilibrium coordinates of vibrational modes that can induce the surprisingly rapid localization of the exciton? The multiplicity of these processes and their occurrence on an ultra-fast scale compared to emission complicate their studies. Finally, we have to explain the results that depend on many factors leading to the versatile and highly efficient emission.
Many new discoveries are expected with carbonic nanostructures. We have entered a new exciting world of knowledge full of intriguing and nontrivial questions for experimental and theoretical work. The prospect is bright, but the road is difficult for a unified understanding of complex quantum phenomena that are in the background of carbonic nanoscale emitters. Based on this understanding, a clear vision should appear, and new areas will be opened for technological developments and applications.
Funding
This research received no external funding.
Acknowledgments
I wish to thank all the members of my research group that contributed their research results discussed in this paper. Special thanks to Mariia Dekaliuk for her enthusiastic activity. Yuriy Maliukin at the Institute of Scintillation Materials in Kharkiv, Ukraine, is recognized for his creative advice and valuable contribution to the development of excitonic concept. The contribution of Joerg Enderlein and his team, particularly of Anna and Alexey Chizhik, at the Gottingen University, Germany, is acknowledged for providing valuable single-molecular studies within the frame of a Ukraine–Germany collaborative project.
Conflicts of Interest
The author declares no conflict of interest.
References
- Sun, Y.P.; Zhou, B.; Lin, Y.; Wang, W.; Fernando, K.A.; Pathak, P.; Meziani, M.J.; Harruff, B.A.; Wang, X.; Wang, H.; et al. Quantum-sized carbon dots for bright and colorful photoluminescence. J. Am. Chem. Soc. 2006, 128, 7756–7757. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ray, R.; Gu, Y.; Ploehn, H.J.; Gearheart, L.; Raker, K.; Scrivens, W.A. Electrophoretic analysis and purification of fluorescent single-walled carbon nanotube fragments. J. Am. Chem. Soc. 2004, 126, 12736–12737. [Google Scholar] [CrossRef] [PubMed]
- Bond, T.C.; Bergstrom, R.W. Light absorption by carbonaceous particles: An investigative review. Aerosol Sci. Technol. 2006, 40, 27–67. [Google Scholar] [CrossRef]
- Krueger, A. New Carbon Materials: Biological Applications of Functionalized Nanodiamond Materials. Chem. Eur. J. 2008, 14, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Schrand, A.M.; Hens, S.A.C.; Shenderova, O.A. Nanodiamond Particles: Properties and Perspectives for Bioapplications. Crit. Rev. Solid State Mater. Sci. 2009, 34, 18–74. [Google Scholar] [CrossRef]
- Neto, A.C.; Guinea, F.; Peres, N.M.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109. [Google Scholar] [CrossRef]
- Barone, P.W.; Baik, S.; Heller, D.A.; Strano, M.S. Near-infrared optical sensors based on single-walled carbon nanotubes. Nat. Mater. 2005, 4, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, Y. Photoluminescence studies on exciton photophysics in carbon nanotubes. J. Mater. Chem. C 2013, 1, 6499–6521. [Google Scholar] [CrossRef]
- Lu, C.-H.; Yang, H.-H.; Zhu, C.-L.; Chen, X.; Chen, G.-N. A Graphene Platform for Sensing Biomolecules. Angew. Chem. Int. Ed. 2009, 48, 4785–4787. [Google Scholar] [CrossRef] [PubMed]
- Esteves da Silva, J.C.G.; Gonçalves, H.M.R. Analytical and bioanalytical applications of carbon dots. TrAC Trends Anal. Chem. 2011, 30, 1327–1336. [Google Scholar] [CrossRef]
- Zhu, S.; Song, Y.; Zhao, X.; Shao, J.; Zhang, J.; Yang, B. The photoluminescence mechanism in carbon dots (graphene quantum dots, carbon nanodots, and polymer dots): Current state and future perspective. Nano Res. 2015, 8, 355–381. [Google Scholar] [CrossRef]
- Cao, L.; Meziani, M.J.; Sahu, S.; Sun, Y.P. Photoluminescence Properties of Graphene versus Other Carbon Nanomaterials. Acc. Chem. Res. 2012, 46, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Lamola, A.A.; Turro, N.J. Energy Transfer and Organic Photochemistry; Interscience Publishers: Geneva, Switzerland; New York, NY, USA, 1969; Volume 14. [Google Scholar]
- Nozik, A.J. Multiple exciton generation in semiconductor quantum dots. Chem. Phys. Lett. 2008, 457, 3–11. [Google Scholar] [CrossRef]
- Reisch, A.; Klymchenko, A.S. Fluorescent polymer nanoparticles based on dyes: Seeking brighter tools for bioimaging. Small 2016, 12, 1968–1992. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P.; Dekaliuk, M.O. The origin of emissive states of carbon nanoparticles derived from ensemble-averaged and single-molecular studies. Nanoscale 2016, 8, 14057–14069. [Google Scholar] [CrossRef] [PubMed]
- Sciortino, A.; Cannizzo, A.; Messina, F. Carbon Nanodots: A Review—From the Current Understanding of the Fundamental Photophysics to the Full Control of the Optical Response. C J. Carbon Res. 2018, 4, 67. [Google Scholar] [CrossRef]
- Schneider, J.; Reckmeier, C.J.; Xiong, Y.; von Seckendorff, M.; Susha, A.S.; Kasák, P.; Rogach, A.L. Molecular Fluorescence in Citric Acid-Based Carbon Dots. J. Phys. Chem. C 2017, 121, 2014–2022. [Google Scholar] [CrossRef]
- Eda, G.; Lin, Y.Y.; Mattevi, C.; Yamaguchi, H.; Chen, H.A.; Chen, I.S.; Chen, C.W.; Chhowalla, M. Blue photoluminescence from chemically derived graphene oxide. Adv. Mater. 2010, 22, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Galande, C.; Mohite, A.D.; Naumov, A.V.; Gao, W.; Ci, L.; Ajayan, A.; Gao, H.; Srivastava, A.; Weisman, R.B.; Ajayan, P.M. Quasi-molecular fluorescence from graphene oxide. Sci. Rep. 2011, 1, 85. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ma, L.; Li, J.; Ai, W.; Yu, T.; Gurzadyan, G.G. The Origin of Fluorescence from Graphene Oxide. Sci. Rep. 2012, 2, 792. [Google Scholar] [CrossRef] [PubMed]
- Selvin, P.R. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Mol. Biol. 2000, 7, 730. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.T.; Goodwin, A.; Zaric, S.; Dai, H. Nano-graphene oxide for cellular imaging and drug delivery. Nano Res. 2008, 1, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guo, B.; Rao, Z.; Zhang, B.; Gong, J.R. Strong two-photon-induced fluorescence from photostable, biocompatible nitrogen-doped graphene quantum dots for cellular and deep-tissue imaging. Nano Lett. 2013, 13, 2436–2441. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, S.; Zhu, C.; Liu, M.; Ji, Y.; Feng, L.; Tao, L.; Wei, Y. Carbon-dots derived from nanodiamond: Photoluminescence tunable nanoparticles for cell imaging. J. Colloid Interface Sci. 2013, 397, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Wen, X.; Toh, Y.-R.; Tang, J. Temperature-Dependent Fluorescence in Carbon Dots. J. Phys. Chem. C 2012, 116, 25552–25557. [Google Scholar] [CrossRef]
- Xiao, L.; Wang, Y.; Huang, Y.; Wong, T.; Sun, H. Self-trapped exciton emission from carbon dots investigated by polarization anisotropy of photoluminescence and photoexcitation. Nanoscale 2017, 9, 12637–12646. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Zhang, Z.L.; Tian, Z.Q.; Zhang, L.; Liu, C.; Lin, Y.; Qi, B.; Pang, D.W. Electrochemical tuning of luminescent carbon nanodots: From preparation to luminescence mechanism. Adv. Mater. 2011, 23, 5801–5806. [Google Scholar] [CrossRef] [PubMed]
- Pacurari, M.; Lowe, K.; Tchounwou, P.B.; Kafoury, R. A Rev. on the respiratory system toxicity of carbon nanoparticles. Int. J. Environ. Res. Public Health 2016, 13, 325. [Google Scholar] [CrossRef] [PubMed]
- Malyukin, Y.; Viagin, O.; Maksimchuk, P.; Dekaliuk, M.; Demchenko, A. Insight into the mechanism of the photoluminescence of carbon nanoparticles derived from cryogenic studies. Nanoscale 2018, 10, 9320–9328. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chizhik, A.M.; Karedla, N.; Dekaliuk, M.O.; Gregor, I.; Schuhmann, H.; Seibt, M.; Bodensiek, K.; Schaap, I.A.; Schulz, O. Photoluminescence of carbon nanodots: Dipole emission centers and electron–phonon coupling. Nano Lett. 2014, 14, 5656–5661. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, V.; Perman, J.A.; Tucek, J.; Zboril, R. Broad Family of Carbon Nanoallotropes: Classification, Chemistry, and Applications of Fullerenes, Carbon Dots, Nanotubes, Graphene, Nanodiamonds, and Combined Superstructures. Chem. Rev. 2015, 115, 4744–4822. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Li, B.; Cui, X.; Wei, Q.; Tajima, K.; Li, L.-S. Independent Tuning of the Band Gap and Redox Potential of Graphene Quantum Dots. J. Phys. Chem. Lett. 2011, 2, 1119–1124. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Zhang, Z.; Zhang, J.; Chen, N. Graphene Quantum Dots: An Emerging Material for the Energy-Related Applications and Beyond. Energy Environ. Sci. 2012, 5, 8869–8890. [Google Scholar]
- Zheng, X.T.; Ananthanarayanan, A.; Luo, K.Q.; Chen, P. Glowing graphene quantum dots and carbon dots: Properties, syntheses, and biological applications. Small 2015, 11, 1620–1636. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Shen, W.; Gao, Z. Carbon quantum dots and their applications. Chem. Soc. Rev. 2015, 44, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Kang, Z.; Liu, Y.; Lee, S.T. Carbon Nanodots: Synthesis, Properties and Applications. J. Mater. Chem. 2012, 22, 24230–24253. [Google Scholar] [CrossRef]
- Dong, X.; Wei, L.; Su, Y.; Li, Z.; Geng, H.; Yang, C.; Zhang, Y. Efficient long lifetime room temperature phosphorescence of carbon dots in a potash alum matrix. J. Mater. Chem. C 2015, 3, 2798–2801. [Google Scholar] [CrossRef]
- Hirata, S. Recent Advances in Materials with Room Temperature Phosphorescence: Photophysics for Triplet Exciton Stabilization. Adv. Opt. Mater. 2017, 5, 1700116. [Google Scholar] [CrossRef]
- Baker, S.N.; Baker, G.A. Luminescent Carbon Nanodots: Emergent Nanolights. Angew. Chem. Int. Ed. 2010, 49, 6726–6744. [Google Scholar] [CrossRef] [PubMed]
- Hola, K.; Zhang, Y.; Wang, Y.; Giannelis, E.P.; Zboril, R.; Rogach, A.L. Carbon dots—Emerging light emitters for bioimaging, cancer therapy and optoelectronics. Nano Today 2014, 9, 590–603. [Google Scholar] [CrossRef]
- Kargbo, O.; Jin, Y.; Ding, S.-N. Recent advances in luminescent carbon dots. Curr. Anal. Chem. 2015, 11, 4–21. [Google Scholar] [CrossRef]
- Reckmeier, C.; Schneider, J.; Susha, A.; Rogach, A. Luminescent colloidal carbon dots: Optical properties and effects of doping [Invited]. Opt. Express 2016, 24, A312–A340. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ye, T.; Mao, C. Fluorescent carbon nanoparticles derived from candle soot. Angew. Chem. Int. Ed. 2007, 46, 6473–6475. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Zhang, P.; Liu, C.; Bai, T.; Li, W.; Dai, L.; Liu, W. Highly luminescent carbon nanodots by microwave-assisted pyrolysis. Chem. Commun. 2012, 48, 7955–7957. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Pathan, S.H.; Mitra, S.; Modha, B.H.; Goswami, A.; Pramanik, P. Tuning of photoluminescence on different surface functionalized carbon quantum dots. RSC Adv. 2012, 2, 3602–3606. [Google Scholar] [CrossRef]
- Hsu, P.C.; Shih, Z.Y.; Lee, C.H.; Chang, H.T. Synthesis and analytical applications of photoluminescent carbon nanodots. Green Chem. 2012, 14, 917–920. [Google Scholar] [CrossRef]
- Zhou, J.; Sheng, Z.; Han, H.; Zou, M.; Li, C. Facile synthesis of fluorescent carbon dots using watermelon peel as a carbon source. Mater. Lett. 2012, 66, 222–224. [Google Scholar] [CrossRef]
- Dong, Y.; Wang, R.; Li, H.; Shao, J.; Chi, Y.; Lin, X.; Chen, G. Polyamine-functionalized carbon quantum dots for chemical sensing. Carbon 2012, 50, 2810–2815. [Google Scholar] [CrossRef]
- Radovic, L.R. (Ed.) Chemistry and Physics of Carbon; CRC Press: Boca Raton, FL, USA, 2012; Volume 31. [Google Scholar]
- Wang, X.; Qu, K.; Xu, B.; Ren, J.; Qu, X. Microwave assisted one-step green synthesis of cell-permeable multicolor photoluminescent carbon dots without surface passivation reagents. J. Mater. Chem. 2011, 21, 2445–2450. [Google Scholar] [CrossRef]
- Deng, Y.; Zhao, D.; Chen, X.; Wang, F.; Song, H.; Shen, D. Long lifetime pure organic phosphorescence based on water soluble carbon dots. Chem. Commun. 2013, 49, 5751–5753. [Google Scholar] [CrossRef] [PubMed]
- Burda, C.; Chen, X.B.; Narayanan, R.; El-Sayed, M.A. Chemical and properties of nanocrystals of different shapes. Chem. Rev. 2005, 105, 1025–1102. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhu, Y.; Yang, X.; Li, C. Graphene quantum dots: Emergent nanolights for bioimaging, sensors, catalysis and photovoltaic devices. Chem. Commun. 2012, 48, 3686–3699. [Google Scholar] [CrossRef] [PubMed]
- Tepliakov, N.V.; Kundelev, E.V.; Khavlyuk, P.D.; Xiong, Y.; Leonov, M.Y.; Zhu, W.; Baranov, A.V.; Fedorov, A.V.; Rogach, A.L.; Rukhlenko, I.D. sp2–sp3-Hybridized atomic domains determine optical features of carbon dots. ACS Nano 2019, 13, 10737–10744. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Saha, A.; Jana, N.R.; Sarkar, R. Fluorescent Carbon Nanoparticle: Synthesis, Characterization and Bio-imaging Application. J. Phys. Chem. C 2009, 113, 18546–18551. [Google Scholar] [CrossRef]
- Yang, Z.C.; Wang, M.; Yong, A.M.; Wong, S.Y.; Zhang, X.H.; Tan, H.; Chang, A.Y.; Li, X.; Wang, J. Intrinsically fluorescent carbon dots with tunable emission derived from hydrothermal treatment of glucose in the presence of monopotassium phosphate. Chem. Commun. 2011, 47, 11615–11617. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wu, G.; Yang, G.; Peng, J.; Zhao, J.; Zhu, J.J. Focusing on luminescent graphene quantum dots: Current status and future perspectives. Nanoscale 2013, 5, 4015–4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, D.; Zhang, J.; Li, Z.; Wu, M. Hydrothermal Route for Cutting Graphene Sheets into Blue-Luminescent Graphene Quantum Dots. Adv. Mater. 2009, 22, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, S.; Dai, L.; Li, L. Nitrogen-doped colloidal graphene quantum dots and their size-dependent electrocatalytic activity for the oxygen reduction reaction. J. Am. Chem. Soc. 2012, 46, 18932–18935. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Liu, Y.; Yang, Y.; Cui, J.; Huang, Z.; Wang, Y.; Yang, L.; Wang, H.; Xiao, Y.; Rong, J. One-step preparation of nitrogen-doped graphene quantum dots from oxidized debris of graphene oxide. J. Mater. Chem. B 2013, 1, 39–42. [Google Scholar] [CrossRef]
- Zhang, M.; Bai, L.; Shang, W.; Xie, W.; Ma, H.; Fu, Y.; Fang, D.; Sun, H.; Fan, L.; Han, M. Facile synthesis of water-soluble, highly fluorescent graphene quantum dots as a robust biological label for stem cells. J. Mater. Chem. 2012, 22, 7461–7467. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, J.; Qiao, C.; Tang, S.; Li, Y.; Yuan, W.; Li, B.; Tian, L.; Liu, F.; Hu, R.; et al. Strongly green-photoluminescent graphene quantum dots for bioimaging applications. Chem. Commun. 2011, 47, 6858–6860. [Google Scholar] [CrossRef] [PubMed]
- Gokus, T.; Nair, R.; Bonetti, A.; Bohmler, M.; Lombardo, A.; Novoselov, K.; Geim, A.; Ferrari, A.; Hartschuh, A. Making graphene luminescent by oxygen plasma treatment. ACS Nano 2009, 3, 3963–3968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Hu, Y.; Zhao, Y.; Shi, G.; Deng, L.; Hou, Y.; Qu, L. An Electrochemical Avenue to Green-Luminescent Graphene Quantum Dots as Potential Electron-Acceptors for Photovoltaics. Adv. Mater. 2011, 23, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, Y.; Cheng, H.; Hu, Y.; Shi, G.; Dai, L.; Qu, L. Nitrogen-Doped Graphene Quantum Dots with Oxygen-Rich Functional Groups. J. Am. Chem. Soc. 2011, 134, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Gao, W.; Gupta, B.K.; Liu, Z.; Romero-Aburto, R.; Ge, L.; Song, L.; Alemany, L.B.; Zhan, X.; Gao, G. Graphene quantum dots derived from carbon fibers. Nano Lett. 2012, 12, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Dekaliuk, M.O.; Viagin, O.; Malyukin, Y.V.; Demchenko, A.P. Fluorescent carbon nanomaterials: “quantum dots” or nanoclusters? Phys. Chem. Chem. Phys. 2014, 16, 16075–16084. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, S.-J.; Wang, H.-Y.; Qu, S.-N.; Zhang, Y.-L.; Zhang, J.-H.; Chen, Q.-D.; Xu, H.-L.; Han, W.; Yang, B. Common origin of green luminescence in carbon nanodots and graphene quantum dots. ACS Nano 2014, 8, 2541–2547. [Google Scholar] [CrossRef] [PubMed]
- Rondeau-Gagné, S.; Morin, J.-F. Preparation of carbon nanomaterials from molecular precursors. Chem. Soc. Rev. 2014, 43, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P.; Dekaliuk, M.O. Novel fluorescent carbonic nanomaterials for sensing and imaging. Methods Appl. Fluoresc. 2013, 1, 042001. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.B.; Gómez-Navarro, C.; Sundaram, R.S.; Burghard, M.; Kern, K. Electrical conduction mechanism in chemically derived graphene monolayers. Nano Lett. 2009, 9, 1787–1792. [Google Scholar] [CrossRef] [PubMed]
- Lavin-Lopez, M.; Valverde, J.; Sanchez-Silva, L.; Romero, A. Solvent-based exfoliation via sonication of graphitic materials for graphene manufacture. Ind. Eng. Chem. Res. 2016, 55, 845–855. [Google Scholar] [CrossRef]
- Björk, J.; Hanke, F.; Palma, C.-A.; Samori, P.; Cecchini, M.; Persson, M. Adsorption of aromatic and anti-aromatic systems on graphene through π-π stacking. J. Phys. Chem. Lett. 2010, 1, 3407–3412. [Google Scholar] [CrossRef]
- Pech, D.; Brunet, M.; Durou, H.; Huang, P.; Mochalin, V.; Gogotsi, Y.; Taberna, P.-L.; Simon, P. Ultrahigh-power micrometre-sized supercapacitors based on onion-like carbon. Nat Nano 2010, 5, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; He, X.; Kang, Z.; Huang, H.; Liu, Y.; Liu, J.; Lian, S.; Tsang, C.H.A.; Yang, X.; Lee, S.T. Water-Soluble Fluorescent Carbon Quantum Dots and Photocatalyst Design. Angew. Chem. Int. Ed. 2010, 49, 4430–4434. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Xiang, C.; Lin, J.; Peng, Z.; Huang, K.; Yan, Z.; Cook, N.P.; Samuel, E.L.; Hwang, C.-C.; Ruan, G. Coal as an abundant source of graphene quantum dots. Nat. Commun. 2013, 4, 2943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhu, J.; Zhai, Y.; Wang, H.; Bai, X.; Dong, B.; Wang, H.; Song, H. A novel mechanism for red emission carbon dots: Hydrogen bond dominated molecular states emission. Nanoscale 2017, 9, 13042–13051. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Sun, Z.; Zhang, H.; Bai, Z. The fluorescence mechanism of carbon dots, and methods for tuning their emission color: A review. Microchim. Acta 2019, 186, 583. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Ehrat, F.; Wang, Y.; Milowska, K.Z.; Reckmeier, C.; Rogach, A.L.; Stolarczyk, J.K.; Urban, A.S.; Feldmann, J. Carbon dots: A unique fluorescent cocktail of polycyclic aromatic hydrocarbons. Nano Lett. 2015, 15, 6030–6035. [Google Scholar] [CrossRef] [PubMed]
- Messina, F.; Sciortino, L.; Popescu, R.; Venezia, A.; Sciortino, A.; Buscarino, G.; Agnello, S.; Schneider, R.; Gerthsen, D.; Cannas, M. Fluorescent nitrogen-rich carbon nanodots with an unexpected β-C3N4 nanocrystalline structure. J. Mater. Chem. C 2016, 4, 2598–2605. [Google Scholar] [CrossRef]
- Sciortino, A.; Cayuela, A.; Soriano, M.; Gelardi, F.; Cannas, M.; Valcárcel, M.; Messina, F. Different natures of surface electronic transitions of carbon nanoparticles. Phys. Chem. Chem. Phys. 2017, 19, 22670–22677. [Google Scholar] [CrossRef] [PubMed]
- Hoeben, F.J.; Jonkheijm, P.; Meijer, E.; Schenning, A.P. About supramolecular assemblies of π-conjugated systems. Chem. Rev. 2005, 105, 1491–1546. [Google Scholar] [CrossRef] [PubMed]
- Sao, S.; Mukherjee, I.; De, P.; Chaudhuri, D. Encapsulation induced aggregation: A self-assembly strategy for weakly pi-stacking chromophores. Chem. Commun. 2017, 53, 3994–3997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Würthner, F. Dipole–dipole interaction driven self-assembly of merocyanine dyes: From dimers to nanoscale objects and supramolecular materials. Acc. Chem. Res. 2016, 49, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Koch, F.; Stolte, M.; Zitzler-Kunkel, A.; Bialas, D.; Steinbacher, A.; Brixner, T.; Würthner, F. Unraveling the structure and exciton coupling for multichromophoric merocyanine dye molecules. Phys. Chem. Chem. Phys. 2017, 19, 6368–6378. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Pang, H.; Yang, H.B.; Guo, C.; Shao, J.; Chi, Y.; Li, C.M.; Yu, T. Carbon-Based Dots Co-doped with Nitrogen and Sulfur for High Quantum Yield and Excitation-Independent Emission. Angew. Chem. Int. Ed. 2013, 52, 7800–7804. [Google Scholar] [CrossRef] [PubMed]
- Krysmann, M.J.; Kelarakis, A.; Dallas, P.; Giannelis, E.P. Formation Mechanism of Carbogenic Nanoparticles with Dual Photoluminescence Emission. J. Am. Chem. Soc. 2011, 134, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Kasprzyk, W.; Bednarz, S.; Żmudzki, P.; Galica, M.; Bogdał, D. Novel efficient fluorophores synthesized from citric acid. RSC Adv. 2015, 5, 34795–34799. [Google Scholar] [CrossRef]
- Zhu, S.; Zhao, X.; Song, Y.; Lu, S.; Yang, B. Beyond bottom-up carbon nanodots: Citric-acid derived organic molecules. Nano Today 2016, 11, 128–132. [Google Scholar] [CrossRef]
- Ehrat, F.; Bhattacharyya, S.; Schneider, J.; Löf, A.; Wyrwich, R.; Rogach, A.L.; Stolarczyk, J.K.; Urban, A.S.; Feldmann, J. Tracking the Source of Carbon Dot Photoluminescence: Aromatic Domains versus Molecular Fluorophores. Nano Lett. 2017, 17, 7710–7716. [Google Scholar] [CrossRef] [PubMed]
- Gude, V.; Das, A.; Chatterjee, T.; Mandal, P.K. Molecular origin of photoluminescence of carbon dots: Aggregation-induced orange-red emission. Phys. Chem. Chem. Phys. 2016, 18, 28274–28280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Yang, J.H.; Zeng, H.B.; Chen, Y.M.; Yang, S.C.; Wu, C.; Zeng, H.; Yoshihito, O.; Zhang, Q. Carbon dots with high fluorescence quantum yield: The fluorescence originates from organic fluorophores. Nanoscale 2016, 8, 14374–14378. [Google Scholar] [CrossRef] [PubMed]
- Reckmeier, C.J.; Schneider, J.; Xiong, Y.; Häusler, J.; Kasák, P.; Schnick, W.; Rogach, A.L. Aggregated Molecular Fluorophores in the Ammonothermal Synthesis of Carbon Dots. Chem. Mater. 2017, 29, 10352–10361. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, X.; Li, Y.; Wang, Z.; Yang, F.; Yang, X. Microwave synthesis of fluorescent carbon nanoparticles with electrochemiluminescence properties. Chem. Commun. 2009, 34, 5118–5120. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Pu, P.; Zhao, J.; Dong, C.; Gao, C.; Chen, Y.; Chen, J.; Liu, Y.; Zhou, H. Preparation of highly photoluminescent sulfur-doped carbon dots for Fe (III) detection. J. Mater. Chem. A 2015, 3, 542–546. [Google Scholar] [CrossRef]
- Zhou, J.; Shan, X.; Ma, J.; Gu, Y.; Qian, Z.; Chen, J.; Feng, H. Facile synthesis of P-doped carbon quantum dots with highly efficient photoluminescence. RSC Adv. 2014, 4, 5465–5468. [Google Scholar] [CrossRef]
- Bourlinos, A.B.; Trivizas, G.; Karakassides, M.A.; Baikousi, M.; Kouloumpis, A.; Gournis, D.; Bakandritsos, A.; Hola, K.; Kozak, O.; Zboril, R. Green and simple route toward boron doped carbon dots with significantly enhanced non-linear optical properties. Carbon 2015, 83, 173–179. [Google Scholar] [CrossRef]
- Choi, Y.; Kang, B.; Lee, J.; Kim, S.; Kim, G.T.; Kang, H.; Lee, B.R.; Kim, H.; Shim, S.-H.; Lee, G. Integrative approach toward uncovering the origin of photoluminescence in dual heteroatom-doped carbon nanodots. Chem. Mater. 2016, 28, 6840–6847. [Google Scholar] [CrossRef]
- Demchenko, A.P. Site-selective Red-Edge effects. Methods Enzymol. 2008, 450, 59–78. [Google Scholar] [PubMed]
- Van Dam, B.; Nie, H.; Ju, B.; Marino, E.; Paulusse, J.M.; Schall, P.; Li, M.; Dohnalová, K. Excitation-Dependent Photoluminescence from Single-Carbon Dots. Small 2017, 13, 1702098. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Choi, Y.; Kwon, O.-H.; Kim, B.-S. Carbon Dots: Bottom-Up Syntheses, Properties, and Light-Harvesting Applications. Chem. Asian J. 2018, 13, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Yu, S.-B.; Wei, J.-S.; Xiong, H.-M. Full-color light-emitting carbon dots with a surface-state-controlled luminescence mechanism. ACS Nano 2015, 10, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Trinchi, A.; Atkin, P.; Cole, I. Tunable photoluminescence across the entire visible spectrum from carbon dots excited by white light. Angew. Chem. Int. Ed. 2015, 54, 2970–2974. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Wei, J.-S.; Zhong, N.; Gao, Q.-Y.; Xiong, H.-M. Highly Efficient Red-Emitting Carbon Dots with Gram-Scale Yield for Bioimaging. Langmuir 2017, 33, 12635–12642. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Zhou, D.; Li, D.; Ji, W.; Jing, P.; Han, D.; Liu, L.; Zeng, H.; Shen, D. Toward Efficient Orange Emissive Carbon Nanodots through Conjugated sp2-Domain Controlling and Surface Charges Engineering. Adv. Mater. 2016, 28, 3516–3521. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Ge, J.; Liu, W.; Niu, G.; Jia, Q.; Wang, H.; Wang, P. Tunable multicolor carbon dots prepared from well-defined polythiophene derivatives and their emission mechanism. Nanoscale 2016, 8, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Sun, S.; Zhang, L.; Lu, Y.; Wu, A.; Cai, C.; Lin, H. Red, green, and blue luminescence by carbon dots: Full-color emission tuning and multicolor cellular imaging. Angew. Chem. Int. Ed. 2015, 54, 5360–5363. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Bai, X.; Bai, J.; Pan, G.; Zhu, Y.; Zhai, Y.; Shao, H.; Chen, X.; Dong, B.; Zhang, H. Emitting color tunable carbon dots by adjusting solvent towards light-emitting devices. Nanotechnology 2018, 29, 085705. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pan, Y.; Fang, Y.; Zhang, L.; Chen, J.; Yi, C. Tuning photoluminescence and surface properties of carbon nanodots for chemical sensing. Nanoscale 2016, 8, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Tetsuka, H.; Nagoya, A.; Asahi, R. Highly luminescent flexible amino-functionalized graphene quantum dots@cellulose nanofiber–clay hybrids for white-light emitting diodes. J. Mater. Chem. C 2015, 3, 3536–3541. [Google Scholar] [CrossRef]
- Righetto, M.; Privitera, A.; Fortunati, I.; Mosconi, D.; Zerbetto, M.; Curri, M.L.; Corricelli, M.; Moretto, A.; Agnoli, S.; Franco, L. Spectroscopic Insights into Carbon Dot Systems. J. Phys. Chem. Lett. 2017, 8, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.-F.; Huang, W.-L.; Chung, C.-J.; Chiang, I.-T.; Chen, L.-C.; Chang, H.-Y.; Su, W.-C.; Cheng, C.; Chen, S.-J.; Teng, H. Elucidating quantum confinement in graphene oxide dots based on excitation-wavelength-independent photoluminescence. J. Phys. Chem. Lett. 2016, 7, 2087–2092. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Liu, X.; Guo, X.; Chu, M.; Zhang, L.; Shen, D. Amplified spontaneous green emission and lasing emission from carbon nanoparticles. Adv. Funct. Mater. 2014, 24, 2689–2695. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, H.; Yu, S.; Yang, H. Observation of Lasing Emission from Carbon Nanodots in Organic Solvents. Adv. Mater. 2012, 24, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, M.; Álvaro, M.; García, H. Graphene as a Quencher of Electronic Excited States of Photochemical Probes. Langmuir 2012, 28, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Matte, H.R.; Subrahmanyam, K.; Rao, K.V.; George, S.J.; Rao, C. Quenching of fluorescence of aromatic molecules by graphene due to electron transfer. Chem. Phys. Lett. 2011, 506, 260–264. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, Z.; Hu, D.; Lin, C.-T.; Li, J.; Lin, Y. Aptamer/graphene oxide nanocomplex for in situ molecular probing in living cells. J. Am. Chem. Soc. 2010, 132, 9274–9276. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, K.; He, Z.; Zhang, Y.; Zhao, Y.; Zhang, H.; Miao, Z. Investigation on Fluorescence Quenching Mechanism of Perylene Diimide Dyes by Graphene Oxide. Molecules 2016, 21, 1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cao, L.; Lu, F.; Meziani, M.J.; Li, H.; Qi, G.; Zhou, B.; Harruff, B.A.; Kermarrec, F.; Sun, Y.-P. Photoinduced electron transfers with carbon dots. Chem. Commun. 2009, 25, 3774–3776. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Wen, X.; Toh, Y.-R.; Lee, Y.-C.; Huang, K.-Y.; Huang, S.; Shrestha, S.; Conibeer, G.; Tang, J. Efficient electron transfer in carbon nanodot–graphene oxide nanocomposites. J. Mater. Chem. C 2014, 2, 2894–2901. [Google Scholar] [CrossRef]
- Tang, L.; Ji, R.; Cao, X.; Lin, J.; Jiang, H.; Li, X.; Teng, K.S.; Luk, C.M.; Zeng, S.; Hao, J. Deep Ultraviolet Photoluminescence of Water-Soluble Self-Passivated Graphene Quantum Dots. ACS Nano 2012, 6, 5102–5110. [Google Scholar] [CrossRef] [PubMed]
- Arshad, F.; Pal, A.; Rahman, M.A.; Ali, M.; Khan, J.A.; Sk, M.P. Insights on the solvatochromic effects in N-doped yellow-orange emissive carbon dots. New J. Chem. 2018, 42, 19837–19843. [Google Scholar] [CrossRef]
- Beddard, G.; Porter, G. Concentration quenching in chlorophyll. Nature 1976, 260, 366. [Google Scholar] [CrossRef]
- An, B.-K.; Kwon, S.-K.; Jung, S.-D.; Park, S.Y. Enhanced emission and its switching in fluorescent organic nanoparticles. J. Am. Chem. Soc. 2002, 124, 14410–14415. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Leung, N.L.; Kwok, R.T.; Lam, J.W.; Tang, B.Z. Aggregation-induced emission: Together we shine, united we soar! Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Lam, J.W.; Tang, B.Z. Aggregation-induced emission. Chem. Soc. Rev. 2011, 40, 5361–5388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, R. The chemistry of excited complexes: A survey of reactions. In Advances in Physical Organic Chemistry; Elsevier: Amsterdam, The Netherlands, 1983; Volume 19, pp. 1–130. [Google Scholar]
- Birks, J.B. Photophysics Aromatic Molecules; Wiley-Interscience: London, UK, 1970; p. 704. [Google Scholar]
- Zhao, Z.; Chen, S.; Lam, J.W.; Wang, Z.; Lu, P.; Mahtab, F.; Sung, H.H.; Williams, I.D.; Ma, Y.; Kwok, H.S. Pyrene-substituted ethenes: Aggregation-enhanced excimer emission and highly efficient electroluminescence. J. Mater. Chem. 2011, 21, 7210–7216. [Google Scholar] [CrossRef]
- Reisch, A.; Trofymchuk, K.; Runser, A.; Fleith, G.; Rawiso, M.; Klymchenko, A.S. Tailoring Fluorescence Brightness and Switching of Nanoparticles through Dye Organization in the Polymer Matrix. ACS Appl. Mater. Interfaces 2017, 9, 43030–43042. [Google Scholar] [CrossRef] [PubMed]
- Doose, S.; Neuweiler, H.; Sauer, M. Fluorescence quenching by photoinduced electron transfer: A reporter for conformational dynamics of macromolecules. ChemPhysChem 2009, 10, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Yu, P.; Toh, Y.R.; Lee, Y.C.; Hsu, A.C.; Tang, J. Near-infrared enhanced carbon nanodots by thermally assisted growth. Appl. Phys. Lett. 2012, 101, 163107. [Google Scholar] [CrossRef]
- Shamsipur, M.; Barati, A.; Karami, S. Long-wavelength, multicolor, and white-light emitting carbon-based dots: Achievements made, challenges remaining, and applications. Carbon 2017, 124, 429–472. [Google Scholar] [CrossRef]
- Lu, W.; Jiao, Y.; Gao, Y.; Qiao, J.; Mozneb, M.; Shuang, S.; Dong, C.; Li, C.-Z. Bright Yellow Fluorescent Carbon Dots as a Multifunctional Sensing Platform for the Label-Free Detection of Fluoroquinolones and Histidine. ACS Appl. Mater. Interfaces 2018, 10, 42915–42924. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chao, D.; Zhou, L.; Li, Y.; Deng, R.; Zhang, H. Yellow emissive carbon dots with quantum yield up to 68.6% from the solvent and manganese ions. Carbon 2018, 135, 253–259. [Google Scholar] [CrossRef]
- Jia, J.; Lu, W.-J.; Li, L.; Jiao, Y.; Gao, Y.-F.; Shuang, S.-M. Orange Luminescent Carbon Dots as Fluorescent Probe for Detection of Nitrite. Chin. J. Anal. Chem. 2019, 47, 560–566. [Google Scholar] [CrossRef]
- Jiao, Y.; Gong, X.; Han, H.; Gao, Y.; Lu, W.; Liu, Y.; Xian, M.; Shuang, S.; Dong, C. Facile synthesis of orange fluorescence carbon dots with excitation independent emission for pH sensing and cellular imaging. Anal. Chim. Acta 2018, 1042, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhang, H.; Lai, J.; Peng, X.; Hu, Y.; Gu, W.; Ye, L. Carbon dots with red-shifted photoluminescence by fluorine doping for optical bio-imaging. Carbon 2018, 128, 78–85. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhai, Y.; Li, Z.; Zhu, P.; Mao, S.; Zhu, C.; Du, D.; Belfiore, L.A.; Tang, J.; Lin, Y. Red carbon dots: Optical property regulations and applications. Mater. Today 2019. [Google Scholar] [CrossRef]
- Li, D.; Jing, P.; Sun, L.; An, Y.; Shan, X.; Lu, X.; Zhou, D.; Han, D.; Shen, D.; Zhai, Y. Near-infrared excitation/emission and multiphoton-induced fluorescence of carbon dots. Adv. Mater. 2018, 30, 1705913. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Zhou, X.; Qin, B.; Zhou, Z.; Zhao, Y. Highly fluorescent near-infrared emitting carbon dots derived from lemon juice and its bioimaging application. J. Lumin. 2019, 211, 298–304. [Google Scholar] [CrossRef]
- Chudakov, D.M.; Matz, M.V.; Lukyanov, S.; Lukyanov, K.A. Fluorescent proteins and their applications in imaging living cells and tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Schneider, J.; Ushakova, E.V.; Rogach, A.L. Influence of molecular fluorophores on the research field of chemically synthesized carbon dots. Nano Today 2018, 23, 124–139. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, L.; Li, B.; Song, Y.; Zhao, X.; Zhang, G.; Zhang, S.; Lu, S.; Zhang, J.; Wang, H. Investigation of photoluminescence mechanism of graphene quantum dots and evaluation of their assembly into polymer dots. Carbon 2014, 77, 462–472. [Google Scholar] [CrossRef]
- Wen, X.; Yu, P.; Toh, Y.R.; Hao, X.; Tang, J. Intrinsic and Extrinsic Fluorescence in Carbon Nanodots: Ultrafast Time-Resolved Fluorescence and Carrier Dynamics. Adv. Opt. Mater. 2013, 1, 173–178. [Google Scholar] [CrossRef]
- Bourlinos, A.B.; Stassinopoulos, A.; Anglos, D.; Zboril, R.; Karakassides, M.; Giannelis, E.P. Surface functionalized carbogenic quantum dots. Small 2008, 4, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Yoo, J.; Lim, B.; Kwon, W.; Rhee, S.-W. Improving the functionality of carbon nanodots: Doping and surface functionalization. J. Mater. Chem. A 2016, 4, 11582–11603. [Google Scholar] [CrossRef]
- Yan, F.; Jiang, Y.; Sun, X.; Bai, Z.; Zhang, Y.; Zhou, X. Surface modification and chemical functionalization of carbon dots: A review. Microchim. Acta 2018, 185, 424. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Sun, J.; Li, X.; Zhou, W.; Wang, Z.; He, P.; Ding, G.; Xie, X.; Kang, Z.; Jiang, M. Large-scale fabrication of heavy doped carbon quantum dots with tunable-photoluminescence and sensitive fluorescence detection. J. Mater. Chem. A 2014, 2, 8660–8667. [Google Scholar] [CrossRef]
- Liu, C.; Bao, L.; Yang, M.; Zhang, S.; Zhou, M.; Tang, B.; Wang, B.; Liu, Y.; Zhang, Z.-L.; Zhang, B. Surface Sensitive Photoluminescence of Carbon Nanodots: Coupling between Carbonyl Group and π-Electron System. J. Phys. Chem. Lett. 2019, 10, 3621–3629. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Hou, Z.; Zhang, C.; Zhang, G.; Zhang, Y.; Dong, C.; Shuang, S. Concentration-dependent multicolor fluorescent carbon dots for colorimetric and fluorescent bimodal detections of Fe3+ and l-ascorbic acid. Anal. Methods 2019, 11, 669–676. [Google Scholar] [CrossRef]
- Mu, Y.; Wang, N.; Sun, Z.; Wang, J.; Li, J.; Yu, J. Carbogenic nanodots derived from organo-templated zeolites with modulated full-color luminescence. Chem. Sci. 2016, 7, 3564–3568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lian, H.; Wei, Y.; He, X.; Chen, Y.; Wang, B.; Zeng, Q.; Lin, J. Concentration-induced multi-colored emissions in carbon dots: Origination from triple fluorescent centers. Nanoscale 2018, 10, 6734–6743. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, X.-C.; Yang, M.; Jiang, P.; Zhao, J.; Jiang, F.-L.; Liu, Y. Concentration-tuned Multicolor Carbon Dots: Microwave-assisted Synthesis, Characterization, Mechanism and Applications. New J. Chem. 2019, 43, 8950–8957. [Google Scholar] [CrossRef]
- Meng, X.; Chang, Q.; Xue, C.; Yang, J.; Hu, S. Full-colour carbon dots: From energy-efficient synthesis to concentration-dependent photoluminescence properties. Chem. Commun. 2017, 53, 3074–3077. [Google Scholar] [CrossRef] [PubMed]
- Fuyuno, N.; Kozawa, D.; Miyauchi, Y.; Mouri, S.; Kitaura, R.; Shinohara, H.; Yasuda, T.; Komatsu, N.; Matsuda, K. Drastic change in photoluminescence properties of graphene quantum dots by chromatographic separation. Adv. Opt. Mater. 2014, 2, 983–989. [Google Scholar] [CrossRef]
- Chen, D.; Gao, H.; Chen, X.; Fang, G.; Yuan, S.; Yuan, Y. Excitation-independent dual-color carbon dots: Surface-state controlling and solid-state lighting. ACS Photonics 2017, 4, 2352–2358. [Google Scholar] [CrossRef]
- Gao, D.; Liu, X.; Jiang, D.; Zhao, H.; Zhu, Y.; Chen, X.; Luo, H.; Fan, H.; Zhang, X. Exploring of multicolor emissive carbon dots with novel double emission mechanism. Sens. Actuators B Chem. 2018, 277, 373–380. [Google Scholar] [CrossRef]
- Wen, Z.-H.; Yin, X.-B. Excitation-independent carbon dots, from photoluminescence mechanism to single-color application. RSC Adv. 2016, 6, 27829–27835. [Google Scholar] [CrossRef]
- Chizhik, A.M.; Stein, S.; Dekaliuk, M.O.; Battle, C.; Li, W.; Huss, A.; Platen, M.; Schaap, I.; Gregor, I.; Demchenko, A.P. Super-resolution optical fluctuation bio-imaging with dual-color carbon nanodots. Nano Lett. 2016, 16, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Xu, H.; Fu, Y. Photon Reabsorption and Nonradiative Energy-Transfer-Induced Quenching of Blue Photoluminescence from Aggregated Graphene Quantum Dots. J. Phys. Chem. C 2016, 120, 29432–29438. [Google Scholar] [CrossRef]
- Liu, X.; Li, H.-B.; Shi, L.; Meng, X.; Wang, Y.; Chen, X.; Xu, H.; Zhang, W.; Fang, X.; Ding, T. Structure and photoluminescence evolution of nanodots during pyrolysis of citric acid: From molecular nanoclusters to carbogenic nanoparticles. J. Mater. Chem. C 2017, 5, 10302–10312. [Google Scholar] [CrossRef]
- Wei, X.; Mei, S.; Yang, D.; Zhang, G.; Xie, F.; Zhang, W.; Guo, R. Surface States Induced Photoluminescence Enhancement of Nitrogen-Doped Carbon Dots via Post-Treatments. Nanoscale Res. Lett. 2019, 14, 172. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Sun, J.; Sun, X.; Song, R.; Xie, Z.; Zhou, S. Precisely Controlled Up/Down-Conversion Liquid and Solid State Photoluminescence of Carbon Dots. Adv. Opt. Mater. 2018, 6, 1800115. [Google Scholar] [CrossRef]
- Reckmeier, C.J.; Wang, Y.; Zboril, R.; Rogach, A.L. Influence of Doping and Temperature on Solvatochromic Shifts in Optical Spectra of Carbon Dots. J. Phys. Chem. C 2016, 120, 10591–10604. [Google Scholar] [CrossRef]
- Sciortino, A.; Marino, E.; Dam, B.v.; Schall, P.; Cannas, M.; Messina, F. Solvatochromism unravels the emission mechanism of carbon nanodots. J. Phys. Chem. Lett. 2016, 7, 3419–3423. [Google Scholar] [CrossRef] [PubMed]
- Jing, P.; Han, D.; Li, D.; Zhou, D.; Zhang, L.; Zhang, H.; Shen, D.; Qu, S. Origin of Anisotropic Photoluminescence in Heteroatom-Doped Carbon Nanodots. Adv. Opt. Mater. 2017, 5, 1601049. [Google Scholar] [CrossRef]
- Sciortino, A.; Madonia, A.; Gazzetto, M.; Sciortino, L.; Rohwer, E.J.; Feurer, T.; Gelardi, F.; Cannas, M.; Cannizzo, A.; Messina, F. The interaction of photoexcited Carbon nanodots with metal ions disclosed down to the femtosecond scale. Nanoscale 2017, 9, 11902–11911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, W.; Levy, D.H. Excimer/exciplex formation in van der Waals dimers of aromatic molecules. J. Phys. Chem. 1996, 100, 11539–11545. [Google Scholar] [CrossRef]
- Stevens, B. Some effects of molecular orientation on fluorescence emission and energy transfer in crystalline aromatic hydrocarbons. Spectrochim. Acta 1962, 18, 439–448. [Google Scholar] [CrossRef]
- Kim, Y.; Bouffard, J.; Kooi, S.E.; Swager, T.M. Highly emissive conjugated polymer excimers. J. Am. Chem. Soc. 2005, 127, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P.; Tang, K.C.; Chou, P.T. Excited-state proton coupled charge transfer modulated by molecular structure and media polarization. Chem. Soc. Rev. 2013, 42, 1379–1408. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P. Collective effects influencing fluorescence emission. Adv. Fluoresc. Report. Chem. Biol. II 2010, 9, 107–132. [Google Scholar]
- Khan, S.; Gupta, A.; Verma, N.C.; Nandi, C.K. Time-resolved emission reveals ensemble of emissive states as the origin of multicolor fluorescence in carbon dots. Nano Lett. 2015, 15, 8300–8305. [Google Scholar] [CrossRef] [PubMed]
- LeCroy, G.E.; Messina, F.; Sciortino, A.; Bunker, C.E.; Wang, P.; Fernando, K.S.; Sun, Y.-P. Characteristic Excitation Wavelength Dependence of Fluorescence Emissions in Carbon “Quantum” Dots. J. Phys. Chem. C 2017, 121, 28180–28186. [Google Scholar] [CrossRef]
- Mishra, K.; Koley, S.; Ghosh, S. Ground-State Heterogeneity along with Fluorescent Byproducts Causes Excitation-Dependent Fluorescence and Time-Dependent Spectral Migration in Citric Acid-Derived Carbon Dots. J. Phys. Chem. Lett. 2019, 10, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Meng, Q.; Wang, L.; Zhang, J.; Song, Y.; Jin, H.; Zhang, K.; Sun, H.; Wang, H.; Yang, B. Highly photoluminescent carbon dots for multicolor patterning, sensors, and bioimaging. Angew. Chem. Int. Ed. 2013, 52, 3953–3957. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Gadly, T.; Gupta, A.; Ballal, A.; Ghosh, S.K.; Kumbhakar, M. Origin of excitation dependent fluorescence in carbon nanodots. J. Phys. Chem. Lett. 2016, 7, 3695–3702. [Google Scholar] [CrossRef] [PubMed]
- Cushing, S.K.; Li, M.; Huang, F.; Wu, N. Origin of strong excitation wavelength dependent fluorescence of graphene oxide. ACS Nano 2013, 8, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P. Weber’s Red-Edge Effect that Changed the Paradigm in Photophysics and Photochemistry. In Perspectives on Fluorescence; Springer: Berlin/Heidelberg, Germany, 2016; pp. 95–141. [Google Scholar]
- Gan, Z.; Xu, H.; Hao, Y. Mechanism for excitation-dependent photoluminescence from graphene quantum dots and other graphene oxide derivates: Consensus, debates and challenges. Nanoscale 2016, 8, 7794–7807. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Shao, J.; Chen, C.; Li, H.; Wang, R.; Chi, Y.; Lin, X.; Chen, G. Blue luminescent graphene quantum dots and graphene oxide prepared by tuning the carbonization degree of citric acid. Carbon 2012, 50, 4738–4743. [Google Scholar] [CrossRef]
- Vinci, J.C.; Ferrer, I.M.; Seedhouse, S.J.; Bourdon, A.K.; Reynard, J.M.; Foster, B.A.; Bright, F.V.; Colón, L.A. Hidden Properties of Carbon Dots Revealed After HPLC Fractionation. J. Phys. Chem. Lett. 2012, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; He, B.; Huang, J. Amphibious fluorescent carbon dots: One-step green synthesis and application for light-emitting polymer nanocomposites. Chem. Commun. 2013, 49, 8078–8080. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zhang, X.; Li, D.; Zhou, D.; Jing, P.; Shen, D.; Qu, S.; Zboril, R.; Rogach, A.L. Full-color inorganic carbon dot phosphors for white-light-emitting diodes. Adv. Opt. Mater. 2017, 5, 1700416. [Google Scholar] [CrossRef]
- Feng, T.; Zeng, Q.; Lu, S.; Yan, X.; Liu, J.; Tao, S.; Yang, M.; Yang, B. Color-tunable carbon dots possessing solid-state emission for full-color light-emitting diodes applications. ACS Photonics 2017, 5, 502–510. [Google Scholar] [CrossRef]
- Mei, S.; Wei, X.; Hu, Z.; Wei, C.; Su, D.; Yang, D.; Zhang, G.; Zhang, W.; Guo, R. Amphipathic carbon dots with solvent-dependent optical properties and sensing application. Opt. Mater. 2019, 89, 224–230. [Google Scholar] [CrossRef]
- Demchenko, A.P. Introduction to Fluorescence Sensing; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Mukherjee, S.; Prasad, E.; Chadha, A. H-Bonding controls the emission properties of functionalized carbon nano-dots. Phys. Chem. Chem. Phys. 2017, 19, 7288–7296. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, A.P.; Yesylevskyy, S.O. Interfacial behavior of fluorescent dyes. In Advance Fluorescence Reporters in Chemical and Biology III; Springer: Berlin/Heidelberg, Germany, 2011; pp. 3–62. [Google Scholar]
- Basu, N.; Mandal, D. Solvatochromic Response of Carbon Dots: Evidence of Solvent Interaction with Different Types of Emission Centers. J. Phys. Chem. C 2018, 122, 18732–18741. [Google Scholar] [CrossRef]
- Das, A.; Roy, D.; De, C.K.; Mandal, P.K. “Where does the fluorescing moiety reside in a carbon dot?”–Investigations based on fluorescence anisotropy decay and resonance energy transfer dynamics. Phys. Chem. Chem. Phys. 2018, 20, 2251–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, B.; Nie, H.; Liu, Z.; Xu, H.; Li, M.; Wu, C.; Wang, H.; Zhang, S.X.-A. Full-colour carbon dots: Integration of multiple emission centres into single particles. Nanoscale 2017, 9, 13326–13333. [Google Scholar] [CrossRef] [PubMed]
- Gharat, P.M.; Chethodil, J.M.; Srivastava, A.P.; Praseetha, P.; Pal, H.; Choudhury, S.D. An insight into the molecular and surface state photoluminescence of carbon dots revealed through solvent-induced modulations in their excitation wavelength dependent emission properties. PhotoChem. Photobiol. Sci. 2019, 18, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; An, X.; Gong, J. Novel pH sensitive N-doped carbon dots with both long fluorescence lifetime and high quantum yield. RSC Adv. 2015, 5, 32319–32322. [Google Scholar] [CrossRef]
- Hou, J.; Wang, W.; Zhou, T.; Wang, B.; Li, H.; Ding, L. Synthesis and formation mechanistic investigation of nitrogen-doped carbon dots with high quantum yields and yellowish-green fluorescence. Nanoscale 2016, 8, 11185–11193. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, Y.; Wagner, W.; Stepanenko, V.; Ren, X.; Ogi, S.; Würthner, F. Near-IR Absorbing J-Aggregate of an Amphiphilic BF2-Azadipyrromethene Dye by Kinetic Cooperative Self-Assembly. Angew. Chem. Int. Ed. 2017, 56, 5729–5733. [Google Scholar] [CrossRef] [PubMed]
- Knox, R.S. Theory Excitons; Academic Press: New York, NY, USA, 1972. [Google Scholar]
- Bardeen, C.J. The structure and dynamics of molecular excitons. Annu. Rev. Phys. Chem. 2014, 65, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Scholes, G.D.; Rumbles, G. Excitons in nanoscale systems. Nat. Mater. 2006, 5, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Takagahara, T.; Takeda, K. Theory of the quantum confinement effect on excitons in quantum dots of indirect-gap materials. Phys. Rev. B 1992, 46, 15578. [Google Scholar] [CrossRef] [PubMed]
- Medintz, I.L.; Uyeda, H.T.; Goldman, E.R.; Mattoussi, H. Quantum dot bioconjugates for imaging, labelling and sensing. Nat. Mater. 2005, 4, 435. [Google Scholar] [CrossRef] [PubMed]
- Sapsford, K.; Pons, T.; Medintz, I.; Mattoussi, H. Biosensing with luminescent semiconductor quantum dots. Sensors 2006, 6, 925–953. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Zhao, Y.; Ullah, N.; Wan, Q.; Zhang, R. Revealing the trap emission in graphene-based nanostructures. Carbon 2019, 5, 5984–5993. [Google Scholar] [CrossRef]
- Feng, J.; Dong, H.; Yu, L.; Dong, L. The optical and electronic properties of graphene quantum dots with oxygen-containing groups: A density functional theory study. J. Mater. Chem. C 2017, 5, 5984–5993. [Google Scholar] [CrossRef]
- Wang, L.; Wang, H.Y.; Wang, Y.; Zhu, S.J.; Zhang, Y.L.; Zhang, J.H.; Chen, Q.D.; Han, W.; Xu, H.L.; Yang, B. Direct Observation of Quantum-Confined Graphene-Like States and Novel Hybrid States in Graphene Oxide by Transient Spectroscopy. Adv. Mater. 2013, 25, 6539–6545. [Google Scholar] [CrossRef] [PubMed]
- Sciortino, A.; Gazzetto, M.; Buscarino, G.; Popescu, R.; Schneider, R.; Giammona, G.; Gerthsen, D.; Rohwer, E.J.; Mauro, N.; Feurer, T. Disentangling size effects and spectral inhomogeneity in carbon nanodots by ultrafast dynamical hole-burning. Nanoscale 2018, 10, 15317–15323. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Song, Y.; Wang, J.; Wan, H.; Zhang, Y.; Ning, Y.; Yang, B. Photoluminescence mechanism in graphene quantum dots: Quantum confinement effect and surface/edge state. Nano Today 2017, 13, 10–14. [Google Scholar] [CrossRef]
- Umari, P.; Mosconi, E.; De Angelis, F. Infrared dielectric screening determines the low exciton binding energy of metal-halide perovskites. J. Phys. Chem. Lett. 2018, 9, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Soavi, G.; Scotognella, F.; Lanzani, G.; Cerullo, G. Ultrafast Photophysics of Single-Walled Carbon Nanotubes. Adv. Opt. Mater. 2016, 4, 1670–1688. [Google Scholar] [CrossRef] [Green Version]
- Chizhik, A.I.; Chizhik, A.M.; Khoptyar, D.; Bär, S.; Meixner, A.J. Excitation isotropy of single CdSe/ZnS nanocrystals. Nano Lett. 2011, 11, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Davydov, A.S. Theory Molecular Excitons; Plenum press: New York, NY, USA, 1971. [Google Scholar]
- Broude, V.L.V.; Rashba, E.I.; Sheka, E.F. Spectroscopy of Molecular Excitons; Springer: Berlin/Heidelberg, Germany, 1985. [Google Scholar]
- Saikin, S.K.; Eisfeld, A.; Valleau, S.; Aspuru-Guzik, A. Photonics meets excitonics: Natural and artificial molecular aggregates. Nanophotonics 2013, 2, 21–38. [Google Scholar] [CrossRef] [Green Version]
- Kasha, M. Energy transfer mechanisms and the molecular exciton model for molecular aggregates. Radiat. Res. 1963, 20, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Kasha, M.; Rawls, H.; Ashraf El-Bayoumi, M. The exciton model in molecular spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. [Google Scholar] [CrossRef] [Green Version]
- McRae, E.G.; Kasha, M. Enhancement of phosphorescence ability upon aggregation of dye molecules. J. Chem. Phys. 1958, 28, 721–722. [Google Scholar] [CrossRef]
- Hochstrasser, R.M.; Kasha, M. Application of the exciton model to mono-molecular lamellar systems. PhotoChem. Photobiol. 1964, 3, 317–331. [Google Scholar] [CrossRef]
- Bricks, J.L.; Slominskii, Y.L.; Panas, I.D.; Demchenko, A.P. Fluorescent J-aggregates of cyanine dyes: Basic research and applications review. Methods Appl. Fluoresc. 2017, 6, 012001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, H.; Ashiba, K. Highly fluorescent organic nanoparticles of thiacyanine dye: A synergetic effect of intermolecular H-aggregation and restricted intramolecular rotation. RSC Adv. 2011, 1, 834–838. [Google Scholar] [CrossRef]
- Rösch, U.; Yao, S.; Wortmann, R.; Würthner, F. Fluorescent H-Aggregates of Merocyanine Dyes. Angew. Chem. 2006, 118, 7184–7188. [Google Scholar] [CrossRef]
- Spano, F.C. The spectral signatures of Frenkel polarons in H-and J-aggregates. Acc. Chem. Res. 2009, 43, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, S.B.; Yakunin, S.; Peng, C.; Vismara, M.V.G.; Graeff, C.F.; Nüesch, F.A.; Jenatsch, S.; Hany, R.; Kovalenko, M.V.; Heier, J. Strongly Red-Shifted Photoluminescence Band Induced by Molecular Twisting in Cyanine (Cy3) Dye Films. J. Phys. Chem. C 2017, 121, 9587–9593. [Google Scholar] [CrossRef]
- Sharma, A.; Gadly, T.; Neogy, S.; Ghosh, S.K.; Kumbhakar, M. Molecular Origin and Self-Assembly of Fluorescent Carbon Nanodots in Polar Solvents. J. Phys. Chem. Lett. 2017, 8, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Spano, F.C.; Silva, C. H-and J-aggregate behavior in polymeric semiconductors. Annu. Rev. Phys. Chem. 2014, 65, 477–500. [Google Scholar] [CrossRef] [PubMed]
- Hestand, N.J.; Spano, F.C. Expanded theory of H-and J-molecular aggregates: The effects of vibronic coupling and intermolecular charge transfer. Chem. Rev. 2018, 118, 7069–7163. [Google Scholar] [CrossRef] [PubMed]
- Brixner, T.; Hildner, R.; Köhler, J.; Lambert, C.; Würthner, F. Exciton Transport in Molecular Aggregates–From Natural Antennas to Synthetic Chromophore Systems. Adv. Energy Mater. 2017, 7, 1700236. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Shen, Y.; Yang, M.; Zhang, X.; Xu, W.; Zhu, Q.; Wu, J.; Tian, Y.; Zhou, H. Novel highly emissive H-aggregates with aggregate fluorescence change in a phenylbenzoxazole-based system. Chem. Commun. 2014, 50, 8723–8726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafov, H.; Lee, B.; Zhou, Q.; Feldman, L.C.; Podzorov, V. Observation of long-range exciton diffusion in highly ordered organic semiconductors. Nat. Mater. 2010, 9, 938–943. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Yuan, W.; Jia, Z.; Liu, G. H- and J-aggregation of fluorene-based chromophores. J. Phys. Chem. B 2014, 118, 14536–14545. [Google Scholar] [CrossRef] [PubMed]
- Lau, V.; Heyne, B. Calix[4]arene sulfonate as a template for forming fluorescent thiazole orange H-aggregates. Chem. Commun. 2010, 46, 3595–3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, A.; Settels, V.; Liu, W.; Würthner, F.; Meier, C.; Fink, R.F.; Schindlbeck, S.; Lochbrunner, S.; Engels, B.; Engel, V. Ultrafast exciton self-trapping upon geometry deformation in perylene-based molecular aggregates. J. Phys. Chem. Lett. 2013, 4, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Spano, F.; Silvestri, L. Multiple mode exciton-vibrational coupling in H-aggregates: Synergistic enhancement of the quantum yield. J. Chem. Phys. 2010, 132, 094704. [Google Scholar] [CrossRef] [PubMed]
- Chenu, A.; Scholes, G.D. Coherence in energy transfer and photosynthesis. Annu. Rev. Phys. Chem. 2015, 66, 69–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholes, G.D.; Fleming, G.R.; Chen, L.X.; Aspuru-Guzik, A.; Buchleitner, A.; Coker, D.F.; Engel, G.S.; Van Grondelle, R.; Ishizaki, A.; Jonas, D.M. Using coherence to enhance function in chemical and biophysical systems. Nature 2017, 543, 647. [Google Scholar] [CrossRef] [PubMed]
- Walschaers, M.; Schlawin, F.; Wellens, T.; Buchleitner, A. Quantum transport on disordered and noisy networks: An interplay of structural complexity and uncertainty. Annu. Rev. Condens. Matter Phys. 2016, 7, 223–248. [Google Scholar] [CrossRef]
- Lloyd, S.; Mohseni, M. Symmetry-enhanced supertransfer of delocalized quantum states. New J. Phys. 2010, 12, 075020. [Google Scholar] [CrossRef]
- Mirkovic, T.; Ostroumov, E.E.; Anna, J.M.; Van Grondelle, R.; Scholes, G.D. Light absorption and energy transfer in the antenna complexes of photosynthetic organisms. Chem. Rev. 2016, 117, 249–293. [Google Scholar] [CrossRef] [PubMed]
- Collini, E.; Scholes, G.D. Coherent intrachain energy migration in a conjugated polymer at room temperature. Science 2009, 323, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Cheng, Y.-C.; Fleming, G.R. Coherence dynamics in photosynthesis: Protein protection of excitonic coherence. Science 2007, 316, 1462–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramavicius, D.; Palmieri, B.; Voronine, D.V.; Sanda, F.; Mukamel, S. Coherent multidimensional optical spectroscopy of excitons in molecular aggregates; quasiparticle versus supermolecule perspectives. Chem. Rev. 2009, 109, 2350–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.; Cichos, F.; Huisken, F.; von Borczyskowski, C. Electron–phonon coupling and localization of excitons in single silicon nanocrystals. Nano Lett. 2008, 8, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, A.; Schmidt, A.; Valencia, V.; Fluegel, B.; Mazumdar, S.; Armstrong, N.; Peyghambarian, N. Evidence for exciton-exciton binding in a molecular aggregate. Phys. Rev. B 1998, 57, R4206. [Google Scholar] [CrossRef]
- D’Avino, G.; Terenziani, F.; Painelli, A. Aggregates of quadrupolar dyes: Giant two-photon absorption from biexciton states. J. Phys. Chem. B 2006, 110, 25590–25592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, L.; Yuan, Y.; Jiang, J.; Yu, S.-H. One-pot gram-scale synthesis of nitrogen and sulfur embedded organic dots with distinctive fluorescence behaviors in free and aggregated states. Chem. Mater. 2016, 28, 4367–4374. [Google Scholar] [CrossRef]
- Agranovich, V.M. Excitations in Organic Solids; OUP Oxford: Oxford, UK, 2009; Volume 142. [Google Scholar]
- Matsui, A.H. Excitonic processes in aromatic molecular crystals of strong exciton-phonon coupling. Pure Appl. Chem. 1995, 67, 429–436. [Google Scholar] [CrossRef]
- Jursenas, S.; Gruodis, A.; Kodis, G.; Chachisvilis, M.; Gulbinas, V.; Silinsh, E.; Valkunas, L. Free and self-trapped charge-transfer excitons in crystals of dipolar molecules of N, N-dimethylaminobenzylidene 1, 3-indandione. J. Phys. Chem. B 1998, 102, 1086–1094. [Google Scholar] [CrossRef]
- Rashba, E.; Sturge, M. (Eds.) Excitons, v. 2; Amsterdam, The Netherlands, 1982. [Google Scholar]
- Song, K.; Williams, R.T. Self-Trapped Excitons; Springer: Berlin/Heidelberg, Germany, 1996; Volume 105. [Google Scholar]
- Rashba, E.I. Theory of strong interactions of electron excitations with lattice vibrations in molecular crystals. 2 Opt. Spektrosk. 1957, 2, 88–98. [Google Scholar]
- Pekar, S.I.; Rashba, E.I.; Sheka, V.I. Free and self-localized Wannier-Mott excitons in ionic-crystals and the activation-energy of their mutual thermal conversion. J. Eksp. Theor. Phys. 1979, 49, 129. [Google Scholar]
- Baessler, H.; Schweitzer, B. Site-selective fluorescence spectroscopy of conjugated polymers and oligomers. Acc. Chem. Res. 1999, 32, 173–182. [Google Scholar] [CrossRef]
- Fidder, H.; Knoester, J.; Wiersma, D.A. Optical properties of disordered molecular aggregates: A numerical study. J. Chem. Phys. 1991, 95, 7880–7890. [Google Scholar] [CrossRef] [Green Version]
- Bialas, D.; Bruening, C.; Schlosser, F.; Fimmel, B.; Thein, J.; Engel, V.; Wuerthner, F. Exciton-Vibrational Couplings in Homo-and Heterodimer Stacks of Perylene Bisimide Dyes within Cyclophanes: Studies on Absorption Properties and Theoretical Analysis. Chem. Eur. J. 2016, 22, 15011–15018. [Google Scholar] [CrossRef] [PubMed]
- Bialas, D.; Zitzler-Kunkel, A.; Kirchner, E.; Schmidt, D.; Würthner, F. Structural and quantum chemical analysis of exciton coupling in homo-and heteroaggregate stacks of merocyanines. Nat. Commun. 2016, 7, 12949. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, J.; Hinderhofer, A.; Broch, K.; Heinemeyer, U.; Kowarik, S.; Vorobiev, A.; Gerlach, A.; Schreiber, F. Structural and Optical Properties of Mixed Diindenoperylene–Perfluoropentacene Thin Films. J. Phys. Chem. C 2012, 116, 10917–10923. [Google Scholar] [CrossRef]
- Winiger, C.B.; Langenegger, S.M.; Calzaferri, G.; Häner, R. Formation of Two Homo-chromophoric H-Aggregates in DNA-Assembled Alternating Dye Stacks. Angew. Chem. Int. Ed. 2015, 54, 3643–3647. [Google Scholar] [CrossRef] [PubMed]
- Franco, I.; Tretiak, S. Electron-vibrational dynamics of photoexcited polyfluorenes. J. Am. Chem. Soc. 2004, 126, 12130–12140. [Google Scholar] [CrossRef] [PubMed]
- Berezin, M.Y.; Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev. 2010, 110, 2641–2684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Xu, M.; Liu, Y.; He, F.; Gao, F.; Su, Y.; Wei, H.; Zhang, Y. Nitrogen-doped, carbon-rich, highly photoluminescent carbon dots from ammonium citrate. Nanoscale 2014, 6, 1890–1895. [Google Scholar] [CrossRef] [PubMed]
- Kalytchuk, S.; Poláková, K.i.; Wang, Y.; Froning, J.P.; Cepe, K.; Rogach, A.L.; Zbořil, R. Carbon dot nanothermometry: Intracellular photoluminescence lifetime thermal sensing. ACS Nano 2017, 11, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Gude, V.; Roy, D.; Chatterjee, T.; De, C.K.; Mandal, P.K. On the molecular origin of photoluminescence of nonblinking carbon dot. J. Phys. Chem. C 2017, 121, 9634–9641. [Google Scholar] [CrossRef]
- Guo, R.; Li, T.; Shi, S. Aggregation-induced emission enhancement of carbon quantum dots and applications in light emitting devices. J. Mater. Chem. C 2019, 7, 5148–5154. [Google Scholar] [CrossRef]
- Long, P.; Feng, Y.; Cao, C.; Li, Y.; Han, J.; Li, S.; Peng, C.; Li, Z.; Feng, W. Self-Protective Room-Temperature Phosphorescence of Fluorine and Nitrogen Codoped Carbon Dots. Adv. Funct. Mater. 2018, 28, 1800791. [Google Scholar] [CrossRef]
- Gao, Y.; Han, H.; Lu, W.; Jiao, Y.; Liu, Y.; Gong, X.; Xian, M.; Shuang, S.; Dong, C. Matrix-free and highly efficient room-temperature phosphorescence of Nitrogen-doped carbon dots. Langmuir 2018, 34, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Ye, Y.; Ren, X.; Zhao, W.; Yue, D. High pH-induced efficient room-temperature phosphorescence from carbon dots in hydrogen-bonded matrices. J. Mater. Chem. C 2018, 6, 7890–7895. [Google Scholar] [CrossRef]
- Gui, R.; Jin, H.; Wang, Z.; Tan, L. Recent advances in optical properties and applications of colloidal quantum dots under two-photon excitation. Coord. Chem. Rev. 2017, 338, 141–185. [Google Scholar] [CrossRef]
- Fassioli, F.; Dinshaw, R.; Arpin, P.C.; Scholes, G.D. Photosynthetic light harvesting: Excitons and coherence. J. R. Soc. Interface 2014, 11, 20130901. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Snaider, J.M.; Yuan, L.; Huang, L. Ultrafast Dynamic Microscopy of Carrier and Exciton Transport. Annu. Rev. Phys. Chem. 2019, 70, 219–244. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Helgeson, R.; Jones, R.M.; McBranch, D.; Whitten, D. Superquenching in cyanine pendant poly (L-lysine) dyes: Dependence on molecular weight, solvent, and aggregation. J. Am. Chem. Soc. 2002, 124, 483–488. [Google Scholar] [CrossRef] [PubMed]
- De Boer, S.; Wiersma, D.A. Optical dynamics of exciton and polaron formation in molecular aggregates. Chem. Phys. 1989, 131, 135–144. [Google Scholar] [CrossRef]
- Hou, Y.; Lu, Q.; Deng, J.; Li, H.; Zhang, Y. One-pot electrochemical synthesis of functionalized fluorescent carbon dots and their selective sensing for mercury ion. Anal. Chim. Acta 2015, 866, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.; Sadhukhan, M. Facile bulk production of highly blue fluorescent graphitic carbon nitride quantum dots and their application as highly selective and sensitive sensors for the detection of mercuric and iodide ions in aqueous media. J. Mater. Chem. 2012, 22, 21832–21837. [Google Scholar] [CrossRef]
- Gao, X.; Du, C.; Zhuang, Z.; Chen, W. Carbon quantum dot-based nanoprobes for metal ion detection. J. Mater. Chem. C 2016, 4, 6927–6945. [Google Scholar] [CrossRef]
- Dutta Chowdhury, A.; Doong, R.-A. Highly sensitive and selective detection of nanomolar ferric ions using dopamine functionalized graphene quantum dots. ACS Appl. Mater. Interfaces 2016, 8, 21002–21010. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Sahu, S.; Anilkumar, P.; Bunker, C.E.; Xu, J.; Fernando, K.S.; Wang, P.; Guliants, E.A.; Tackett, K.N.; Sun, Y.-P. Carbon nanoparticles as visible-light photocatalysts for efficient CO2 conversion and beyond. J. Am. Chem. Soc. 2011, 133, 4754–4757. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Sahu, S.; Cao, L.; Bunker, C.E.; Peng, G.; Liu, Y.; Fernando, K.S.; Wang, P.; Guliants, E.A.; Meziani, M.J. Efficient Fluorescence Quenching in Carbon Dots by Surface-Doped Metals-Disruption of Excited State Redox Processes and Mechanistic Implications. Langmuir 2012, 28, 16141–16147. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Yu, J.; Wu, C.; Szymanski, C.; McNeill, J. Amplified energy transfer in conjugated polymer nanoparticle tags and sensors. Nanoscale 2010, 2, 1999–2011. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; McNeill, J. Light-harvesting and amplified energy transfer in conjugated polymer nanoparticles. Chem. Rev. 2016, 117, 838–859. [Google Scholar] [CrossRef] [PubMed]
- Lebedenko, A.N.; Grynyov, R.S.; Guralchuk, G.Y.; Sorokin, A.V.; Yefimova, S.L.; Malyukin, Y.V. Coherent mechanism of exciton transport in disordered J-aggregates. J. Phys. Chem. C 2009, 113, 12883–12887. [Google Scholar] [CrossRef]
- Jones, R.M.; Bergstedt, T.S.; Buscher, C.T.; McBranch, D.; Whitten, D. Superquenching and its applications in J-aggregated cyanine polymers. Langmuir 2001, 17, 2568–2571. [Google Scholar] [CrossRef]
- Xie, X.S.; Trautman, J.K. Optical studies of single molecules at room temperature. Annu. Rev. Phys. Chem. 1998, 49, 441–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, D.; Zheng, M.; Zhang, L.; Zhao, H.; Xie, Z.; Jing, X.; Haddad, R.E.; Fan, H.; Sun, Z. Formation mechanism and optimization of highly luminescent N-doped graphene quantum dots. Sci. Rep. 2014, 4, 5294. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Liu, Y.; Yeom, S.; Kim, D.Y.; Richards, C.I. Single-Particle Fluorescence Intensity Fluctuations of Carbon Nanodots. Nano Lett. 2014, 14, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Damm, C.; Walter, J.; Nacken, T.J.; Peukert, W. Photobleaching and stabilization of carbon nanodots produced by solvothermal synthesis. Phys. Chem. Chem. Phys. 2016, 18, 466–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Verma, N.C.; Gupta, A.; Nandi, C.K. Reversible photoswitching of carbon dots. Sci. Rep. 2015, 5, 11423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitten, D.; Chen, L.; Jones, R.; Bergstedt, T.; Heeger, P.; McBranch, D. From superquenching to biodetection: Building sensors based on fluorescent polyelectrolytes. Mol. Supramol. Photochem. 2001, 7, 189–208. [Google Scholar]
- Fathi, P.; Khamo, J.S.; Huang, X.; Srivastava, I.; Esch, M.B.; Zhang, K.; Pan, D. Bulk-state and single-particle imaging are central to understanding carbon dot photo-physics and elucidating the effects of precursor composition and reaction temperature. Carbon 2019, 145, 572–585. [Google Scholar] [CrossRef]
- Misra, S.K.; Srivastava, I.; Khamo, J.S.; Krishnamurthy, V.V.; Sar, D.; Schwartz-Duval, A.S.; Soares, J.A.; Zhang, K.; Pan, D. Carbon dots with induced surface oxidation permits imaging at single-particle level for intracellular studies. Nanoscale 2018, 10, 18510–18519. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.C.; Rao, C.; Nandi, C.K. Nitrogen-doped biocompatible carbon dot as a fluorescent probe for STORM nanoscopy. J. Phys. Chem. C 2018, 122, 4704–4709. [Google Scholar] [CrossRef]
- Khan, S.; Li, W.; Karedla, N.; Thiart, J.; Gregor, I.; Chizhik, A.M.; Enderlein, J.; Nandi, C.K.; Chizhik, A.I. Charge-driven fluorescence blinking in carbon nanodots. J. Phys. Chem. Lett. 2017, 8, 5751–5757. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Novotny, L. Robustness of quantum dot power-law blinking. Nano Lett. 2011, 11, 2137–2141. [Google Scholar] [CrossRef] [PubMed]
- Frantsuzov, P.A.; Volkán-Kacsó, S.N.; Jankó, B.R. Universality of the fluorescence intermittency in nanoscale systems: Experiment and theory. Nano Lett. 2013, 13, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Porter, G.; Sadkowski, P.; Tredwell, C. Picosecond rotational diffusion in kinetic and steady state fluorescence spectroscopy. Chem. Phys. Lett. 1977, 49, 416–420. [Google Scholar] [CrossRef]
- Chipem, F.A.; Krishnamoorthy, G. Temperature effect on dual fluorescence of 2-(2′-Hydroxyphenyl) benzimidazole and its nitrogen substituted analogues. J. Phys. Chem. B. 2013, 117, 14079–14088. [Google Scholar] [CrossRef] [PubMed]
- Elsaesser, T.; Kaiser, W. Vibrational and vibronic relaxation of large polyatomic molecules in liquids. Annu. Rev. Phys. Chem. 1991, 42, 83–107. [Google Scholar] [CrossRef]
- Pandey, A.; Guyot-Sionnest, P. Slow electron cooling in colloidal quantum dots. Science 2008, 322, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Fu, J.; Xu, Q.; Sum, T.C. Slow Hot-Carrier Cooling in Halide Perovskites: Prospects for Hot-Carrier Solar Cells. Adv. Mater. 2019, 1802486. [Google Scholar] [CrossRef] [PubMed]
- Patra, D.; Gregor, I.; Enderlein, J. Image analysis of defocused single-molecule images for three-dimensional molecule orientation studies. J. Phys. Chem. A 2004, 108, 6836–6841. [Google Scholar] [CrossRef] [Green Version]
- Agranovich, V.; Zakhidov, A. Interaction of charge transfer excitons with phonons in molecular crystals. Chem. Phys. Lett. 1977, 50, 278–281. [Google Scholar] [CrossRef]
- Hoffmann, M.; Schmidt, K.; Fritz, T.; Hasche, T.; Agranovich, V.M.; Leo, K. The lowest energy Frenkel and charge-transfer excitons in quasi-one-dimensional structures: Application to MePTCDI and PTCDA crystals. Chem. Phys. 2000, 258, 73–96. [Google Scholar] [CrossRef]
- Luo, P.G.; Yang, F.; Yang, S.-T.; Sonkar, S.K.; Yang, L.; Broglie, J.J.; Liu, Y.; Sun, Y.-P. Carbon-based quantum dots for fluorescence imaging of cells and tissues. Rsc Adv. 2014, 4, 10791–10807. [Google Scholar] [CrossRef]
- Cao, L.; Wang, X.; Meziani, M.J.; Lu, F.; Wang, H.; Luo, P.G.; Lin, Y.; Harruff, B.A.; Veca, L.M.; Murray, D.; et al. Carbon dots for multiphoton bioimaging. J. Am. Chem. Soc. 2007, 129, 11318–11319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, G.; Wang, J.; Wang, R.; Guo, X.; He, L.; Qiu, F.; Wang, G.; Zhu, B.; Zhu, X.; Liu, T. Amorphous carbon dots with high two-photon fluorescence for cellular imaging passivated by hyperbranched poly (amino amine). J. Mater. Chem. B 2015, 3, 700–706. [Google Scholar] [CrossRef]
- He, G.S.; Tan, L.-S.; Zheng, Q.; Prasad, P.N. Multiphoton absorbing materials: Molecular designs, characterizations, and applications. Chem. Rev. 2008, 108, 1245–1330. [Google Scholar] [CrossRef] [PubMed]
- Makarov, N.S.; Drobizhev, M.; Rebane, A. Two-photon absorption standards in the 550–1600 nm excitation wavelength range. Opt. Express 2008, 16, 4029–4047. [Google Scholar] [CrossRef] [PubMed]
- Spano, F.C.; Mukamel, S. Excitons in confined geometries: Size scaling of nonlinear susceptibilities. J. Chem. Phys. 1991, 95, 7526–7540. [Google Scholar] [CrossRef] [Green Version]
- Collini, E. Cooperative effects to enhance two-photon absorption efficiency: Intra-versus inter-molecular approach. Phys. Chem. Chem. Phys. 2012, 14, 3725–3736. [Google Scholar] [CrossRef] [PubMed]
- Maurin, M.; Vurth, L.; Vial, J.-C.; Baldeck, P.; Marder, S.R.; Van der Sanden, B.; Stephan, O. Fluorescent Pluronic nanodots for in vivo two-photon imaging. Nanotechnology 2009, 20, 235102. [Google Scholar] [CrossRef] [PubMed]
- Cepraga, C.; Gallavardin, T.; Marotte, S.; Lanoë, P.-H.; Mulatier, J.-C.; Lerouge, F.; Parola, S.; Lindgren, M.; Baldeck, P.L.; Marvel, J. Biocompatible well-defined chromophore–polymer conjugates for photodynamic therapy and two-photon imaging. Polym. Chem. 2013, 4, 61–67. [Google Scholar] [CrossRef]
- Drobizhev, M.; Stepanenko, Y.; Dzenis, Y.; Karotki, A.; Rebane, A.; Taylor, P.N.; Anderson, H.L. Extremely strong near-IR two-photon absorption in conjugated porphyrin dimers: Quantitative description with three-essential-states model. J. Phys. Chem. B 2005, 109, 7223–7236. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Yan, L.; Zhao, Z.; Ma, X.; Guo, Q.; Jia, M.; Lu, P.; Ramos-Ortiz, G.; Maldonado, J.L.; Rodriguez, M. Gigantic two-photon absorption cross sections and strong two-photon excited fluorescence in pyrene core dendrimers with fluorene/carbazole as dendrons and acetylene as linkages. J. Phys. Chem. B 2010, 114, 11737–11745. [Google Scholar] [CrossRef] [PubMed]
- Ahn, T.K.; Kim, K.S.; Kim, D.Y.; Noh, S.B.; Aratani, N.; Ikeda, C.; Osuka, A.; Kim, D. Relationship between two-photon absorption and the π-conjugation pathway in porphyrin arrays through dihedral angle control. J. Am. Chem. Soc. 2006, 128, 1700–1704. [Google Scholar] [CrossRef] [PubMed]
- Mukamel, S. Principles Nonlinear Optical Spectroscopy; Oxford University Press: New York, NY, USA, 1995; Volume 29. [Google Scholar]
- Ariese, F.; Bader, A.N.; Gooijer, C. Fluorescence line-narrowing spectroscopy for probing purposes in bioanalytical and environmental chemistry. TrAC Trends Anal. Chem. 2008, 27, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Sciortino, A.; Cannas, M.; Messina, F. Temperature-Dependence of Solvent-Induced Stokes Shift and Fluorescence Tunability in Carbon Nanodots. C 2019, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Oncul, S.; Demchenko, A.P. The effects of thermal quenching on the excited-state intramolecular proton transfer reaction in 3-hydroxyflavones. Spectrochim. Acta Part Mol. Biomol. Spectrosc. 2006, 65, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Barragán, I.; Costa-Fernández, J.; Sanz-Medel, A.; Valledor, M.; Campo, J. Room-temperature phosphorescence (RTP) for optical sensing. TrAC Trends Anal. Chem. 2006, 25, 958–967. [Google Scholar] [CrossRef]
- Köhler, A.; Bässler, H. Triplet states in organic semiconductors. Mater. Sci. Eng. R Rep. 2009, 66, 71–109. [Google Scholar] [CrossRef]
- Lakowicz, J.R.; Weber, G. Quenching of fluorescence by oxygen. Probe for structural fluctuations in macromolecules. Biochemical 1973, 12, 4161–4170. [Google Scholar] [CrossRef] [PubMed]
- Lower, S.; El-Sayed, M. The triplet state and molecular electronic processes in organic molecules. Chem. Rev. 1966, 66, 199–241. [Google Scholar] [CrossRef]
- Tan, J.; Zou, R.; Zhang, J.; Li, W.; Zhang, L.; Yue, D. Large-scale synthesis of N-doped carbon quantum dots and their phosphorescence properties in a polyurethane matrix. Nanoscale 2016, 8, 4742–4747. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Wang, Y.; Gao, X.; Cai, C.; Lin, H. Facile, Quick, and Gram-Scale Synthesis of Ultralong-Lifetime Room-Temperature-Phosphorescent Carbon Dots by Microwave Irradiation. Angew. Chem. Int. Ed. 2018, 57, 6216–6220. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, M.; Yang, M.; Yang, Q.; Zhang, Z.; Shi, J. Induction of long-lived room temperature phosphorescence of carbon dots by water in hydrogen-bonded matrices. Nat. Commun. 2018, 9, 734. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wang, S.; Ji, Y.; Chen, R.; Zhu, Q.; Wang, Y.; Zheng, C.; Tao, Y.; Fan, Q.; Huang, W. Invoking ultralong room temperature phosphorescence of purely organic compounds through H-aggregation engineering. Mater. Horiz. 2019. [Google Scholar] [CrossRef]
- Lanzani, G.; Stagira, S.; Cerullo, G.; De Silvestri, S.; Comoretto, D.; Moggio, I.; Cuniberti, C.; Musso, G.; Dellepiane, G. Triplet exciton generation and decay in a red polydiacetylene studied by femtosecond spectroscopy. Chem. Phys. Lett. 1999, 313, 525–532. [Google Scholar] [CrossRef]
- An, Z.; Zheng, C.; Tao, Y.; Chen, R.; Shi, H.; Chen, T.; Wang, Z.; Li, H.; Deng, R.; Liu, X. Stabilizing triplet excited states for ultralong organic phosphorescence. Nat. Mater. 2015, 14, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Lucenti, E.; Forni, A.; Botta, C.; Carlucci, L.; Giannini, C.; Marinotto, D.; Previtali, A.; Righetto, S.; Cariati, E. H-Aggregates Granting Crystallization-Induced Emissive Behavior and Ultralong Phosphorescence from a Pure Organic Molecule. J. Phys. Chem. Lett. 2017, 8, 1894–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepien, M.; Gonka, E.; Żyła, M.; Sprutta, N. Heterocyclic nanographenes and other polycyclic heteroaromatic compounds: Synthetic routes, properties, and applications. Chem. Rev. 2016, 117, 3479–3716. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Z.; Zhang, X. Amphiphilic building blocks for self-assembly: From amphiphiles to supra-amphiphiles. Acc. Chem. Res. 2012, 45, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lohr, A.; Saha-Möller, C.R.; Würthner, F. Self-assembled π-stacks of functional dyes in solution: Structural and thermodynamic features. Chem. Soc. Rev. 2009, 38, 564–584. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Rao, K.V.; George, S.J. Supramolecular charge transfer nanostructures. Phys. Chem. Chem. Phys. 2014, 16, 1300–1313. [Google Scholar] [CrossRef] [PubMed]
- Guldi, D.M.; Prato, M. Electrostatic interactions by design. Versatile methodology towards multifunctional assemblies/nanostructured electrodes. Chem. Commun. 2004, 2517–2525. [Google Scholar]
- Würthner, F.; Saha-Möller, C.R.; Fimmel, B.; Ogi, S.; Leowanawat, P.; Schmidt, D. Perylene bisimide dye assemblies as archetype functional supramolecular materials. Chem. Rev. 2015, 116, 962–1052. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, H.; da Silva, J.C.E. Fluorescent carbon dots capped with PEG 200 and mercaptosuccinic acid. J. Fluoresc. 2010, 20, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Strauss, V.; Kahnt, A.; Zolnhofer, E.M.; Meyer, K.; Maid, H.; Placht, C.; Bauer, W.; Nacken, T.J.; Peukert, W.; Etschel, S.H. Assigning electronic states in carbon nanodots. Adv. Funct. Mater. 2016, 26, 7975–7985. [Google Scholar] [CrossRef]
- Hutton, G.A.; Martindale, B.C.; Reisner, E. Carbon dots as photosensitisers for solar-driven catalysis. Chem. Soc. Rev. 2017, 46, 6111–6123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, M.; Zhu, S.; Lu, S.; Song, Y.; Feng, T.; Tao, S.; Liu, J.; Yang, B. Recent progress on the photocatalysis of carbon dots: Classification, mechanism and applications. Nano Today 2018, 19, 201–218. [Google Scholar] [CrossRef]
- Hestand, N.J.; Spano, F.C. Interference between Coulombic and CT-mediated couplings in molecular aggregates: H-to J-aggregate transformation in perylene-based π-stacks. J. Chem. Phys. 2015, 143, 244707. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of three types of C-dots with different surface groups and their typical fluorescence excitation and emission spectra [68]. The ‘‘violet’’ C-dots (a) are with nitrogen- and oxygen-containing groups, ‘‘blue’’ (b) with oxygen-containing groups and cations, and ‘‘green’’ (c) C-dots contain oxygen groups only. Excitation wavelengths λex = 315 and 350 nm (a), 350 nm (b), 425 nm (c). Emission wavelengths λem = 405 nm (a), 440 nm (b), 530 nm (c).
Figure 1.
Schematic representation of three types of C-dots with different surface groups and their typical fluorescence excitation and emission spectra [68]. The ‘‘violet’’ C-dots (a) are with nitrogen- and oxygen-containing groups, ‘‘blue’’ (b) with oxygen-containing groups and cations, and ‘‘green’’ (c) C-dots contain oxygen groups only. Excitation wavelengths λex = 315 and 350 nm (a), 350 nm (b), 425 nm (c). Emission wavelengths λem = 405 nm (a), 440 nm (b), 530 nm (c).

Figure 2.
High-resolution transmission electron microscopy (HRTEM) images of individual C-dots possessing various crystal structures and shapes. These nanoparticles are localized as bright spots on fluorescence images (a,b). The images (c–h) were obtained in the areas where the bright spots corresponding to individual luminescent nanoparticles were detected by fluorescence microscopy. The white bars at the right bottom corners are the 5 nm scale bars. Reproduced with permission from Ref. [31].
Figure 2.
High-resolution transmission electron microscopy (HRTEM) images of individual C-dots possessing various crystal structures and shapes. These nanoparticles are localized as bright spots on fluorescence images (a,b). The images (c–h) were obtained in the areas where the bright spots corresponding to individual luminescent nanoparticles were detected by fluorescence microscopy. The white bars at the right bottom corners are the 5 nm scale bars. Reproduced with permission from Ref. [31].

Figure 3.
The analysis of TEM images of single C-dots. The crystal structures shown in Figure 1c,g of this article are presented in enlarged form as (a,d). Fourier transform (FT) of these areas are shown as (b,e). (c,f) are the cross-sections through the images (a,d) along the white lines. FT and cross-sections through TEM images show the lattice constant values of 0.35 and 0.25 nm for crystal structures I and II, respectively. Reproduced with permission from Ref. [31].
Figure 3.
The analysis of TEM images of single C-dots. The crystal structures shown in Figure 1c,g of this article are presented in enlarged form as (a,d). Fourier transform (FT) of these areas are shown as (b,e). (c,f) are the cross-sections through the images (a,d) along the white lines. FT and cross-sections through TEM images show the lattice constant values of 0.35 and 0.25 nm for crystal structures I and II, respectively. Reproduced with permission from Ref. [31].

Figure 4.
The absorption (Abs) and emission (Em) spectra under excitation with light of different wavelengths (see the inset legends) of C-dot fractions emitting light of different colors, blue (A), green (B), yellow (C) and red (D). Reproduced with permission from Ref. [103].
Figure 4.
The absorption (Abs) and emission (Em) spectra under excitation with light of different wavelengths (see the inset legends) of C-dot fractions emitting light of different colors, blue (A), green (B), yellow (C) and red (D). Reproduced with permission from Ref. [103].

Figure 5.
The excitation bands and the emission spectra at indicated excitation wavelengths of C-dots obtained by pyrolysis from polyethylene glycol. (a) Passivated with polyethylene glycol. (b) Additionally passivated with EDA. Reproduced with permission from Ref. [110].
Figure 5.
The excitation bands and the emission spectra at indicated excitation wavelengths of C-dots obtained by pyrolysis from polyethylene glycol. (a) Passivated with polyethylene glycol. (b) Additionally passivated with EDA. Reproduced with permission from Ref. [110].

Figure 6.
Normalized fluorescence spectra of oxidized carbon dots and species doped with nitrogen, sulfur, and selenium (a) and the relation of the observed effect to the electronegativity of doped atoms (b). Reproduced with permission from Ref. [150].
Figure 6.
Normalized fluorescence spectra of oxidized carbon dots and species doped with nitrogen, sulfur, and selenium (a) and the relation of the observed effect to the electronegativity of doped atoms (b). Reproduced with permission from Ref. [150].

Figure 7.
Sketch of the processes in carbon dots that may contribute to the observed emission. The sp2 hybridized core is shown as graphene or graphite flakes embedded in an amorphous sp3-hybridized carbon matrix. Dopant heteroatoms are shown as colored dots at the edge and within functional groups. Fluorophores are attached to the surface (stars). The shown processes are (1) π–π* charge transfer from the inner to outer part of the sp2 hybridized core; (2) n–π* transitions from the edge (white arrow) or from surface groups into the core; (3) charge transfer from the edge into the surface states and corresponding sub-bandgaps (orange arrow) or vice versa (blue arrow); both processes mentioned in (2) and (3) can also involve fluorophores if a transition pathway is possible; (4) intrinsic surface state transitions. These include sub-bandgaps created by functional surface groups; (5) hydrogen bonding interactions between the solvent and the surface. Fluorophores are found to be solvent sensitive and, therefore, assumed to accumulate at the surface. The drawing is not to scale. Reproduced with permission from Ref. [166].
Figure 7.
Sketch of the processes in carbon dots that may contribute to the observed emission. The sp2 hybridized core is shown as graphene or graphite flakes embedded in an amorphous sp3-hybridized carbon matrix. Dopant heteroatoms are shown as colored dots at the edge and within functional groups. Fluorophores are attached to the surface (stars). The shown processes are (1) π–π* charge transfer from the inner to outer part of the sp2 hybridized core; (2) n–π* transitions from the edge (white arrow) or from surface groups into the core; (3) charge transfer from the edge into the surface states and corresponding sub-bandgaps (orange arrow) or vice versa (blue arrow); both processes mentioned in (2) and (3) can also involve fluorophores if a transition pathway is possible; (4) intrinsic surface state transitions. These include sub-bandgaps created by functional surface groups; (5) hydrogen bonding interactions between the solvent and the surface. Fluorophores are found to be solvent sensitive and, therefore, assumed to accumulate at the surface. The drawing is not to scale. Reproduced with permission from Ref. [166].

Figure 8.
The illustration of the exciton model suggested by Michael Kasha for the variations of energy on electronic transitions of molecular dimers. The H-type dimers formed by side-to-side associations demonstrate the highly increased energy separation between the absorbing and emitting states, and, due to the forbidden character, their fluorescence is low. In contrast, in a J-type dimer, the transition dipoles are in line. They exhibit a strong decrease in transition energy and an increase in the transition dipole moment enhancing the probability of light absorption together with the red shifts in the absorption and emission bands. For simplicity, the ground state energies are shown on the same levels for monomer and dimers [220].
Figure 8.
The illustration of the exciton model suggested by Michael Kasha for the variations of energy on electronic transitions of molecular dimers. The H-type dimers formed by side-to-side associations demonstrate the highly increased energy separation between the absorbing and emitting states, and, due to the forbidden character, their fluorescence is low. In contrast, in a J-type dimer, the transition dipoles are in line. They exhibit a strong decrease in transition energy and an increase in the transition dipole moment enhancing the probability of light absorption together with the red shifts in the absorption and emission bands. For simplicity, the ground state energies are shown on the same levels for monomer and dimers [220].

Figure 9.
Different cases of excitation energy transfer in molecular chromophore systems. (1) Coherent exciton delocalization in stacked aromatic fluorophores. (2) Super-transfer among such systems of delocalized excitons absorbing and emitting in different colors. (3) FRET between exciton and molecular dye, in which the dye emitting at short wavelength (blue) is the energy donor. (4) FRET in which the excitonic system is the donor and molecular dye absorbing and emitting at longer wavelengths (red) is the acceptor.
Figure 9.
Different cases of excitation energy transfer in molecular chromophore systems. (1) Coherent exciton delocalization in stacked aromatic fluorophores. (2) Super-transfer among such systems of delocalized excitons absorbing and emitting in different colors. (3) FRET between exciton and molecular dye, in which the dye emitting at short wavelength (blue) is the energy donor. (4) FRET in which the excitonic system is the donor and molecular dye absorbing and emitting at longer wavelengths (red) is the acceptor.

Figure 10.
The simplified Jablonski diagram for the energy transformations on electronic excitation in the case of molecules and their H-aggregates. The colored (from violet to red) arrows illustrate the variation of energies of absorbing and emitting quanta. The vibrational modes of intramolecular and intermolecular origin and the coupled nuclear transformations as well as non-emissive decay processes are not shown.
Figure 10.
The simplified Jablonski diagram for the energy transformations on electronic excitation in the case of molecules and their H-aggregates. The colored (from violet to red) arrows illustrate the variation of energies of absorbing and emitting quanta. The vibrational modes of intramolecular and intermolecular origin and the coupled nuclear transformations as well as non-emissive decay processes are not shown.

Figure 11.
On–off intermittency and bleaching of carbon dots [16]. (a) The photoluminescence time traces (1–2) of two individual C-dots with “green” emission [31]. The emission intermittency varies on a scale from short off-states near several milliseconds, to longer ones on the order of hundreds of milliseconds. (b) Representative traces of a reduced particle showing one emission intensity level and single-step bleaching (1), and another one demonstrating multiple fluorescence intensity levels (2) before bleaching [286]. (c) The traces observed for CDs showing single-step bleaching in the presence of ascorbic acid, an electron donor (1), methyl viologen, and an electron acceptor (2) [288].
Figure 11.
On–off intermittency and bleaching of carbon dots [16]. (a) The photoluminescence time traces (1–2) of two individual C-dots with “green” emission [31]. The emission intermittency varies on a scale from short off-states near several milliseconds, to longer ones on the order of hundreds of milliseconds. (b) Representative traces of a reduced particle showing one emission intensity level and single-step bleaching (1), and another one demonstrating multiple fluorescence intensity levels (2) before bleaching [286]. (c) The traces observed for CDs showing single-step bleaching in the presence of ascorbic acid, an electron donor (1), methyl viologen, and an electron acceptor (2) [288].

Figure 12.
Fluorescence images of the individual CDs spin coated on the glass surface. (a–e) Fluorescence excitation patterns; double white arrows show the orientation of the excitation transition dipole moment (TDM). (f–j) Defocused images and assigned orientation (double white arrows) of the emission TDM. Reproduced with permission from Ref. [31].
Figure 12.
Fluorescence images of the individual CDs spin coated on the glass surface. (a–e) Fluorescence excitation patterns; double white arrows show the orientation of the excitation transition dipole moment (TDM). (f–j) Defocused images and assigned orientation (double white arrows) of the emission TDM. Reproduced with permission from Ref. [31].

Figure 13.
Photoluminescence spectra of C-dots in the broad temperature range down to 10 K at λexc = 340 nm (a) and the decay curves obtained at the short-wavelength edge of the emission spectrum (λem = 410 nm) (b) [30].
Figure 13.
Photoluminescence spectra of C-dots in the broad temperature range down to 10 K at λexc = 340 nm (a) and the decay curves obtained at the short-wavelength edge of the emission spectrum (λem = 410 nm) (b) [30].

Figure 14.
The proposed model of excitonic H-aggregate as the light-emissive functionality of C-dots, illustrating the mechanism of their emission. The fluorescent H-aggregate is formed by side-to-side stacking of aromatic groups resulting in their π–electronic coupling at the particle shell. The major sequence of events is (1) absorption of light quantum by H-aggregate as an integral quantum system, (2) exciton propagation and transition to self-localized state, and (3) the emissive electron–hole recombination at the self-trapped site.
Figure 14.
The proposed model of excitonic H-aggregate as the light-emissive functionality of C-dots, illustrating the mechanism of their emission. The fluorescent H-aggregate is formed by side-to-side stacking of aromatic groups resulting in their π–electronic coupling at the particle shell. The major sequence of events is (1) absorption of light quantum by H-aggregate as an integral quantum system, (2) exciton propagation and transition to self-localized state, and (3) the emissive electron–hole recombination at the self-trapped site.

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Demchenko, A.P. Excitons in Carbonic Nanostructures. C 2019, 5, 71. https://doi.org/10.3390/c5040071
AMA Style
Demchenko AP. Excitons in Carbonic Nanostructures. C. 2019; 5(4):71. https://doi.org/10.3390/c5040071
Chicago/Turabian StyleDemchenko, Alexander P. 2019. "Excitons in Carbonic Nanostructures" C 5, no. 4: 71. https://doi.org/10.3390/c5040071
APA StyleDemchenko, A. P. (2019). Excitons in Carbonic Nanostructures. C, 5(4), 71. https://doi.org/10.3390/c5040071
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.