Application of Experimental Design to Hydrogen Storage: Optimisation of Lignin-Derived Carbons

 , and
, and
Abstract
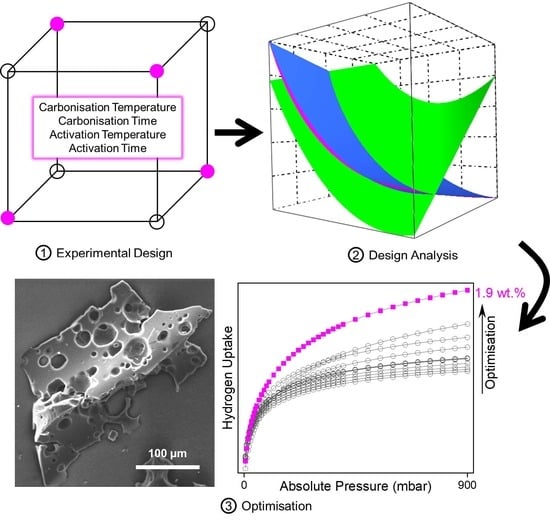
:
1. Introduction
2. Materials and Methods
2.1. Activated Carbon Preparation
Experimental Design
2.2. Activated Carbon Characterisation
3. Results and Discussion
3.1. Influence of Factors on Lignin-Derived Carbon Properties
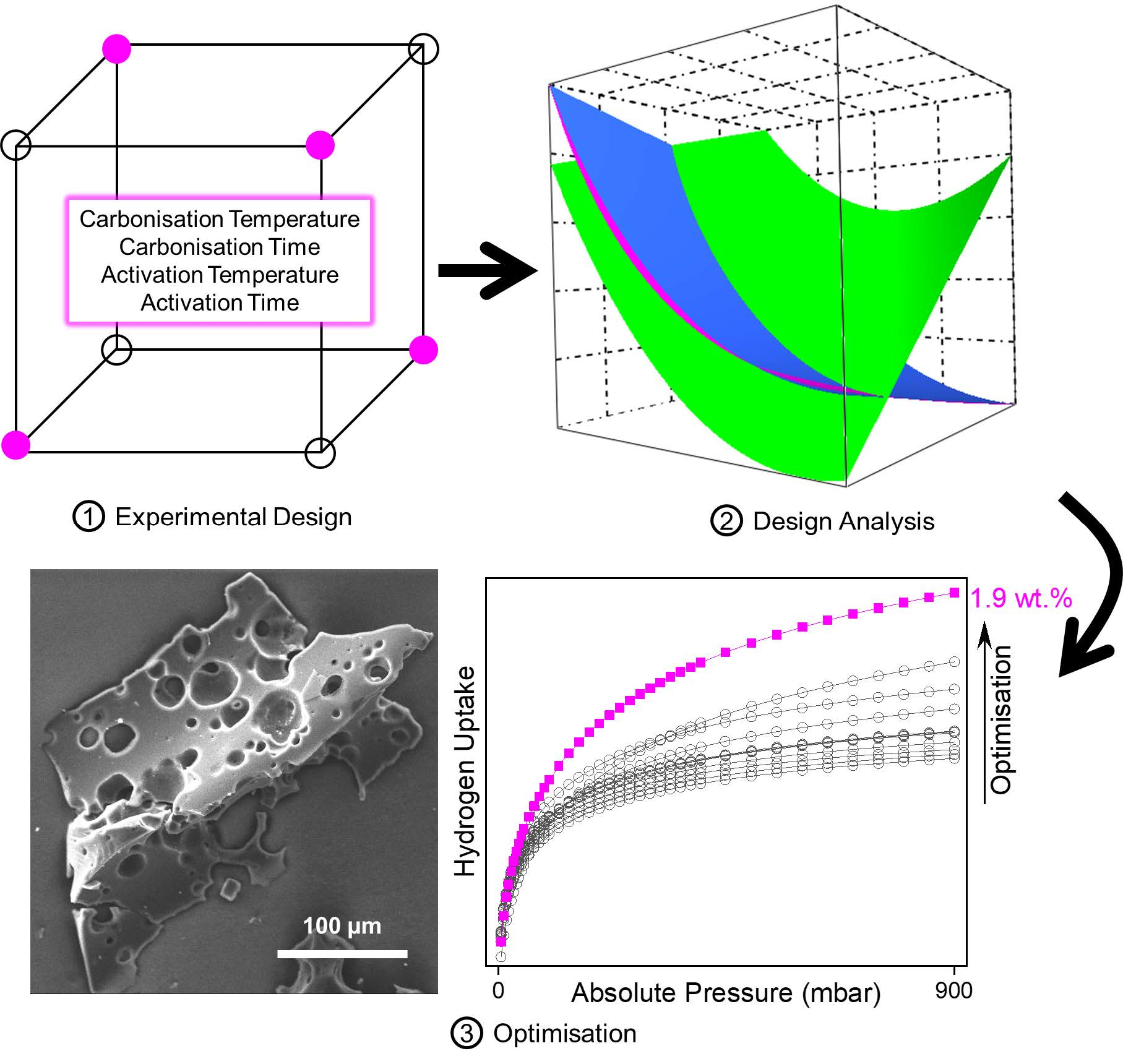
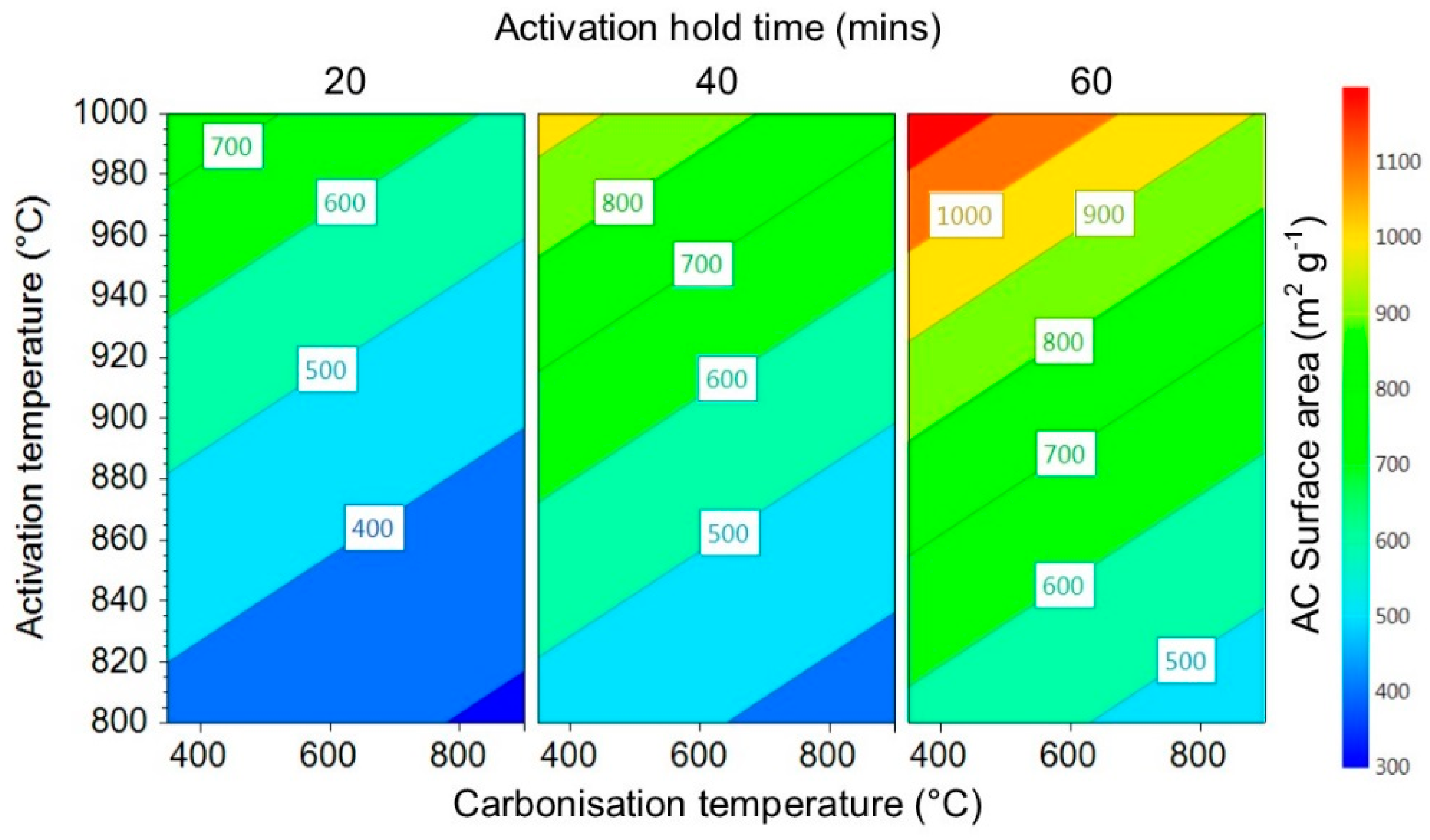
3.1.1. Activated Carbon Surface Area
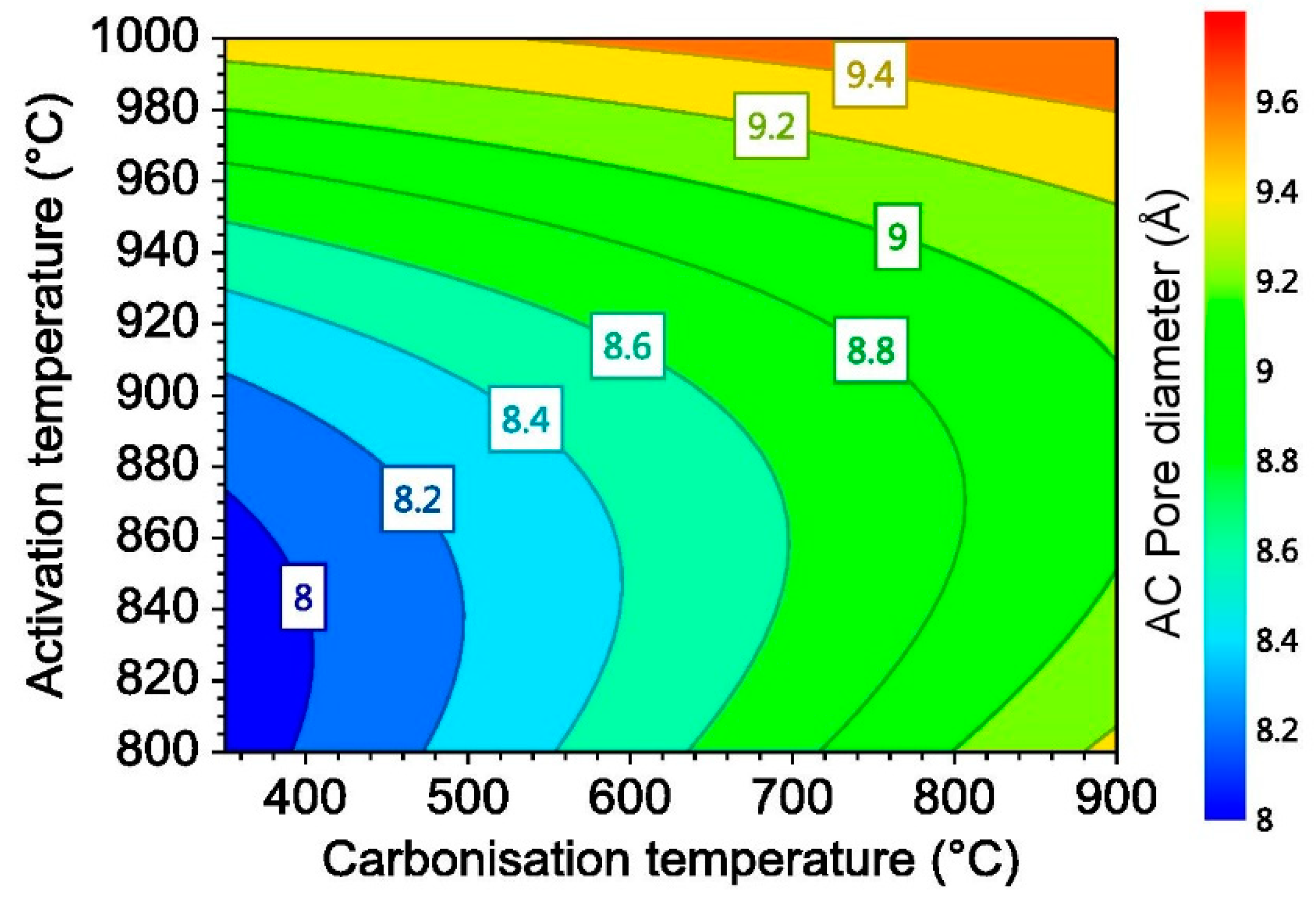
3.1.2. Activated Carbon Average Pore Size
3.1.3. Activated Carbon Hydrogen Uptake
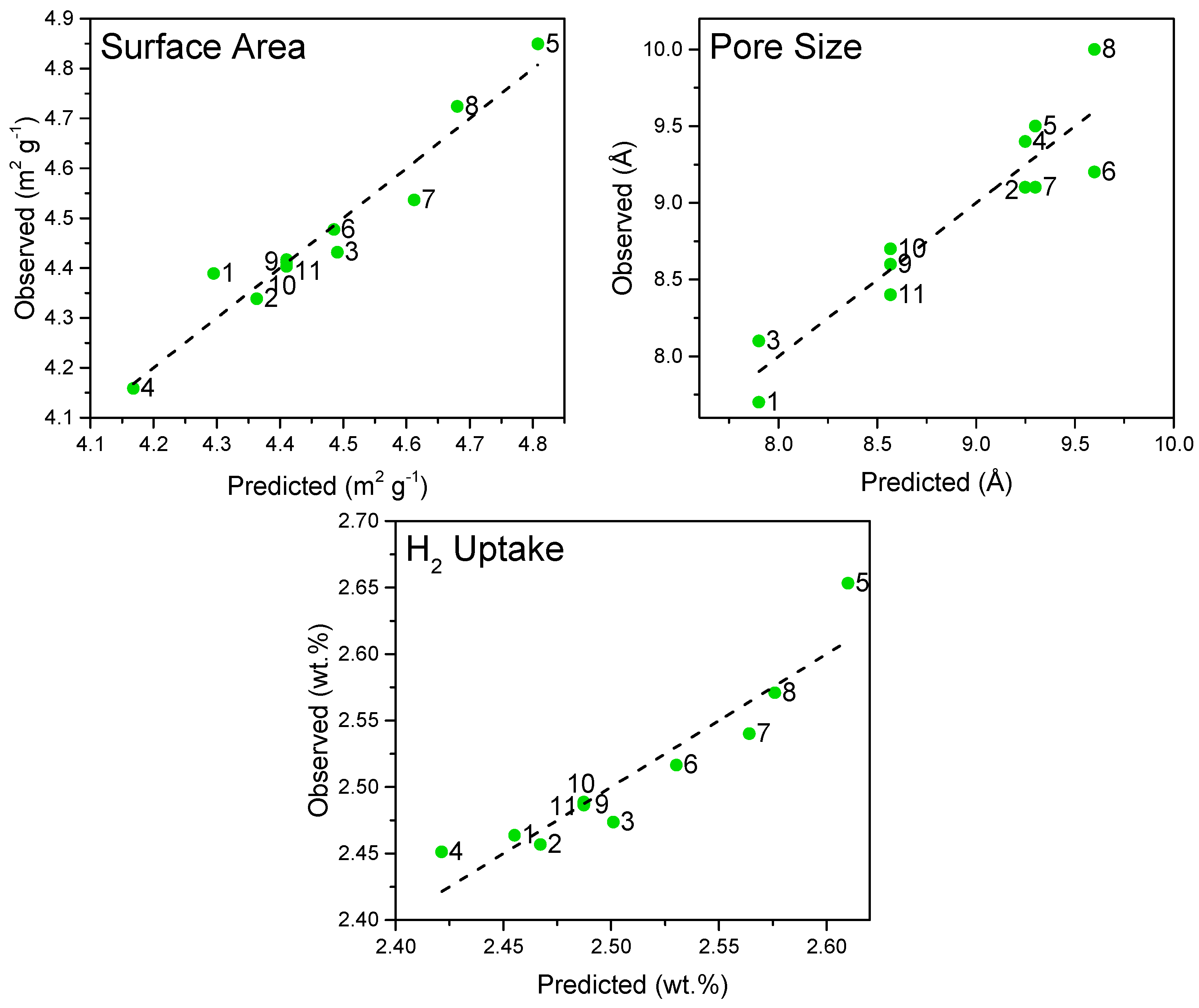
3.1.4. Analysis of Model Fit
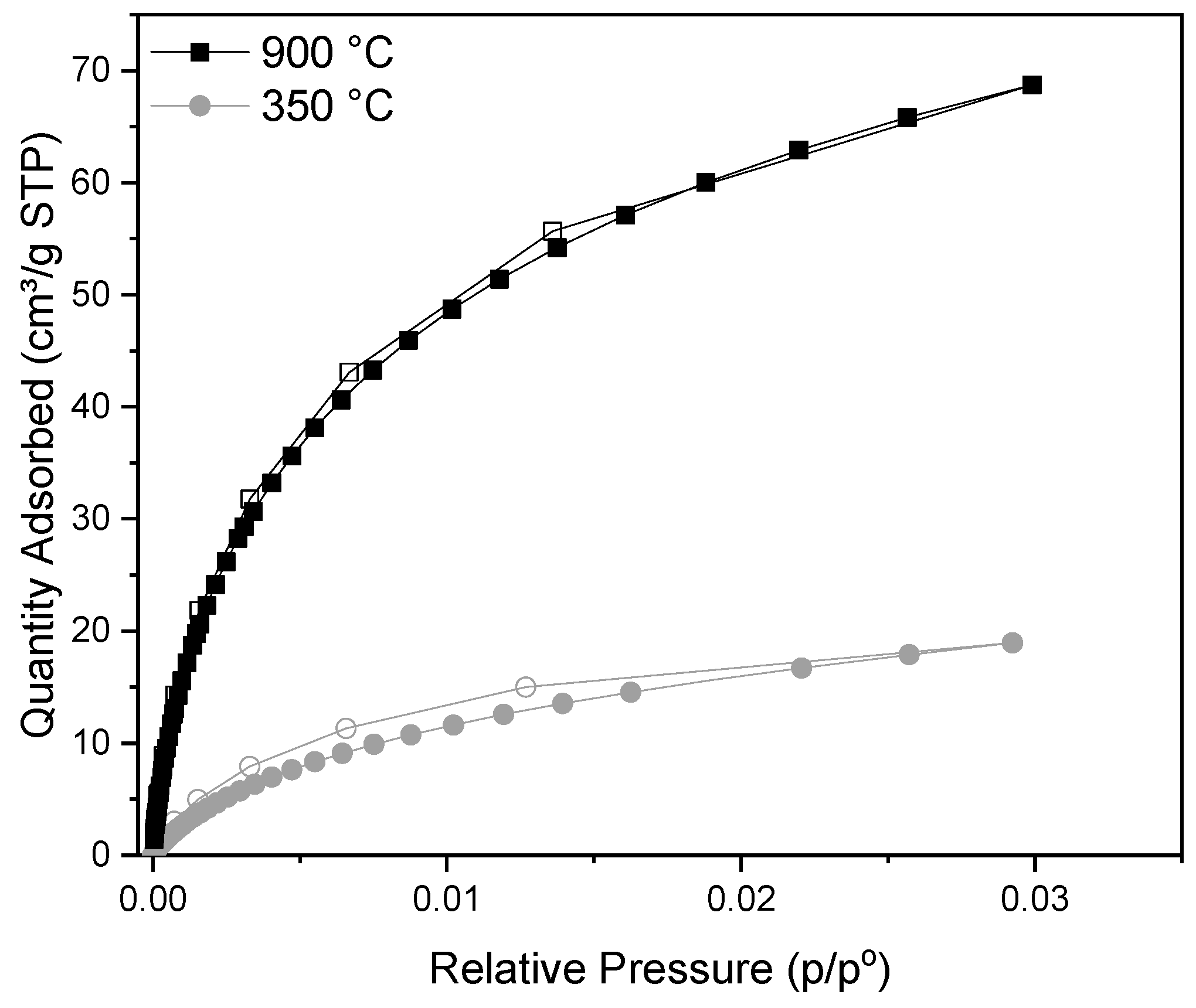
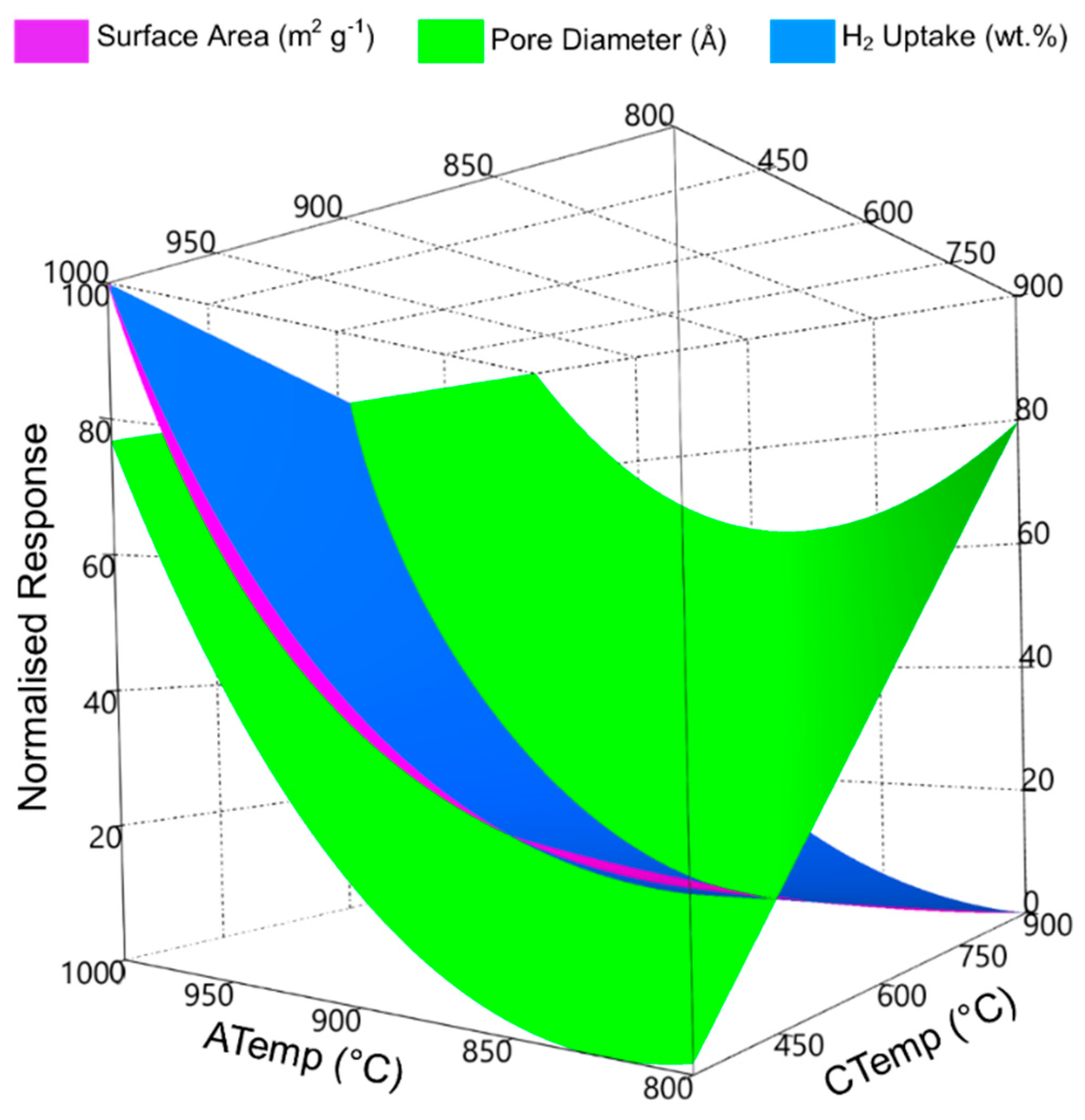
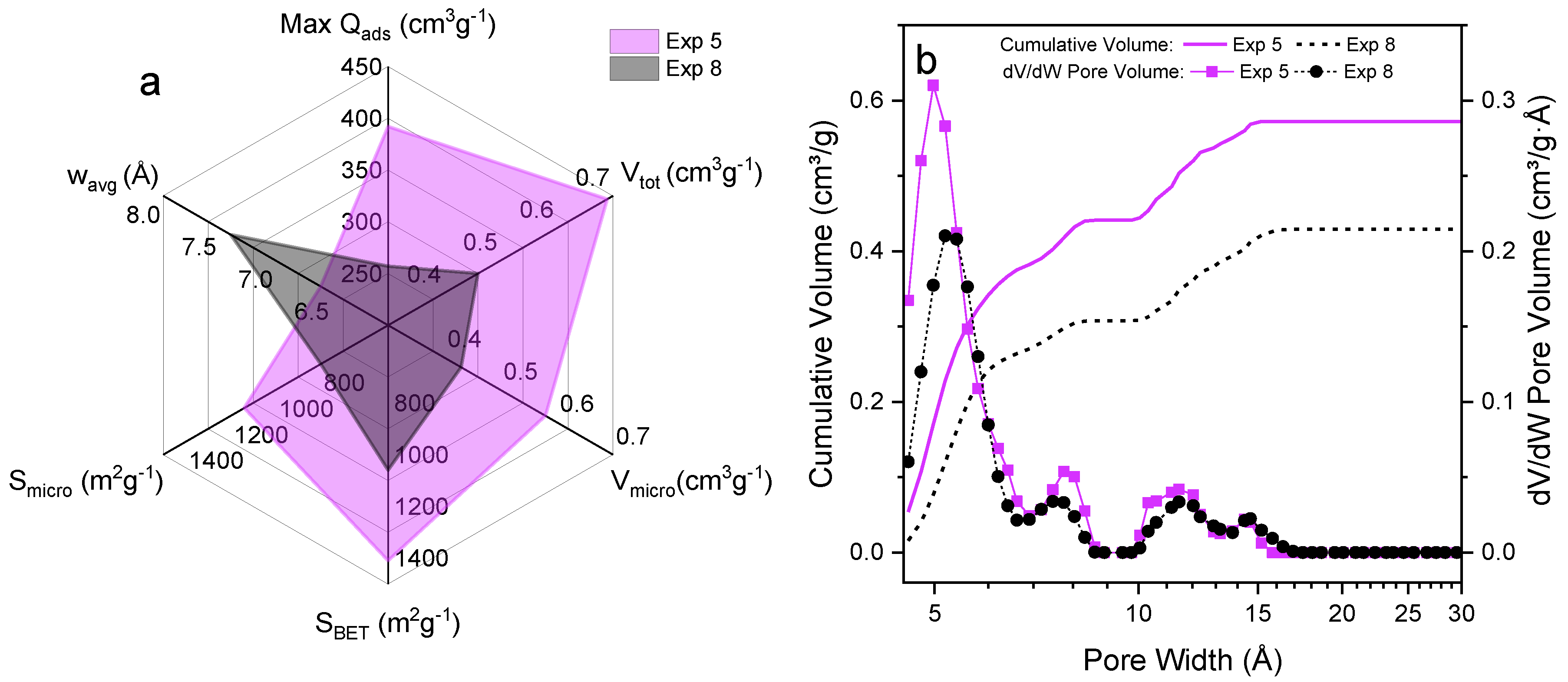
3.2. Adsorptive Characteristics of Optimal Activated Carbon
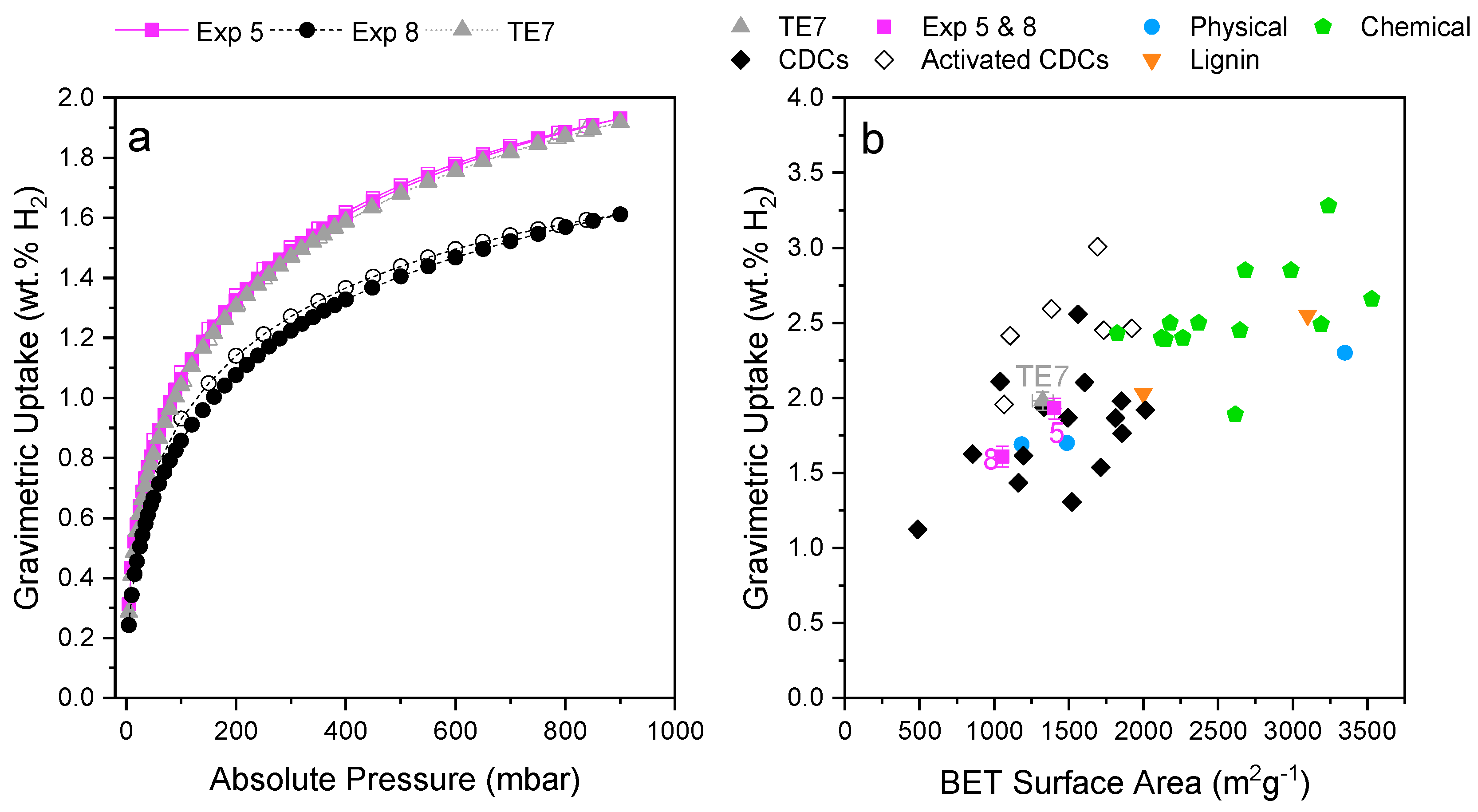
3.3. Hydrogen Uptake of Optimal Activated Carbons
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as renewable raw material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Wu, K.C.-W.; Saha, B. Emerging strategies for breaking the 3D amorphous network of lignin. Catal. Sci. Technol. 2014, 4, 3785–3799. [Google Scholar] [CrossRef]
- Meister, J.J. Modification of Lignin. J. Macromol. Sci. Part C Polym. Rev. 2007, 42, 235–289. [Google Scholar] [CrossRef]
- Carrott, P.J.M.; Carrott, M.R. Lignin-from natural adsorbent to activated carbon: A review. Bioresour. Technol. 2007, 98, 2301–2312. [Google Scholar] [CrossRef]
- Snowdon, M.R.; Mohanty, A.K.; Misra, M. A Study of Carbonized Lignin as an Alternative to Carbon Black. ACS Sustain. Chem. Eng. 2014, 2, 1257–1263. [Google Scholar] [CrossRef]
- de Wild, P.J.; Huijgen, W.J.J.; Heeres, H.J. Pyrolysis of wheat straw-derived organosolv lignin. J. Anal. Appl. Pyrolysis 2012, 93, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Ntuli, V.; Hapazari, I. Sustainable waste management by production of activated carbon from agroforestry residues. S. Afr. J. Sci. 2013, 109, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Busquets, R.; Kozynchenko, O.P.; Whitby, R.L.D.; Tennison, S.R.; Cundy, A.B. Phenolic carbon tailored for the removal of polar organic contaminants from water: A solution to the metaldehyde problem? Water Res. 2014, 61, 46–56. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, J.E.; Bimbo, N.; Ting, V.P.; Rechain, B.; Joubert, E.; Mays, T.J. Modelling the potential of adsorbed hydrogen for use in aviation. Microporous Mesoporous Mater. 2015, 209, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Tang, Z.; Sun, D.; Ouyang, L.; Zhu, M. Recent advances and remaining challenges of nanostructured materials for hydrogen storage applications. Prog. Mater. Sci. 2017, 88, 1–48. [Google Scholar] [CrossRef]
- Sevilla, M.; Mokaya, R. Energy storage applications of activated carbons: Supercapacitors and hydrogen storage. Energy Environ. Sci. 2014, 7, 1250–1280. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Senkovska, I.; Kaskel, S.; Liu, Q. Chemically activated fungi-based porous carbons for hydrogen storage. Carbon 2014, 75, 372–380. [Google Scholar] [CrossRef]
- Yang, R.; Liu, G.; Li, M.; Zhang, J.; Hao, X. Preparation and N2, CO2 and H2 adsorption of super activated carbon derived from biomass source hemp (Cannabis sativa L.) stem. Microporous Mesoporous Mater. 2012, 158, 108–116. [Google Scholar] [CrossRef]
- Bansal, R.C.; Donet, J.B.; Stoeckli, H.F. Active Carbon; Marcel Dekker: New York, NY, USA, 1988. [Google Scholar]
- Marsh, H.; Rodríguez-Reinoso, F. Activated Carbon; Elsevier: London, UK, 2006. [Google Scholar] [CrossRef]
- Świątkowski, A. Industrial Carbon Adsorbents. Stud. Surf. Catal. 1999, 120, 69–94. [Google Scholar] [CrossRef]
- Guo, S.; Peng, J.; Li, W.; Yang, K.; Zhang, L.; Zhang, S.; Xia, H. Effects of CO2 activation on porous structures of coconut shell-based activated carbons. Appl. Surf. Sci. 2009, 255, 8443–8449. [Google Scholar] [CrossRef]
- Yang, K.; Peng, J.; Xia, H.; Zhang, L.; Srinivasakannan, C.; Guo, S. Textural characteristics of activated carbon by single step CO2 activation from coconut shells. J. Taiwan Inst. Chem. Eng. 2010, 41, 367–372. [Google Scholar] [CrossRef]
- Arena, N.; Lee, J.; Clift, R. Life Cycle Assessment of activated carbon production from coconut shells. J. Clean. Prod. 2016, 125, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Hjaila, K.; Baccar, R.; Sarrà, M.; Gasol, C.M.; Blánquez, P. Environmental impact associated with activated carbon preparation from olive-waste cake via life cycle assessment. J. Environ. Manag. 2013, 130, 242–247. [Google Scholar] [CrossRef]
- Li, Y.; Li, D.; Rao, Y.; Zhao, X.; Wu, M. Superior CO2, CH4, and H2 uptakes over ultrahigh-surface-area carbon spheres prepared from sustainable biomass-derived char by CO2activation. Carbon 2016, 105, 454–462. [Google Scholar] [CrossRef]
- Wang, H.; Gao, Q.; Hu, J. High hydrogen storage capacity of porous carbons prepared by using activated carbon. J. Am. Chem. Soc. 2009, 131, 7016–7022. [Google Scholar] [CrossRef]
- Bader, N.; Abdelmottaleb, O.N. CO2 activation of olive bagasse for hydrogen storage. Environ. Prog. Sustain. Energy 2017, 36, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Yang, Z.; Zhu, Y. Porous carbon-based materials for hydrogen storage: Advancement and challenges. J. Mater. Chem. A 2013, 1, 9365–9381. [Google Scholar] [CrossRef]
- Masika, E.; Mokaya, R. Hydrogen storage in high surface area carbons with identical surface areas but different pore sizes: Direct demonstration of the effects of pore size. J. Phys. Chem. C 2012, 116, 25734–25740. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Portet, C.; Osswald, S.; Simmons, J.M.; Yildirim, T.; Laudisio, G.; Fischer, J.E. Importance of pore size in high-pressure hydrogen storage by porous carbons. Int. J. Hydrog. Energy 2009, 34, 6314–6319. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Dash, R.K.; Yushin, G.; Yildirim, T.; Laudisio, G.; Fischer, J.E. Tailoring of nanoscale porosity in carbide-derived carbons for hydrogen storage. J. Am. Chem. Soc. 2005, 127, 16006–16007. [Google Scholar] [CrossRef]
- Sethia, G.; Sayari, A. Activated carbon with optimum pore size distribution for hydrogen storage. Carbon 2016, 99, 289–294. [Google Scholar] [CrossRef]
- Ting, V.P.; Ramirez-Cuesta, A.J.; Bimbo, N.; Sharpe, J.E.; Noguera-Diaz, A.; Presser, V.; Rudic, S.; Mays, T.J. Direct Evidence for Solid-like Hydrogen in a Nanoporous Carbon Hydrogen Storage Material at Supercritical Temperatures. ACS Nano 2015, 9, 8249–8254. [Google Scholar] [CrossRef] [Green Version]
- Loloie, Z.; Mozaffarian, M.; Soleimani, M.; Asassian, N. Carbonization and CO2 activation of scrap tires: Optimization of specific surface area by the Taguchi method. Korean J. Chem. Eng. 2017, 34, 366–375. [Google Scholar] [CrossRef]
- Sumathi, S.; Bhatia, S.; Lee, K.T.; Mohamed, A.R. Optimization of microporous palm shell activated carbon production for flue gas desulphurization: Experimental and statistical studies. Bioresour. Technol. 2009, 100, 1614–1621. [Google Scholar] [CrossRef]
- Jia, Q.; Lua, A.C. Effects of pyrolysis conditions on the physical characteristics of oil-palm-shell activated carbons used in aqueous phase phenol adsorption. J. Anal. Appl. Pyrolysis 2008, 83, 175–179. [Google Scholar] [CrossRef]
- Lua, A.C.; Lau, F.Y.; Guo, J. Influence of pyrolysis conditions on pore development of oil-palm-shell activated carbons. J. Anal. Appl. Pyrolysis 2006, 76, 96–102. [Google Scholar] [CrossRef]
- Bouchelta, C.; Medjram, M.S.; Bertrand, O.; Bellat, J.P. Preparation and characterization of activated carbon from date stones by physical activation with steam. J. Anal. Appl. Pyrolysis 2008, 82, 70–77. [Google Scholar] [CrossRef]
- Eriksson, L.; Johansson, E.; Kettaneh-Wold, N.; Wikström, C.; Wold, S. Design of Experiments: Principles and Applications; UMetrics AB: Umeå, Sweden, 2000. [Google Scholar]
- Montgomery, D.C. Design and Analysis of Experiments, 7th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Wahid, Z.; Nadir, N. Improvement of one factor at a time through design of experiments. World Appl. Sci. J. 2013, 21, 56–61. [Google Scholar]
- Czitrom, V. One-factor-at-a-time versus designed experiments. Am. Stat. 1999, 53, 126–131. [Google Scholar] [CrossRef]
- Baçaoui, A.; Dahbi, A.; Yaacoubi, A.; Bennouna, C.; Maldonado-Hódar, F.; Rivera-Utrilla, J.; Carrasco-Marín, F.; Moreno-Castilla, C. Experimental Design To Optimize Preparation of Activated Carbons for Use in Water Treatment. Environ. Sci. Technol. 2002, 36, 3844–3849. [Google Scholar] [CrossRef]
- Zhao, W.; Fierro, V.; Zlotea, C.; Aylon, E.; Izquierdo, M.T.; Latroche, M.; Celzard, A. Optimization of activated carbons for hydrogen storage. Int. J. Hydrog. Energy 2011, 36, 11746–11751. [Google Scholar] [CrossRef]
- Mussatto, S.; Fernandes, M.; Rocha, G. Production, characterization and application of activated carbon from brewer’s spent grain lignin. Bioresour. Technol. 2010, 101, 2450–2457. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Liang, J.; Zhao, J.; Tao, Z.; Chen, J. Biomass waste-derived microporous carbons with controlled texture and enhanced hydrogen uptake. Chem. Mater. 2008, 20, 1889–1895. [Google Scholar] [CrossRef]
- Nor, N.M.; Lau, L.C.; Lee, K.T.; Mohamed, A.R. Synthesis of activated carbon from lignocellulosic biomass and its applications in air pollution control—A review. J. Environ. Chem. Eng. 2013, 1, 658–666. [Google Scholar] [CrossRef]
- Daud, W.M.; Ali, W.S. Comparison on pore development of activated carbon produced from palm shell and coconut shell. Bioresour. Technol. 2004, 93, 63–69. [Google Scholar] [CrossRef]
- Laudisio, G.; Dash, R.K.; Singer, J.P.; Yushin, G.; Gogotsi, Y.; Fischer, J.E. Carbide-Derived Carbons: A Comparative Study of Porosity Based on Small-Angle Scattering and Adsorption Isotherms. Langmuir 2006, 22, 8945–8950. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.; Chmiola, J.; Yushin, G.; Gogotsi, Y.; Laudisio, G.; Singer, J.; Fischer, J.; Kucheyev, S. Titanium carbide derived nanoporous carbon for energy-related applications. Carbon 2006, 44, 2489–2497. [Google Scholar] [CrossRef] [Green Version]
- Yeon, S.H.; Osswald, S.; Gogotsi, Y.; Singer, J.P.; Simmons, J.M.; Fischer, J.E.; Lillo-Ródenas, M.A.; Linares-Solano, Á. Enhanced methane storage of chemically and physically activated carbide-derived carbon. J. Power Sources 2009, 191, 560–567. [Google Scholar] [CrossRef]
- ISO. Pore Size Distribution and Porosity of Solid Materials by Mercury Porosimetry and Gas Adsorption—Part 3: Analysis of micropores by Gas Adsorption; 2007; ISO 15901-3:2007. Available online: https://www.iso.org/standard/40364.html (accessed on 6 December 2019).
- Quantachrome. Powder Tech Note 35: Micropore Size Analysis of Porous Carbons, (n.d.) 1–4. Available online: https://www.quantachrome.com/technotes.html (accessed on 3 October 2017).
- ISO. Determination of the specific Surface Area of Solids by Gas Adsorption: BET Method; 2010; ISO 9277: 2010. Available online: https://www.iso.org/standard/44941.html (accessed on 6 December 2019).
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Putnam, C.D.; Hammel, M.; Hura, G.L.; Tainer, J.A. X-Ray Scattering (SAXS) Combined with Crystallography and Computation: Defining Accurate Macromolecular Structures, Conformations and Assemblies in Solution. Biophys. J. 2011. [Google Scholar] [CrossRef] [PubMed]
- Schnablegger, H.; Singh, Y. The SAXS Guide: Getting Acquainted with the Principles; Anton Paar GmbH: Graz, Austria, 2011. [Google Scholar]
- Kalliat, M.; Kwak, C.Y.; Schmidt, P.W. Small-Angle X-Ray Investigation of the Porosity in Coals. In New Approaches Coal Chemistry; American Chemical Society: Washington, DC, USA, 1981; pp. 3–22. [Google Scholar] [CrossRef] [Green Version]
- Gibaud, A.; Xue, J.S.; Dahn, J.R. A small angle X-ray scattering study of carbons made from pyrolyzed sugar. Carbon 1996, 34, 499–503. [Google Scholar] [CrossRef]
- Rowlandson, J.L.; Woodman, T.J.; Tennison, S.R.; Edler, K.J.; Ting, V.P. Influence of Aromatic Structure on the Thermal Behaviour of Lignin. Waste Biomass Valori. 2018, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Ergun, S. Kinetics of the reaction of carbon with carbon dioxide. J. Phys. Chem. 1956, 60, 480–485. [Google Scholar] [CrossRef]
- Teng, H.; Lin, H. Activated carbon production from low ash subbituminous coal with CO2 activation. AIChE J. 1998, 44, 1170–1177. [Google Scholar] [CrossRef]
- Yang, T.; Lua, A.C. Characteristics of activated carbons prepared from pistachio-nut shells by physical activation. J. Colloid Interface Sci. 2003, 267, 408–417. [Google Scholar] [CrossRef]
- Zhang, T.; Walawender, W.P.; Fan, L.T.; Fan, M.; Daugaard, D.; Brown, R.C. Preparation of activated carbon from forest and agricultural residues through CO2 activation. Chem. Eng. J. 2004, 105, 53–59. [Google Scholar] [CrossRef]
- Gañán, J.; González, J.F.; González-García, C.M.; Ramiro, A.; Sabio, E.; Román, S. Air-activated carbons from almond tree pruning: Preparation and characterization. Appl. Surf. Sci. 2006, 252, 5988–5992. [Google Scholar] [CrossRef]
- Rodríguez-Mirasol, J.; Cordero, T.; Rodríguez, J.J. Preparation and characterization of activated carbons from eucalyptus kraft lignin. Carbon 1993, 31, 87–95. [Google Scholar] [CrossRef]
- Lua, A.C.; Yang, T.; Guo, J. Effects of pyrolysis conditions on the properties of activated carbons prepared from pistachio-nut shells. J. Anal. Appl. Pyrolysis 2004, 72, 279–287. [Google Scholar] [CrossRef]
- Guo, J.; Lua, A.C. Characterization of chars pyrolyzed from oil palm stones for the preparation of activated carbons. J. Anal. Appl. Pyrolysis 1998, 46, 113–125. [Google Scholar] [CrossRef]
- Daud, W.M.; Ali, W.S.; Sulaiman, M.Z. Effects of carbonization temperature on pore development in palm-shell-based activated carbon. Carbon 2000, 38, 1925–1932. [Google Scholar] [CrossRef]
- Rodriguez-Reinoso, F. Controlled Gasification of Carbon and Pore Structure Development. In Fundamental Issues in Control of Carbon Gasification Reactivity; Springer: Dordrecht, The Netherlands, 1991; pp. 533–571. [Google Scholar] [CrossRef]
- Baklanova, O.N.; Plaksin, G.V.; Drozdov, V.A.; Duplyakin, V.K.; Chesnokov, N.V.; Kuznetsov, B.N. Preparation of microporous sorbents from cedar nutshells and hydrolytic lignin. Carbon 2003, 41, 1793–1800. [Google Scholar] [CrossRef]
- Ismailova, M.G. Influence of the carbonization conditions on the formation of the porous structure of activated carbon from cotton lignin. Prot. Met. Phys. Chem. Surf. 2009, 45, 212–215. [Google Scholar] [CrossRef]
- Rodriguez-Mirasol, J.; Cordero, T.; Rodriguez, J.J. Activated carbons from carbon dioxide partial gasification of eucalyptus kraft lignin. Energy Fuels 1993, 7, 133–138. [Google Scholar] [CrossRef]
- Fu, K.; Yue, Q.; Gao, B.; Sun, Y.; Zhu, L. Preparation, characterization and application of lignin-based activated carbon from black liquor lignin by steam activation. Chem. Eng. J. 2013, 228, 1074–1082. [Google Scholar] [CrossRef]
- Carrott, P.J.M.; Carrott, M.R.; Guerrero, C.I.; Delgado, L.A. Reactivity and porosity development during pyrolysis and physical activation in CO2 or steam of kraft and hydrolytic lignins. J. Anal. Appl. Pyrolysis 2008, 82, 264–271. [Google Scholar] [CrossRef] [Green Version]
- Bimbo, N.; Ting, V.P.; Sharpe, J.E.; Mays, T.J. Analysis of optimal conditions for adsorptive hydrogen storage in microporous solids. Colloids Surf. A Physicochem. Eng. Asp. 2013, 437, 113–119. [Google Scholar] [CrossRef]
- Hruzewicz-Kołodziejczyk, A.; Ting, V.P.; Bimbo, N.; Mays, T.J. Improving comparability of hydrogen storage capacities of nanoporous materials. Int. J. Hydrog. Energy 2012, 37, 2728–2736. [Google Scholar] [CrossRef]
- Armandi, M.; Bonelli, B.; Geobaldo, F.; Garrone, E. Nanoporous carbon materials obtained by sucrose carbonization in the presence of KOH. Microporous Mesoporous Mater. 2010, 132, 414–420. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B.; Mokaya, R. High density hydrogen storage in superactivated carbons from hydrothermally carbonized renewable organic materials. Energy Environ. Sci. 2011, 4, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Zhang, Y.; Wang, S. Enhancing thermoelectric properties of organic composites through hierarchical nanostructures. Sci. Rep. 2013, 3, 3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balathanigaimani, M.S.; Shim, W.-G.; Kim, T.-H.; Cho, S.-J.; Lee, J.-W.; Moon, H. Hydrogen storage on highly porous novel corn grain-based carbon monoliths. Catal. Today 2009, 146, 234–240. [Google Scholar] [CrossRef]
- Heo, Y.-J.; Park, S.-J. Synthesis of activated carbon derived from rice husks for improving hydrogen storage capacity. J. Ind. Eng. Chem. 2015, 31, 330–334. [Google Scholar] [CrossRef]
- Yushin, G.; Dash, R.; Jagiello, J.; Fischer, J.E.; Gogotsi, Y. Carbide-derived carbons: Effect of pore size on hydrogen uptake and heat of adsorption. Adv. Funct. Mater. 2006, 16, 2288–2293. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Factor Abbreviation | Low Level | Corresponding Low Value | High Level | Corresponding High Value | Mid-Point | Corresponding Mid-Point Value |
|---|---|---|---|---|---|---|---|
| Carbonisation temperature | CTemp | −1 | 350 °C | +1 | 900 °C | 0 | 625 °C |
| Carbonisation dwell time | CTime | −1 | 30 min | +1 | 360 min | 0 | 195 min |
| Activation temperature | ATemp | −1 | 800 °C | +1 | 1000 °C | 0 | 900 °C |
| Activation dwell time | ATime | −1 | 20 min | +1 | 60 min | 0 | 40 min |
| Experiment Number (Exp) | Experimental Parameters | Response Parameters | |||||
|---|---|---|---|---|---|---|---|
| Carbonisation | Activation | SBET (m2 g−1) | wavg: SAXS (Å) | H2 Uptake (wt.%) | |||
| CTemp (°C) | CTime (min) | ATemp (°C) | ATime (min) | ||||
| 1 | 350 | 30 | 800 | 20 | 486 | 7.7 | 1.20 |
| 2 | 900 | 30 | 800 | 60 | 432 | 9.1 | 1.18 |
| 3 | 350 | 360 | 800 | 60 | 536 | 8.1 | 1.24 |
| 4 | 900 | 360 | 800 | 20 | 284 | 9.4 | 1.16 |
| 5 | 350 | 30 | 1000 | 60 | 1409 | 9.3 | 1.93 |
| 6 | 900 | 30 | 1000 | 20 | 596 | 9.2 | 1.39 |
| 7 | 350 | 360 | 1000 | 20 | 684 | 9.1 | 1.48 |
| 8 | 900 | 360 | 1000 | 60 | 1055 | 10.0 | 1.61 |
| 9 | 625 | 195 | 900 | 40 | 512 | 8.6 | 1.28 |
| 10 | 625 | 195 | 900 | 40 | 518 | 8.7 | 1.29 |
| 11 | 625 | 195 | 900 | 40 | 502 | 8.4 | 1.29 |
| Response | R2 | Q2 | p-Value | |
|---|---|---|---|---|
| BET Surface Area | 0.94 | 0.81 | <0.01 | 0.01 |
| Average Pore Size | 0.88 | 0.65 | 0.01 | 0.17 |
| H2 Uptake | 0.88 | 0.79 | 0.01 | >0.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rowlandson, J.L.; Coombs OBrien, J.; Edler, K.J.; Tian, M.; Ting, V.P. Application of Experimental Design to Hydrogen Storage: Optimisation of Lignin-Derived Carbons. C 2019, 5, 82. https://doi.org/10.3390/c5040082
Rowlandson JL, Coombs OBrien J, Edler KJ, Tian M, Ting VP. Application of Experimental Design to Hydrogen Storage: Optimisation of Lignin-Derived Carbons. C. 2019; 5(4):82. https://doi.org/10.3390/c5040082
Chicago/Turabian StyleRowlandson, Jemma L., James Coombs OBrien, Karen J. Edler, Mi Tian, and Valeska P. Ting. 2019. "Application of Experimental Design to Hydrogen Storage: Optimisation of Lignin-Derived Carbons" C 5, no. 4: 82. https://doi.org/10.3390/c5040082
APA StyleRowlandson, J. L., Coombs OBrien, J., Edler, K. J., Tian, M., & Ting, V. P. (2019). Application of Experimental Design to Hydrogen Storage: Optimisation of Lignin-Derived Carbons. C, 5(4), 82. https://doi.org/10.3390/c5040082