Characterization of Carbon Materials for Hydrogen Storage and Compression

 , ,
, ,  ,
, 
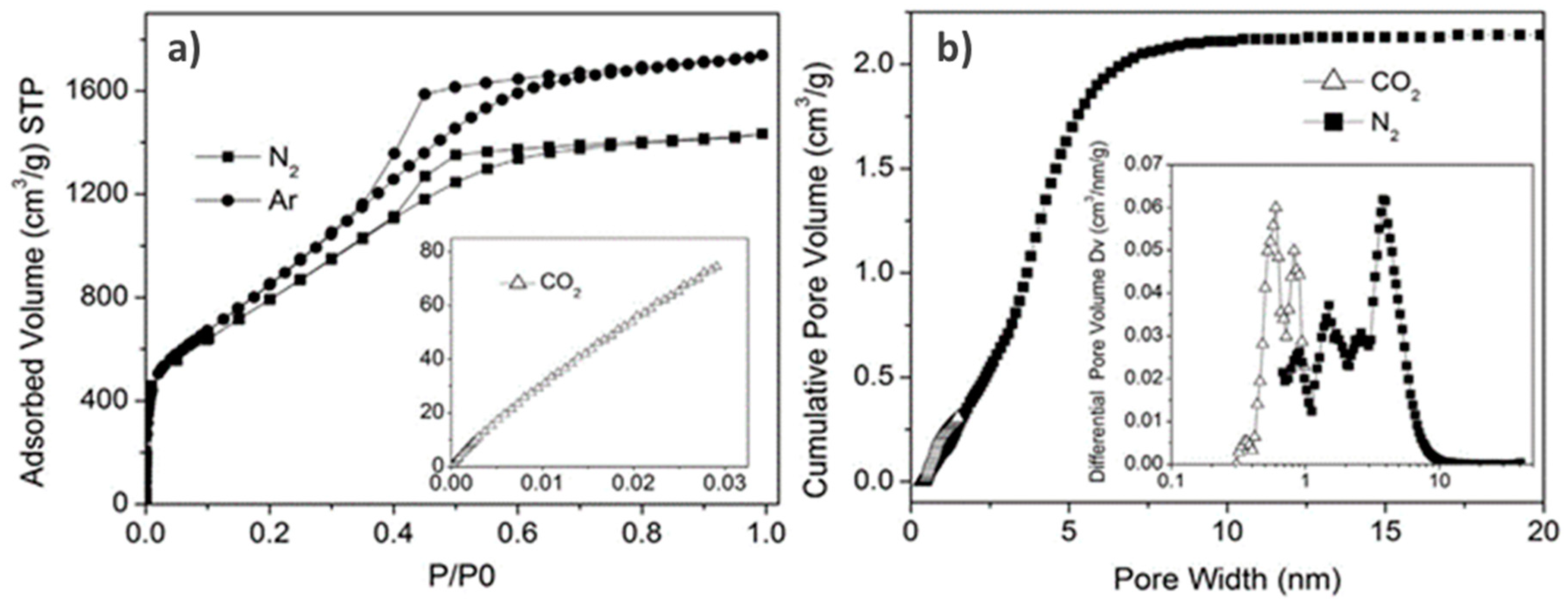{kind=link}
{kind=link}
{kind=link}
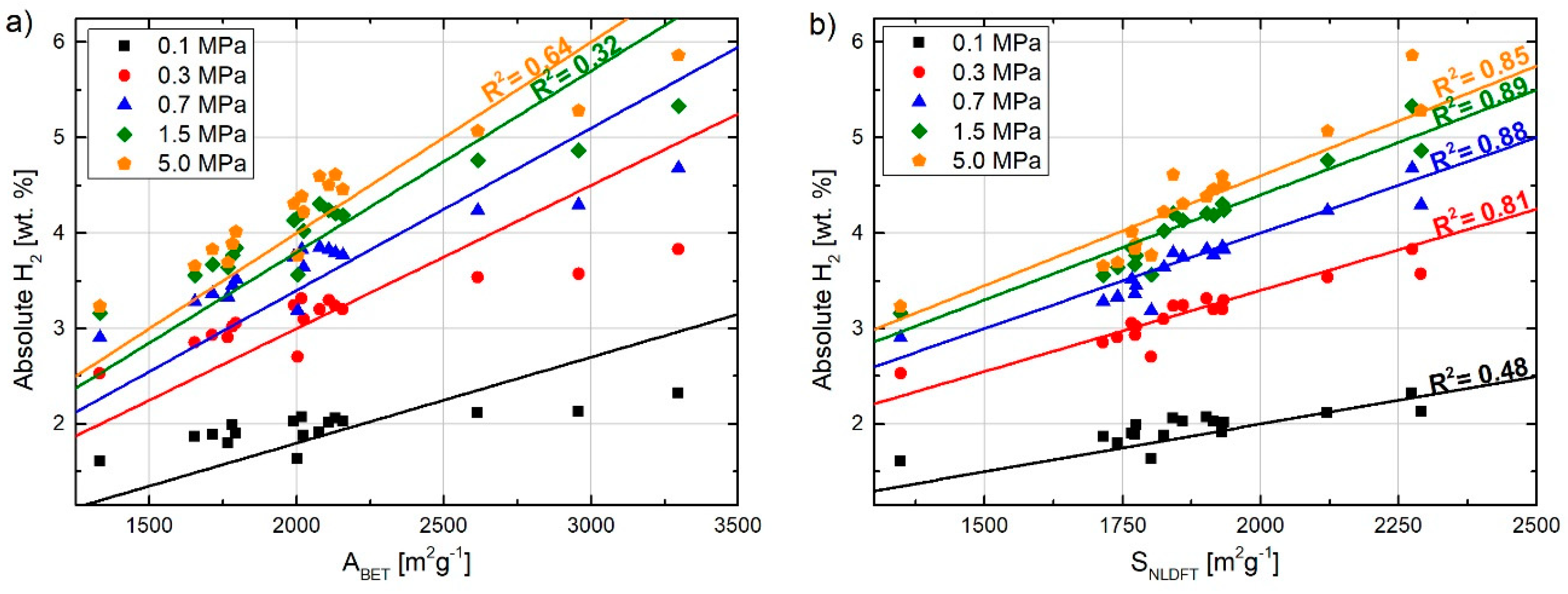
{kind=link}
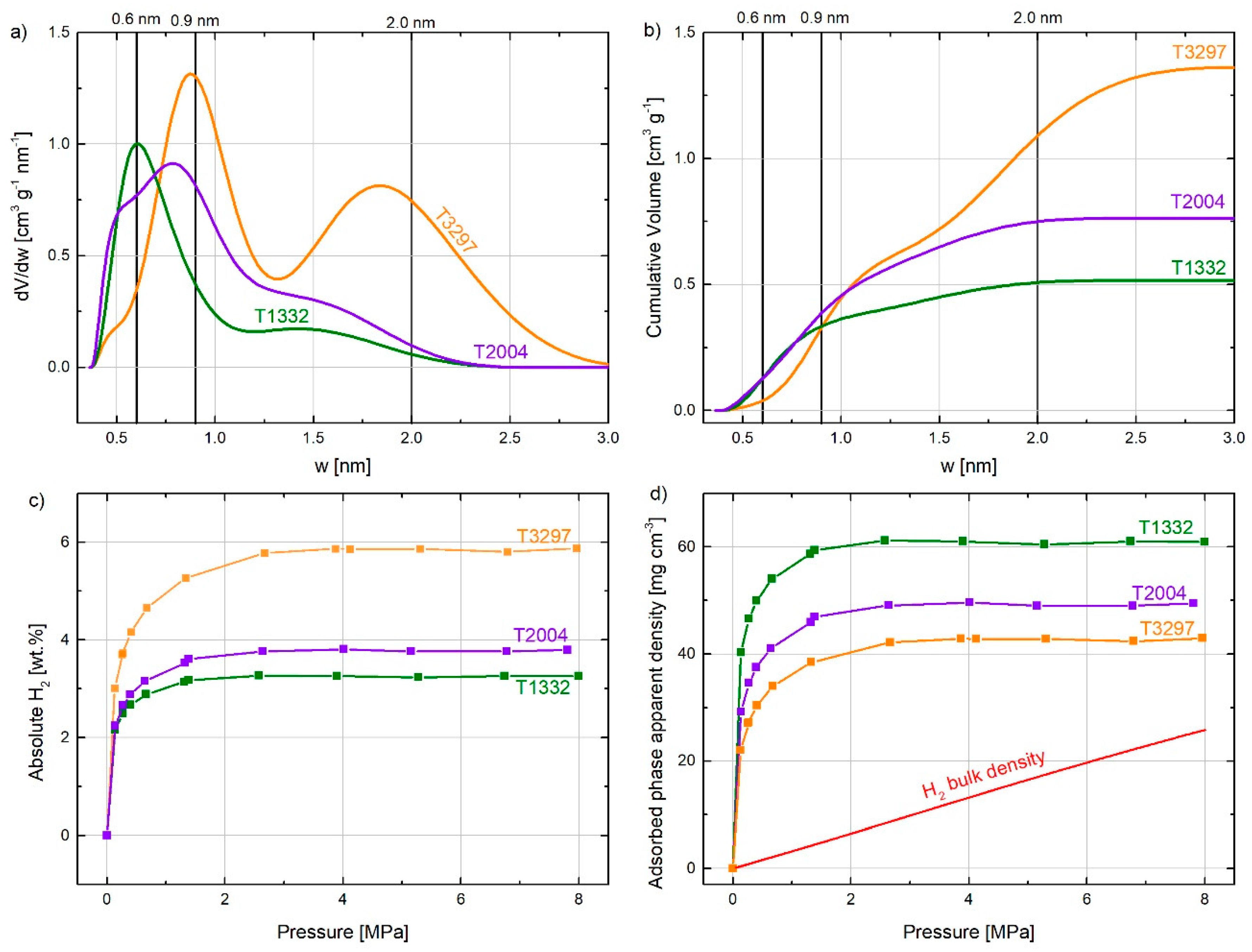
{kind=link}
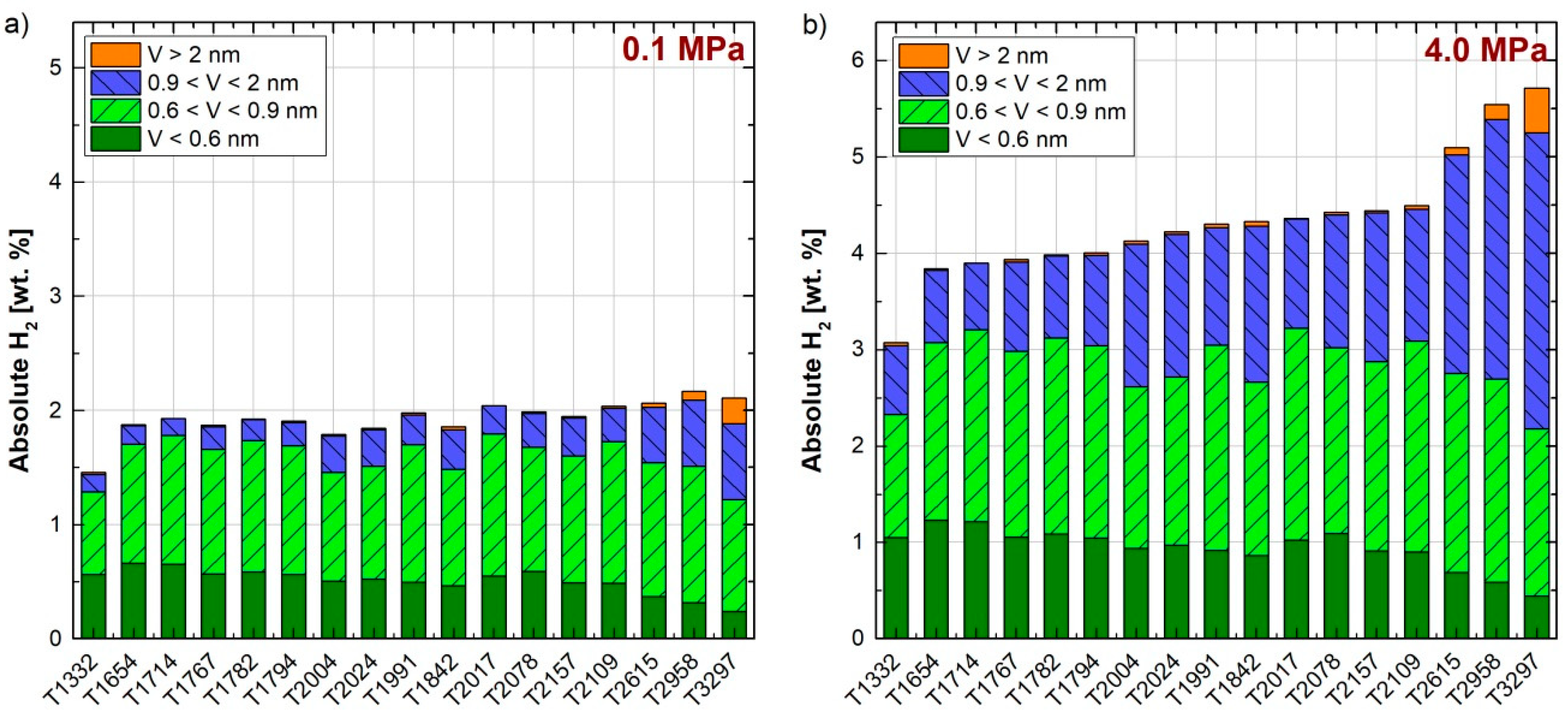
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Physical Adsorption: Characterization of Nanoporous Carbons
2.1. General
2.2. Choice of Adsorptive
2.2.1. Standard Adsorptives
2.2.2. Other Adsorptives for Carbon Characterization
2.3. Aspects of Surface Area Assessment
2.3.1. BET Method
2.3.2. Other Methods for Surface Area Assessment
2.4. Pore Size and Porosity Analysis
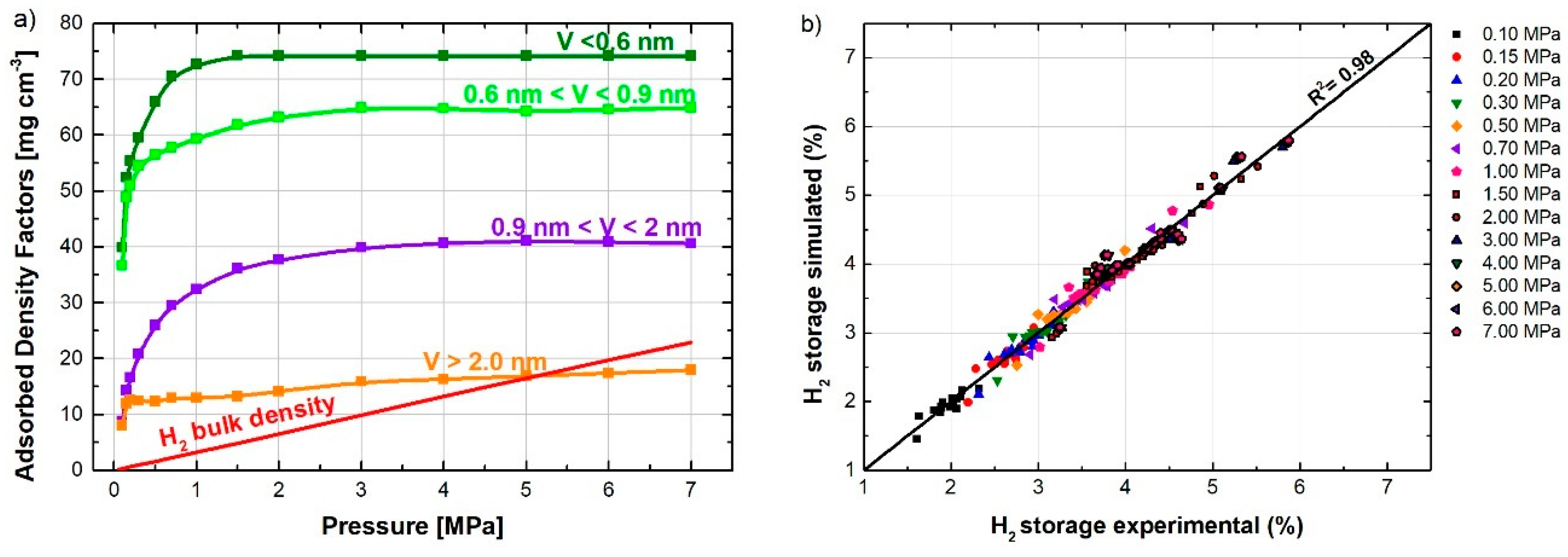
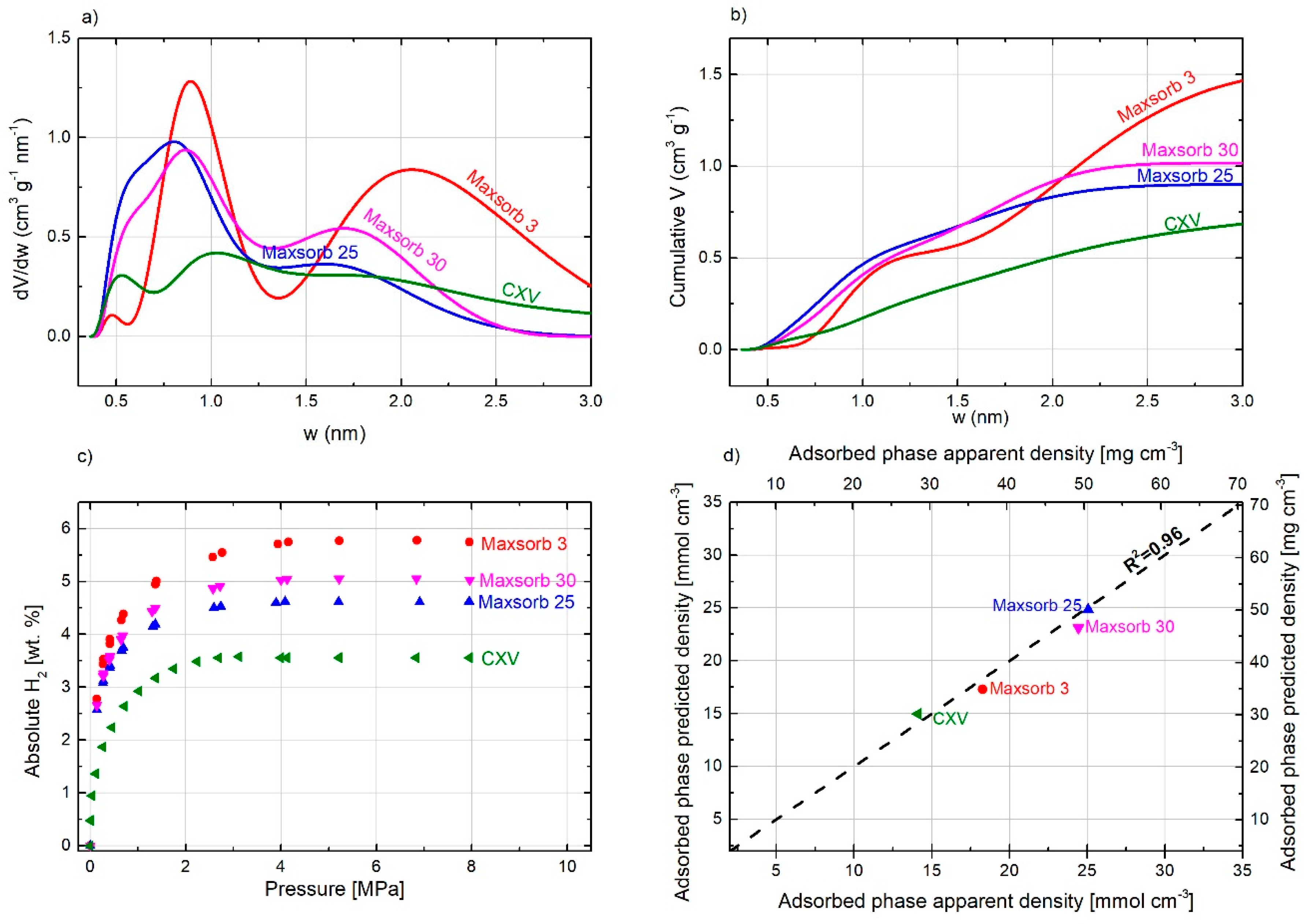
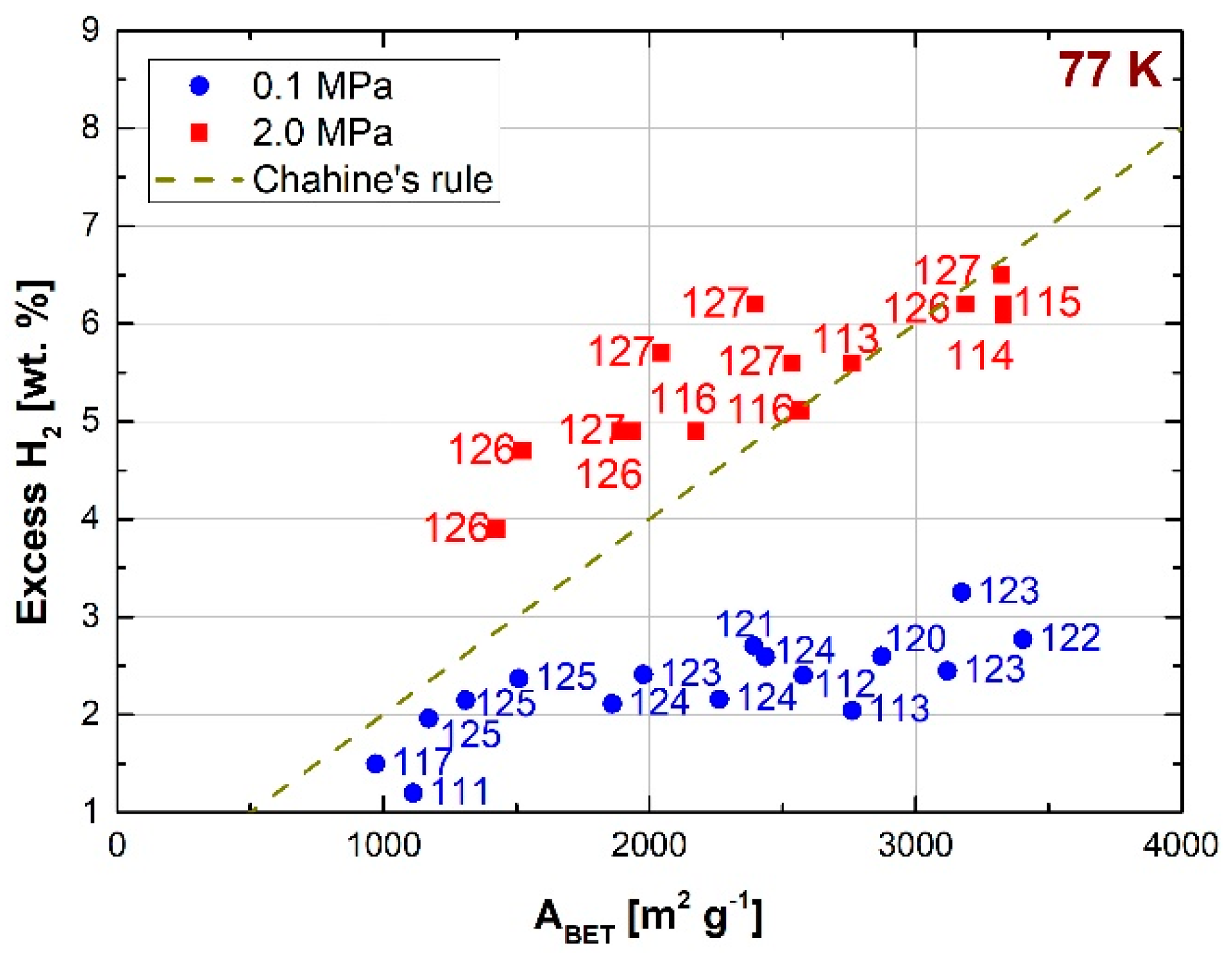
2.5. Effect of Pore Size Distribution on Hydrogen Storage
3. Carbon-Based Materials for Hydrogen Storage
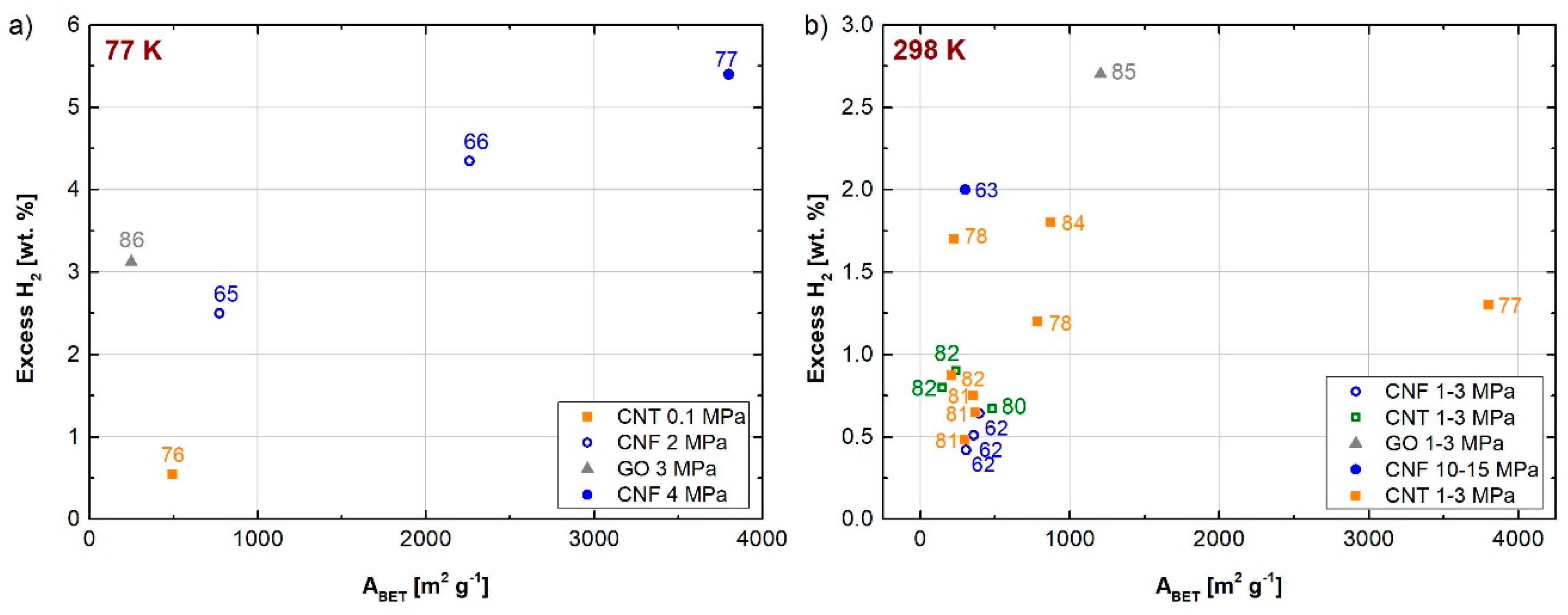
3.1. Graphene-Derived Carbons: Carbon Nanotubes and Nanofibers
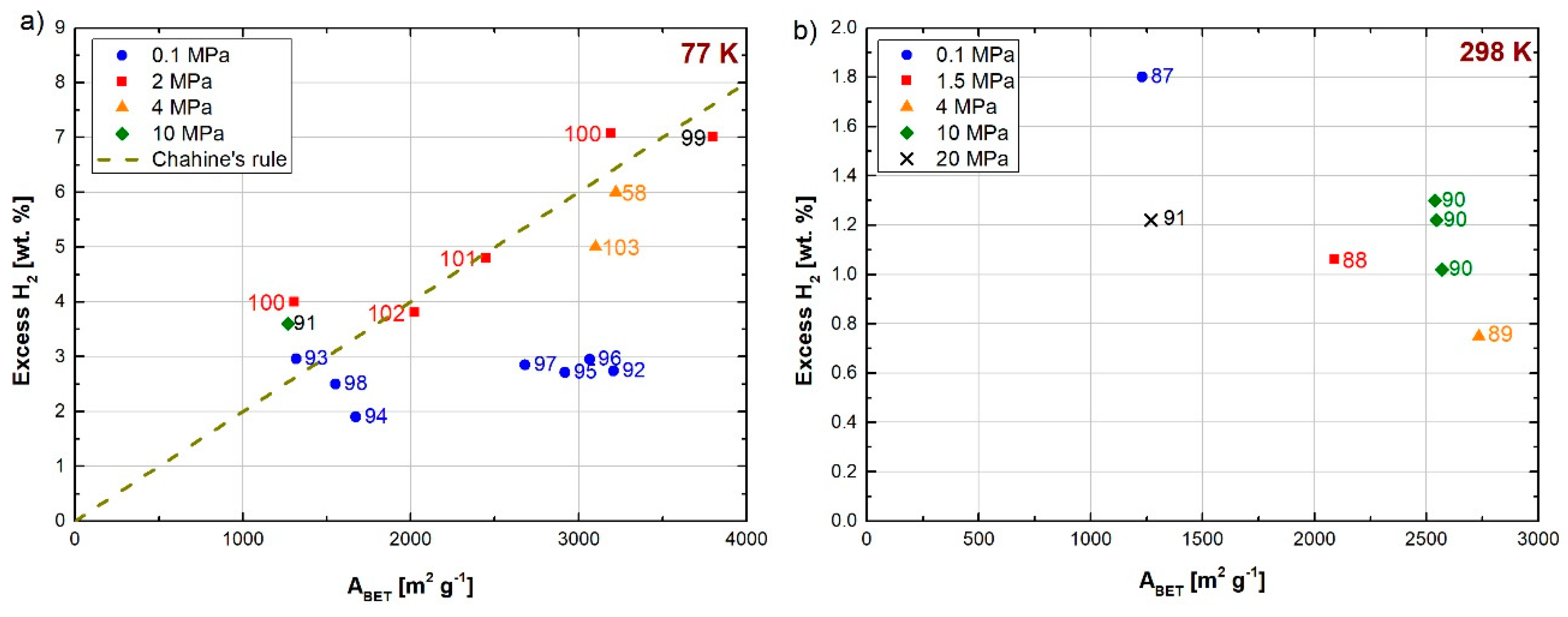
3.2. Activated Carbons
3.3. Templated Carbons
3.4. Other Carbon Materials
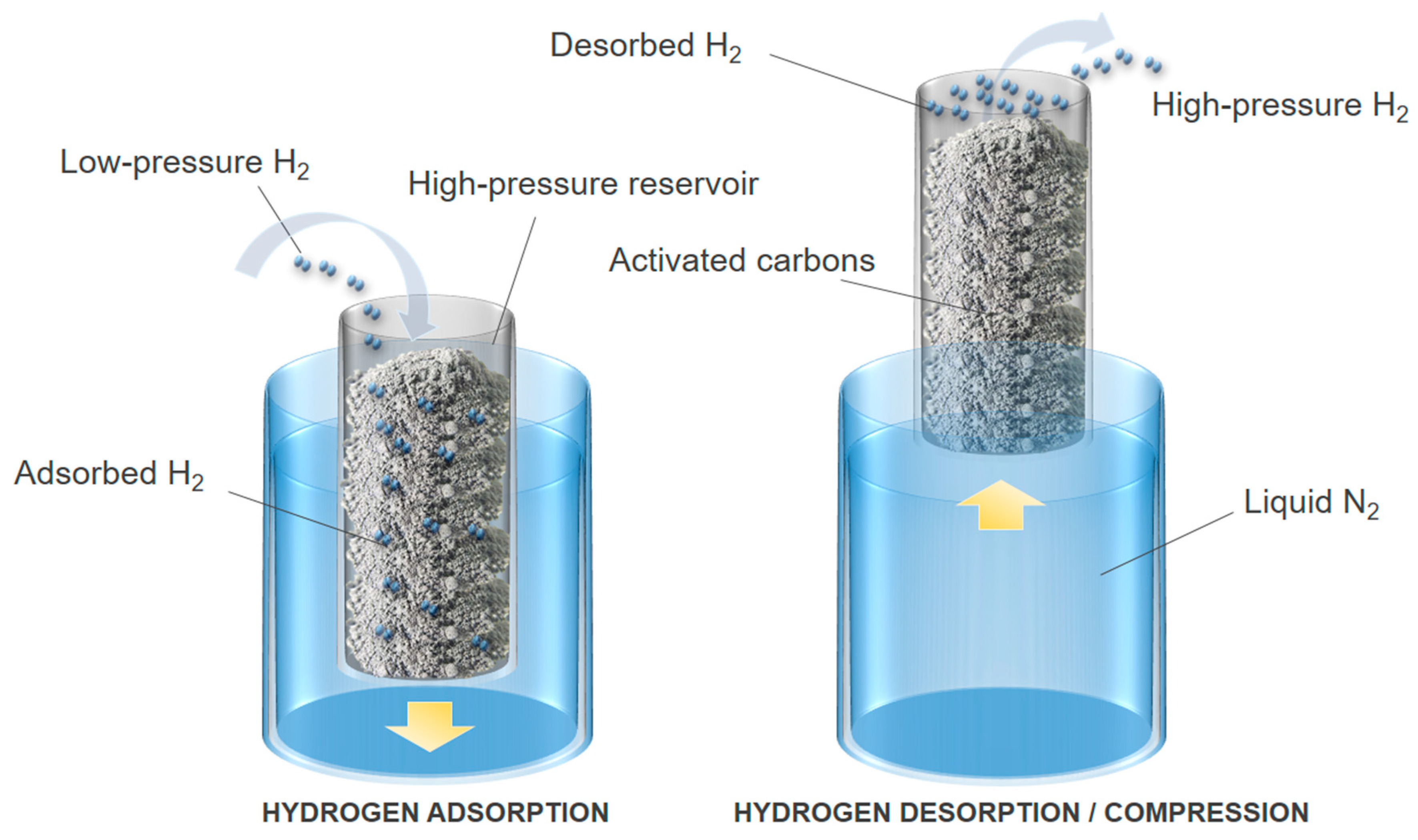
4. Carbon Materials for Hydrogen Compression
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Züttel, A.; Sudan, P.; Mauron, P.H.; Kiyobayashi, T.; Emmenegger, C.H.; Schlapbach, L. Hydrogen storage in carbon nanostructures. Int. J. Hydrogen Energy 2002, 27, 203–212. [Google Scholar] [CrossRef]
- Kadono, K.; Kajiura, H.; Shiraishi, M. Dense hydrogen adsorption on carbon subnanopores at 77 K. Appl. Phys. Lett. 2003, 83, 3392–3394. [Google Scholar] [CrossRef]
- Poirier, E.; Dailly, A. On the nature of the adsorbed hydrogen phase in microporous metal−organic frameworks at supercritical temperatures. Langmuir 2009, 25, 12169–12176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohnke, E. On the High Density Hydrogen Films Adsorbed in Carbon Nanospaces. Ph.D. Thesis, University of Missouri, Columbia, MO, USA, 2015. [Google Scholar]
- Fierro, V.; Zhao, W.; Izquierdo, M.T.; Aylon, E.; Celzard, A. Adsorption and compression contributions to hydrogen storage in activated anthracites. Int. J. Hydrogen Energy 2010, 35, 9038–9045. [Google Scholar] [CrossRef]
- De la Casa-Lillo, M.A.; Lamari-Darkrim, F.; Cazorla-Amorós, D.; Linares-Solano, A. Hydrogen storage in activated carbons and activated carbon fibers. J. Phys. Chem. B 2002, 106, 10930–10934. [Google Scholar] [CrossRef]
- Haul, R.S.J.; Gregg, K.S.W. Sing: Adsorption, surface area and porosity. Ber. Bunsenges. Phys. Chem. 1982, 86, 957. [Google Scholar] [CrossRef]
- Rouquerol, J.; Rouquerol, F.; Llewellyn, P.; Maurin, G.; Sing, K.S.W. Adsorption by Powders and Porous Solids: Principles, Methodology and Applications; Academic Press: Cambridge, MA, USA, 2013; ISBN 978-0-08-097036-3. [Google Scholar]
- Lowell, S.; Shields, J.E.; Thomas, M.A.; Thommes, M. Characterization of Porous Solids and Powders: Surface Area, Pore Size and Density; Particle Technology Series; Springer: Dordrecht, The Netherlands, 2004; ISBN 978-1-4020-2302-6. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Monson, P.A. Understanding adsorption/desorption hysteresis for fluids in mesoporous materials using simple molecular models and classical density functional theory. Microporous Mesoporous Mater. 2012, 160, 47–66. [Google Scholar] [CrossRef]
- Landers, J.; Gor, G.Y.; Neimark, A.V. Density functional theory methods for characterization of porous materials. Colloids Surf. Physicochem. Eng. Asp. 2013, 437, 3–32. [Google Scholar] [CrossRef]
- Cychosz, K.A.; Guillet-Nicolas, R.; García-Martínez, J.; Thommes, M. Recent advances in the textural characterization of hierarchically structured nanoporous materials. Chem. Soc. Rev. 2017, 46, 389–414. [Google Scholar] [CrossRef]
- Thommes, M.; Cychosz, K.A.; Neimark, A.V. Advanced physical adsorption characterization of nanoporous carbons. In Novel Carbon Adsorbent; Elsevier: Oxford, UK, 2012; pp. 107–145. [Google Scholar]
- Prehal, C.; Grätz, S.; Krüner, B.; Thommes, M.; Borchardt, L.; Presser, V.; Paris, O. Comparing pore structure models of nanoporous carbons obtained from small angle X-ray scattering and gas adsorption. Carbon 2019, 152, 416–423. [Google Scholar] [CrossRef]
- Garrido, J.; Linares-Solano, A.; Martin-Martinez, J.M.; Molina-Sabio, M.; Rodriguez-Reinoso, F.; Torregrosa, R. Use of nitrogen vs. carbon dioxide in the characterization of activated carbons. Langmuir 1987, 3, 76–81. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, D.; Xu, D.; Asahina, S.; Cychosz, K.A.; Agrawal, K.V.; Wahedi, Y.A.; Bhan, A.; Hashimi, S.A.; Terasaki, O.; et al. Synthesis of self-pillared zeolite nanosheets by repetitive branching. Science 2012, 336, 1684–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagiello, J.; Ania, C.; Parra, J.B.; Cook, C. Dual gas analysis of microporous carbons using 2D-NLDFT heterogeneous surface model and combined adsorption data of N2 and CO2. Carbon 2015, 91, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Jagiello, J.; Kenvin, J.; Celzard, A.; Fierro, V. Enhanced resolution of ultra micropore size determination of biochars and activated carbons by dual gas analysis using N2 and CO2 with 2D-NLDFT adsorption models. Carbon 2019, 144, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Dantas, S.; Struckhoff, K.C.; Thommes, M.; Neimark, A.V. Phase behavior and capillary condensation hysteresis of carbon dioxide in mesopores. Langmuir 2019, 35, 11291–11298. [Google Scholar] [CrossRef]
- Jagiello, J.; Ansón, A.; Martínez, M.T. DFT-based prediction of high-pressure H2 adsorption on porous carbons at ambient temperatures from low-pressure adsorption data measured at 77 K. J. Phys. Chem. B 2006, 110, 4531–4534. [Google Scholar] [CrossRef]
- Jagiello, J.; Thommes, M. Comparison of DFT characterization methods based on N2, Ar, CO2, and H2 adsorption applied to carbons with various pore size distributions. Carbon 2004, 42, 1227–1232. [Google Scholar] [CrossRef]
- Jagiello, J.; Kenvin, J.; Ania, C.O.; Parra, J.B.; Celzard, A.; Fierro, V. Exploiting the adsorption of simple gases O2 and H2 with minimal quadrupole moments for the dual gas characterization of nanoporous carbons using 2D-NLDFT models. Carbon 2020, 160, 164–175. [Google Scholar] [CrossRef]
- Kuwabara, H.; Suzuki, T.; Kaneko, K. Ultramicropores in microporous carbon fibres evidenced by helium adsorption at 4.2 K. J. Chem. Soc. Faraday Trans. 1991, 87, 1915–1916. [Google Scholar] [CrossRef]
- Suzuki, M. Fundamentals of Adsorption; Elsevier: Amsterdam, The Netherlands, 1993; ISBN 978-0-08-088772-2. [Google Scholar]
- Fierro, V.; Bosch, G.; Siperstein, F.R. Pore size distribution in microporous carbons obtained from molecular modeling and density functional theory. Stud. Surf. Sci. Catal. 2007, 519–526. [Google Scholar] [CrossRef]
- Sdanghi, G.; Schaefer, S.; Maranzana, G.; Celzard, A.; Fierro, V. Application of the modified Dubinin-Astakhov equation for a better understanding of high-pressure hydrogen adsorption on activated carbons. Int. J. Hydrogen Energy 2019. [Google Scholar] [CrossRef] [Green Version]
- Poirier, E.; Chahine, R.; Bose, T.K. Hydrogen adsorption in carbon nanostructures. Int. J. Hydrogen Energy 2001, 26, 831–835. [Google Scholar] [CrossRef]
- Rouquerol, J.; Llewellyn, P.; Rouquerol, F. Is the bet equation applicable to microporous adsorbents? In Studies in Surface Science and Catalysis; Llewellyn, P.L., Rodriquez-Reinoso, F., Rouqerol, J., Seaton, N., Eds.; Characterization of Porous Solids VII; Elsevier: Amsterdam, The Netherlands, 2007; Volume 160, pp. 49–56. [Google Scholar]
- Keii, T.; Takagi, T.; Kanetaka, S. A new plotting of the BET method. Anal. Chem. 1961, 33, 1965. [Google Scholar] [CrossRef]
- Kaneko, K.; Itoh, T.; Fujimori, T. Collective interactions of molecules with an interfacial solid. Chem. Lett. 2012, 41, 466–475. [Google Scholar] [CrossRef] [Green Version]
- Centeno, T.A.; Stoeckli, F. The assessment of surface areas in porous carbons by two model-independent techniques, the DR equation and DFT. Carbon 2010, 48, 2478–2486. [Google Scholar] [CrossRef] [Green Version]
- Horváth, G.; Kawazoe, K. Method for the calculation of effective pore size distribution in molecular sieve carbon. J. Chem. Eng. Jpn. 1983, 16, 470–475. [Google Scholar] [CrossRef] [Green Version]
- Tarazona, P. Solid-fluid transition and interfaces with density functional approaches. Surf. Sci. 1995, 331–333, 989–994. [Google Scholar] [CrossRef]
- Lastoskie, C.; Gubbins, K.E.; Quirke, N. Pore size distribution analysis of microporous carbons: A density functional theory approach. J. Phys. Chem. 1993, 97, 4786–4796. [Google Scholar] [CrossRef]
- Olivier, J.P.; Conklin, W.B.; Szombathely, M.V. Determination of pore size distribution from density functional theory: A comparison of nitrogen and argon results. In Studies in Surface Science and Catalysis; Rouquerol, J., Rodríguez-Reinoso, F., Sing, K.S.W., Unger, K.K., Eds.; Characterization of Porous Solids III; Elsevier: Amsterdam, The Netherlands, 1994; Volume 87, pp. 81–89. [Google Scholar]
- Neimark, A.V. The method of indeterminate Lagrange multipliers in nonlocal density functional theory. Langmuir 1995, 11, 4183–4184. [Google Scholar] [CrossRef]
- Olivier, J.P. Improving the models used for calculating the size distribution of micropore volume of activated carbons from adsorption data. Carbon 1998, 36, 1469–1472. [Google Scholar] [CrossRef]
- Bandosz, T.J.; Biggs, M.J.; Gubbins, K.E.; Hattori, Y.; Iiyama, T.; Kaneko, K.; Pikunic, J.; Thomson, K.T. Molecular models of porous carbons. Chem. Phys. Carbon 2003, 28, 41–228. [Google Scholar]
- Gor, G.Y.; Thommes, M.; Cychosz, K.A.; Neimark, A.V. Quenched solid density functional theory method for characterization of mesoporous carbons by nitrogen adsorption. Carbon 2012, 50, 1583–1590. [Google Scholar] [CrossRef]
- Cychosz, K.A.; Guo, X.; Fan, W.; Cimino, R.; Gor, G.Y.; Tsapatsis, M.; Neimark, A.V.; Thommes, M. Characterization of the pore structure of three-dimensionally ordered mesoporous carbons using high resolution gas sorption. Langmuir 2012, 28, 12647–12654. [Google Scholar] [CrossRef] [PubMed]
- Jagiello, J.; Olivier, J.P. A simple two-dimensional NLDFT model of gas adsorption in finite carbon pores. Application to pore structure analysis. J. Phys. Chem. C 2009, 113, 19382–19385. [Google Scholar] [CrossRef]
- Hu, X.; Radosz, M.; Cychosz, K.A.; Thommes, M. CO2-filling capacity and selectivity of carbon nanopores: Synthesis, texture, and pore-size distribution from Quenched-Solid Density Functional Theory (QSDFT). Environ. Sci. Technol. 2011, 45, 7068–7074. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, S.; Fierro, V.; Izquierdo, M.T.; Celzard, A. Assessment of hydrogen storage in activated carbons produced from hydrothermally treated organic materials. Int. J. Hydrogen Energy 2016, 41, 12146–12156. [Google Scholar] [CrossRef] [Green Version]
- Rzepka, M.; Lamp, P.; de la Casa-Lillo, M.A. Physisorption of hydrogen on microporous carbon and carbon nanotubes. J. Phys. Chem. B 1998, 102, 10894–10898. [Google Scholar] [CrossRef] [Green Version]
- Cabria, I.; López, M.J.; Alonso, J.A. The optimum average nanopore size for hydrogen storage in carbon nanoporous materials. Carbon 2007, 45, 2649–2658. [Google Scholar] [CrossRef]
- Zhao, W.; Fierro, V.; Zlotea, C.; Aylon, E.; Izquierdo, M.T.; Latroche, M.; Celzard, A. Activated carbons with appropriate micropore size distribution for hydrogen adsorption. Int. J. Hydrogen Energy 2011, 36, 5431–5434. [Google Scholar] [CrossRef]
- Jhung, S.H.; Kim, H.-K.; Yoon, J.W.; Chang, J.-S. Low-temperature adsorption of hydrogen on nanoporous Aluminophosphates: Effect of pore size. J. Phys. Chem. B 2006, 110, 9371–9374. [Google Scholar] [CrossRef] [PubMed]
- Tellez-Juárez, M.C.; Fierro, V.; Zhao, W.; Fernández-Huerta, N.; Izquierdo, M.T.; Reguera, E.; Celzard, A. Hydrogen storage in activated carbons produced from coals of different ranks: Effect of oxygen content. Int. J. Hydrogen Energy 2014, 39, 4996–5002. [Google Scholar] [CrossRef]
- Kostoglou, N.; Koczwara, C.; Prehal, C.; Terziyska, V.; Babic, B.; Matovic, B.; Constantinides, G.; Tampaxis, C.; Charalambopoulou, G.; Steriotis, T.; et al. Nanoporous activated carbon cloth as a versatile material for hydrogen adsorption, selective gas separation and electrochemical energy storage. Nano Energy 2017, 40, 49–64. [Google Scholar] [CrossRef]
- Kostoglou, N.; Tarat, A.; Walters, I.; Ryzhkov, V.; Tampaxis, C.; Charalambopoulou, G.; Steriotis, T.; Mitterer, C.; Rebholz, C. Few-layer graphene-like flakes derived by plasma treatment: A potential material for hydrogen adsorption and storage. Microporous Mesoporous Mater. 2016, 225, 482–487. [Google Scholar] [CrossRef]
- Zhao, W.; Fierro, V.; Zlotea, C.; Aylon, E.; Izquierdo, M.T.; Latroche, M.; Celzard, A. Optimization of activated carbons for hydrogen storage. Int. J. Hydrogen Energy 2011, 36, 11746–11751. [Google Scholar] [CrossRef]
- Nelder, J.A.; Mead, R. A simplex method for function minimization. Comput. J. 1964, 7, 308–313. [Google Scholar] [CrossRef]
- Patchkovskii, S.; Tse, J.S.; Yurchenko, S.N.; Zhechkov, L.; Heine, T.; Seifert, G. Graphene nanostructures as tunable storage media for molecular hydrogen. Proc. Natl. Acad. Sci. USA 2005, 102, 10439–10444. [Google Scholar] [CrossRef] [Green Version]
- Georgiev, P.A.; Ross, D.K.; Albers, P.; Ramirez-Cuesta, A.J. The rotational and translational dynamics of molecular hydrogen physisorbed in activated carbon: A direct probe of microporosity and hydrogen storage performance. Carbon 2006, 44, 2724–2738. [Google Scholar] [CrossRef]
- Celzard, A.; Fierro, V.; Marêché, J.F.; Furdin, G. Advanced preparative strategies for activated carbons designed for the adsorptive storage of hydrogen. Adsorpt. Sci. Technol. 2007, 25, 129–142. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Portet, C.; Osswald, S.; Simmons, J.M.; Yildirim, T.; Laudisio, G.; Fischer, J.E. Importance of pore size in high-pressure hydrogen storage by porous carbons. Int. J. Hydrogen Energy 2009, 34, 6314–6319. [Google Scholar] [CrossRef]
- Fierro, V.; Szczurek, A.; Zlotea, C.; Marêché, J.F.; Izquierdo, M.T.; Albiniak, A.; Latroche, M.; Furdin, G.; Celzard, A. Experimental evidence of an upper limit for hydrogen storage at 77 K on activated carbons. Carbon 2010, 48, 1902–1911. [Google Scholar] [CrossRef]
- Sdanghi, G.; Maranzana, G.; Celzard, A.; Fierro, V. Hydrogen adsorption on nanotextured carbon materials. In Hyrdogen Storage Technologies; Wiley: Hoboken, NJ, USA, 2018; pp. 263–297. ISBN 978-1-119-46057-2. [Google Scholar]
- Chambers, A.; Park, C.; Baker, R.T.K.; Rodriguez, N.M. Hydrogen storage in graphite nanofibers. J. Phys. Chem. B 1998, 102, 4253–4256. [Google Scholar] [CrossRef]
- Rzepka, M.; Bauer, E.; Reichenauer, G.; Schliermann, T.; Bernhardt, B.; Bohmhammel, K.; Henneberg, E.; Knoll, U.; Maneck, H.E.; Braue, W. Hydrogen storage capacity of catalytically grown carbon nanofibers. J. Phys. Chem. B 2005, 109, 14979–14989. [Google Scholar] [CrossRef]
- Jaybhaye, S.; Sharon, M.; Sharon, M.; Sathiyamoorthy, D.; Dasgupta, K. Semiconducting carbon nanofibers and hydrogen storage. Synth. React. Inorg. Met. Org. Nano-Met. Chem. 2007, 37, 473–476. [Google Scholar] [CrossRef]
- Kim, B.-J.; Park, S.-J. Preparation of nanoporous carbons from graphite nanofibres. Nanotechnology 2006, 17, 4395–4398. [Google Scholar] [CrossRef]
- Kim, B.-J.; Lee, Y.-S.; Park, S.-J. A study on the hydrogen storage capacity of Ni-plated porous carbon nanofibers. Int. J. Hydrogen Energy 2008, 33, 4112–4115. [Google Scholar] [CrossRef]
- Yadav, A.; Faisal, M.; Subramaniam, A.; Verma, N. Nickel nanoparticle-doped and steam-modified multiscale structure of carbon micro-nanofibers for hydrogen storage: Effects of metal, surface texture and operating conditions. Int. J. Hydrogen Energy 2017, 42, 6104–6117. [Google Scholar] [CrossRef]
- Hwang, S.-H.; Choi, W.M.; Lim, S.K. Hydrogen storage characteristics of carbon fibers derived from rice straw and paper mulberry. Mater. Lett. 2016, 167, 18–21. [Google Scholar] [CrossRef]
- Ullah Rather, S. Preparation, characterization and hydrogen storage studies of carbon nanotubes and their composites: A review. Int. J. Hydrogen Energy 2020, 45, 4653–4672. [Google Scholar] [CrossRef]
- Darkrim, F.L.; Malbrunot, P.; Tartaglia, G.P. Review of hydrogen storage by adsorption in carbon nanotubes. Int. J. Hydrogen Energy 2002, 27, 193–202. [Google Scholar] [CrossRef]
- Dillon, A.C.; Jones, K.M.; Bekkedahl, T.A.; Kiang, C.H.; Bethune, D.S.; Heben, M.J. Storage of hydrogen in single-walled carbon nanotubes. Nature 1997, 386, 377–379. [Google Scholar] [CrossRef]
- Chen, P.; Wu, X.; Lin, J.; Tan, K.L. High H2 uptake by alkali-doped carbon nanotubes under ambient pressure and moderate temperatures. Science 1999, 285, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.T. Hydrogen storage by alkali-doped carbon nanotubes-revisited. Carbon 2000, 38, 623–626. [Google Scholar] [CrossRef]
- Minami, D.; Ohkubo, T.; Kuroda, Y.; Sakai, K.; Sakai, H.; Abe, M. Structural optimization of arranged carbon nanotubes for hydrogen storage by grand canonical Monte Carlo simulation. Int. J. Hydrogen Energy 2010, 35, 12398–12404. [Google Scholar] [CrossRef]
- Saito, Y.; Yoshikawa, T.; Bandow, S.; Tomita, M.; Hayashi, T. Interlayer spacings in carbon nanotubes. Phys. Rev. B 1993, 48, 1907–1909. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Padmanabhan, V. Hydrogen storage capacity of bundles of single-walled carbon nanotubes with defects. Int. J. Energy Res. 2017, 41, 1108–1117. [Google Scholar] [CrossRef]
- Wen, Y.W.; Liu, H.J.; Pan, L.; Tan, X.J.; Lv, H.Y.; Shi, J.; Tang, X.F. A triplet form of (5,0) carbon nanotube with higher hydrogen storage capacity. J. Phys. Chem. C 2011, 115, 9227–9231. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Park, S.-J. Influence of the pore size in multi-walled carbon nanotubes on the hydrogen storage behaviors. J. Solid State Chem. 2012, 194, 307–312. [Google Scholar] [CrossRef]
- Adeniran, B.; Mokaya, R. Low temperature synthesized carbon nanotube superstructures with superior CO2 and hydrogen storage capacity. J. Mater. Chem. A 2015, 3, 5148–5161. [Google Scholar] [CrossRef] [Green Version]
- Lyu, J.; Kudiiarov, V.; Lider, A. An overview of the recent progress in modifications of carbon nanotubes for hydrogen adsorption. Nanomaterials 2020, 10, 255. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.-H.; Li, S.-J.; Su, Y.; Chen, G. Theoretical study of hydrogen storage by spillover on porous carbon materials. Int. J. Hydrogen Energy 2020. [Google Scholar] [CrossRef]
- Elyassi, M.; Rashidi, A.; Hantehzadeh, M.R.; Elahi, S.M. Hydrogen storage behaviors by adsorption on multi-walled carbon nanotubes. J. Inorg. Organomet. Polym. Mater. 2017, 27, 285–295. [Google Scholar] [CrossRef]
- Lee, S.Y.; Soo, J.P. Hydrogen storage behaviors of Ni-loaded activated carbon nanotubes. Adv. Mater. Res. 2010, 123–125, 695–698. [Google Scholar] [CrossRef]
- Rather, S.U.; Hwang, S.-W. Comparative hydrogen uptake study on titanium-MWCNTs composite prepared by two different methods. Int. J. Hydrogen Energy 2016, 41, 18114–18120. [Google Scholar] [CrossRef]
- Sharma, A. Investigation on platinum loaded multi-walled carbon nanotubes for hydrogen storage applications. Int. J. Hydrogen Energy 2020, 45, 2967–2974. [Google Scholar] [CrossRef]
- Ariharan, A.; Viswanathan, B.; Nandhakumar, V. Nitrogen-incorporated carbon nanotube derived from polystyrene and polypyrrole as hydrogen storage material. Int. J. Hydrogen Energy 2018, 43, 5077–5088. [Google Scholar] [CrossRef]
- Yuan, W.; Li, B.; Li, L. A green synthetic approach to graphene nanosheets for hydrogen adsorption. Appl. Surf. Sci. 2011, 257, 10183–10187. [Google Scholar] [CrossRef]
- Singh, S.B.; De, M. Thermally exfoliated graphene oxide for hydrogen storage. Mater. Chem. Phys. 2020, 239, 122102. [Google Scholar] [CrossRef]
- Rowlandson, J.L.; Edler, K.J.; Tian, M.; Ting, V.P. Toward process-resilient lignin-derived activated carbons for hydrogen storage applications. ACS Sustain. Chem. Eng. 2020, 8, 2186–2195. [Google Scholar] [CrossRef]
- Samantaray, S.S.; Mangisetti, S.R.; Ramaprabhu, S. Investigation of room temperature hydrogen storage in biomass derived activated carbon. J. Alloys Compd. 2019, 789, 800–804. [Google Scholar] [CrossRef]
- Zhao, W.; Luo, L.; Chen, T.; Li, Z.; Zhang, Z.; Wang, H.; Rao, J.; Feo, L.; Fan, M. Synthesis and characterization of Pt-N-doped activated biocarbon composites for hydrogen storage. Compos. Part B Eng. 2019, 161, 464–472. [Google Scholar] [CrossRef]
- Wang, L.; Yang, R.T. Hydrogen storage properties of carbons doped with ruthenium, platinum, and nickel nanoparticles. J. Phys. Chem. C 2008, 112, 12486–12494. [Google Scholar] [CrossRef]
- Bader, N.; Ouederni, A. Optimization of biomass-based carbon materials for hydrogen storage. J. Energy Storage 2016, 5, 77–84. [Google Scholar] [CrossRef]
- Zhao, W.; Luo, L.; Wang, H.; Fan, M. Synthesis of bamboo-based activated carbons with super-high specific surface area for hydrogen storage. BioResources 2017, 12, 1246–1262. [Google Scholar] [CrossRef] [Green Version]
- Sethia, G.; Sayari, A. Activated carbon with optimum pore size distribution for hydrogen storage. Carbon 2016, 99, 289–294. [Google Scholar] [CrossRef]
- Zhao, W.; Luo, L.; Fan, M. Preparation and characterization of nitrogen-containing cellular activated carbon for CO2 and H2 adsorption. Nano 2017, 12, 1750007. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, L.; Xu, F.; Zhou, H.; Peng, X.; Sun, D.; Wang, J.; Du, Y. Nitrogen-doped porous carbons with high performance for hydrogen storage. Int. J. Hydrogen Energy 2016, 41, 8489–8497. [Google Scholar] [CrossRef]
- Wróbel-Iwaniec, I.; Díez, N.; Gryglewicz, G. Chitosan-based highly activated carbons for hydrogen storage. Int. J. Hydrogen Energy 2015, 40, 5788–5796. [Google Scholar] [CrossRef]
- Heo, Y.-J.; Park, S.-J. Synthesis of activated carbon derived from rice husks for improving hydrogen storage capacity. J. Ind. Eng. Chem. 2015, 31, 330–334. [Google Scholar] [CrossRef]
- Choi, Y.-K.; Park, S.-J. Preparation and characterization of sucrose-based microporous carbons for increasing hydrogen storage. J. Ind. Eng. Chem. 2015, 28, 32–36. [Google Scholar] [CrossRef]
- Blankenship, T.S., II; Balahmar, N.; Mokaya, R. Oxygen-rich microporous carbons with exceptional hydrogen storage capacity. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Gao, Q.; Hu, J. High hydrogen storage capacity of porous carbons prepared by using activated carbon. J. Am. Chem. Soc. 2009, 131, 7016–7022. [Google Scholar] [CrossRef]
- Zhao, W.; Fierro, V.; Zlotea, C.; Izquierdo, M.T.; Chevalier-César, C.; Latroche, M.; Celzard, A. Activated carbons doped with Pd nanoparticles for hydrogen storage. Int. J. Hydrogen Energy 2012, 37, 5072–5080. [Google Scholar] [CrossRef]
- Zhao, W.; Fierro, V.; Fernández-Huerta, N.; Izquierdo, M.T.; Celzard, A. Hydrogen uptake of high surface area-activated carbons doped with nitrogen. Int. J. Hydrogen Energy 2013, 38, 10453–10460. [Google Scholar] [CrossRef] [Green Version]
- Romanos, J.; Beckner, M.; Prosniewski, M.; Rash, T.; Lee, M.; Robertson, J.D.; Firlej, L.; Kuchta, B.; Pfeifer, P. Boron-neutron capture on activated carbon for hydrogen storage. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yahya, M.A.; Al-Qodah, Z.; Ngah, C.W.Z. Agricultural bio-waste materials as potential sustainable precursors used for activated carbon production: A review. Renew. Sustain. Energy Rev. 2015, 46, 218–235. [Google Scholar] [CrossRef]
- Demiral, H.; Demiral, İ.; Karabacakoğlu, B.; Tümsek, F. Production of activated carbon from olive bagasse by physical activation. Chem. Eng. Res. Des. 2011, 89, 206–213. [Google Scholar] [CrossRef]
- Loloie, Z.; Mozaffarian, M.; Soleimani, M.; Asassian, N. Carbonization and CO2 activation of scrap tires: Optimization of specific surface area by the Taguchi method. Korean J. Chem. Eng. 2016, 2, 366–375. [Google Scholar] [CrossRef]
- Melouki, R.; Llewellyn, P.L.; Tazibet, S.; Boucheffa, Y. Hydrogen adsorption on activated carbons prepared from olive waste: Effect of activation conditions on uptakes and adsorption energies. J. Porous Mater. 2017, 24, 1–11. [Google Scholar] [CrossRef]
- Bader, N.; Zacharia, R.; Abdelmottaleb, O.; Cossement, D. How the activation process modifies the hydrogen storage behavior of biomass-derived activated carbons. J. Porous Mater. 2018, 25, 221–234. [Google Scholar] [CrossRef]
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Nesterenko, E.P.; Nesterenko, P.N.; Brabazon, D.; Zhou, L.; Glennon, J.D.; Luong, J.H.T.; Paull, B. Fabrication and characterization of nanotemplated carbon monolithic material. ACS Appl. Mater. Interfaces 2013, 5, 8572–8580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musyoka, N.M.; Ren, J.; Langmi, H.W.; North, B.C.; Mathe, M. A comparison of hydrogen storage capacity of commercial and fly ash-derived zeolite X together with their respective templated carbon derivatives. Int. J. Hydrogen Energy 2015, 40, 12705–12712. [Google Scholar] [CrossRef]
- Yang, Z.; Xia, Y.; Mokaya, R. Enhanced hydrogen storage capacity of high surface area zeolite-like carbon materials. J. Am. Chem. Soc. 2007, 129, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yang, Z.; Gou, X.; Zhu, Y. A simple method for the production of highly ordered porous carbon materials with increased hydrogen uptake capacities. Int. J. Hydrogen Energy 2013, 38, 5039–5052. [Google Scholar] [CrossRef]
- Masika, E.; Mokaya, R. Preparation of ultrahigh surface area porous carbons templated using zeolite 13X for enhanced hydrogen storage. Prog. Nat. Sci. Mater. Int. 2013, 23, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Li, L.; Lv, X.; Yang, C.; Zhao, X. Large surface area ordered porous carbons via nanocasting zeolite 10X and high performance for hydrogen storage application. ACS Appl. Mater. Interfaces 2014, 6, 167–175. [Google Scholar] [CrossRef]
- Yang, Z.; Xiong, W.; Wang, J.; Zhu, Y.; Xia, Y. A systematic study on the preparation and hydrogen storage of zeolite 13X-templated microporous carbons. Eur. J. Inorg. Chem. 2016, 2152–2158. [Google Scholar] [CrossRef] [Green Version]
- Carraro, P.M.; García Blanco, A.A.; Lener, G.; Barrera, D.; Amaya-Roncancio, S.; Chanquía, C.; Troiani, H.; Oliva, M.I.; Eimer, G.A. Nanostructured carbons modified with nickel as potential novel reversible hydrogen storage materials: Effects of nickel particle size. Microporous Mesoporous Mater. 2019, 273, 50–59. [Google Scholar] [CrossRef]
- Juárez, J.M.; Costa, M.G.; Anunziata, O.A. Direct synthesis of ordered mesoporous carbon applied in hydrogen storage. J. Porous Mater. 2018, 25, 1359–1363. [Google Scholar] [CrossRef]
- Attia, N.F.; Lee, S.M.; Kim, H.J.; Geckeler, K.E. Nanoporous carbon-templated silica nanoparticles: Preparation, effect of different carbon precursors, and their hydrogen storage adsorption. Microporous Mesoporous Mater. 2013, 173, 139–146. [Google Scholar] [CrossRef]
- Liu, B.; Shioyama, H.; Akita, T.; Xu, Q. Metal-organic framework as a template for porous carbon synthesis. J. Am. Chem. Soc. 2008, 130, 5390–5391. [Google Scholar] [CrossRef]
- Segakweng, T.; Musyoka, N.M.; Ren, J.; Crouse, P.; Langmi, H.W. Comparison of MOF-5-and Cr-MOF-derived carbons for hydrogen storage application. Res. Chem. Intermed. 2016, 42, 4951–4961. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.-L.; Liu, B.; Lan, Y.-Q.; Kuratani, K.; Akita, T.; Shioyama, H.; Zong, F.; Xu, Q. From metal–organic framework to nanoporous carbon: Toward a very high surface area and hydrogen uptake. J. Am. Chem. Soc. 2011, 133, 11854–11857. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Kim, T.; Im, J.H.; Kim, Y.S.; Lee, K.; Jung, H.; Park, C.R. MOF-derived hierarchically porous carbon with exceptional porosity and hydrogen storage capacity. Chem. Mater. 2012, 24, 464–470. [Google Scholar] [CrossRef]
- Wang, Q.; Xia, W.; Guo, W.; An, L.; Xia, D.; Zou, R. Functional zeolitic-Imidazolate-framework-templated porous carbon materials for CO2 capture and enhanced capacitors. Chem. Asian J. 2013, 8, 1879–1885. [Google Scholar] [CrossRef]
- Pachfule, P.; Biswal, B.P.; Banerjee, R. Control of porosity by using Isoreticular Zeolitic Imidazolate Frameworks (IRZIFs) as a template for porous carbon synthesis. Chem. Eur. J. 2012, 18, 11399–11408. [Google Scholar] [CrossRef]
- Almasoudi, A.; Mokaya, R. Preparation and hydrogen storage capacity of templated and activated carbons nanocast from commercially available zeolitic Imidazolate framework. J. Mater. Chem. 2011, 22, 146–152. [Google Scholar] [CrossRef]
- Almasoudi, A.; Mokaya, R. Porosity modulation of activated ZIF-templated carbons via compaction for hydrogen and CO2 storage applications. J. Mater. Chem. A 2014, 2, 10960–10968. [Google Scholar] [CrossRef]
- Shi, J.; Li, W.; Li, D. Synthesis, nickel decoration, and hydrogen adsorption of silica-templated mesoporous carbon material with high surface area. J. Phys. Chem. C 2015, 119, 23430–23435. [Google Scholar] [CrossRef]
- Konwar, R.J.; De, M. Synthesis of high surface area silica gel templated carbon for hydrogen storage application. J. Anal. Appl. Pyrolysis 2014, 107, 224–232. [Google Scholar] [CrossRef]
- Kruk, M.; Jaroniec, M.; Ryoo, R.; Joo, S.H. Characterization of ordered mesoporous carbons synthesized using MCM-48 Silicas as templates. J. Phys. Chem. B 2000, 104, 7960–7968. [Google Scholar] [CrossRef]
- Kaneti, Y.V.; Tang, J.; Salunkhe, R.R.; Jiang, X.; Yu, A.; Wu, K.C.-W.; Yamauchi, Y. Nanoarchitectured design of porous materials and nanocomposites from metal-organic frameworks. Adv. Mater. 2017, 29, 1604898. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Suh, K.; Kim, Y.; Yoon, M.; Park, H.; Dybtsev, D.N.; Kim, K. Porous carbon materials with a controllable surface area synthesized from metal-organic frameworks. Chem. Commun. 2012, 48, 7447–7449. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, D.; Rao, Y.; Zhao, X.; Wu, M. Superior CO2, CH4, and H2 uptakes over ultrahigh-surface-area carbon spheres prepared from sustainable biomass-derived char by CO2 activation. Carbon 2016, 105, 454–462. [Google Scholar] [CrossRef]
- Baca, M.; Cendrowski, K.; Kukulka, W.; Bazarko, G.; Moszyński, D.; Michalkiewicz, B.; Kalenczuk, R.J.; Zielinska, B. A comparison of hydrogen storage in Pt, Pd and Pt/Pd alloys loaded disordered mesoporous hollow carbon spheres. Nanomaterials 2018, 8, 639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Bhatnagar, A.; Dixit, V.; Shukla, V.; Shaz, M.A.; Sinha, A.S.K.; Srivastava, O.N.; Sekkar, V. Synthesis, characterization and hydrogen storage characteristics of ambient pressure dried carbon aerogel. Int. J. Hydrogen Energy 2016, 41, 3561–3570. [Google Scholar] [CrossRef]
- Zhang, C.; Kong, R.; Wang, X.; Xu, Y.; Wang, F.; Ren, W.; Wang, Y.; Su, F.; Jiang, J.-X. Porous carbons derived from hypercrosslinked porous polymers for gas adsorption and energy storage. Carbon 2017, 114, 608–618. [Google Scholar] [CrossRef]
- Lee, J.-S.M.; Briggs, M.E.; Hasell, T.; Cooper, A.I. Hyperporous carbons from hypercrosslinked polymers. Adv. Mater. 2016, 28, 9804–9810. [Google Scholar] [CrossRef]
- Qin, Q.; Sun, T.; Wang, H.; Brault, P.; An, H.; Xie, L.; Peng, Q. Adsorption and diffusion of hydrogen in carbon honeycomb. Nanomaterials 2020, 10, 344. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xiao, Y.; Dong, H.; Zheng, M.; Liu, Y. Polyacrylonitrile-based highly porous carbon materials for exceptional hydrogen storage. Int. J. Hydrogen Energy 2019, 44, 23210–23215. [Google Scholar] [CrossRef]
- Sdanghi, G.; Maranzana, G.; Celzard, A.; Fierro, V. Towards non-mechanical hybrid hydrogen compression for decentralized hydrogen facilities. Energies 2020, 13, 3145. [Google Scholar] [CrossRef]
- Panella, B.; Hirscher, M.; Siegmar, R. Hydrogen adsorption in different carbon nanostructures. Carbon 2005, 43, 2209–2214. [Google Scholar] [CrossRef]
- Suh, M.P.; Park, H.J.; Prasad, T.K.; Lim, D.-W. Hydrogen storage in metal-organic frameworks. Chem. Rev. 2012, 112, 782–835. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, D.; Mason, J.A.; James, B.D.; Houchins, C.; Long, J.R.; Veenstra, M. Techno-economic analysis of metal–organic frameworks for hydrogen and natural gas storage. Energy Fuels 2017, 31, 2024–2032. [Google Scholar] [CrossRef]
- Ghanem, B.S.; Msayib, K.J.; McKeown, N.B.; Harris, K.D.; Pan, Z.; Budd, P.M.; Butler, A.; Selbie, J.; Book, D.; Walton, A. A triptycene-based polymer of intrinsic microposity that displays enhanced surface area and hydrogen adsorption. Chem. Commun. 2007, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Makhseed, S.; Samuel, J. Hydrogen adsorption in microporous organic framework polymer. Chem. Commun. 2008, 4342–4344. [Google Scholar] [CrossRef]
- Thomas, K.M. Hydrogen adsorption and storage on porous materials. Catal. Today 2007, 120, 389–398. [Google Scholar] [CrossRef]
- Dwivedi, S.N. Design considerations for high-pressure reciprocating compressors for refinery services. In Proceedings of the International Compressor Engineering Conference 1990, West Lafayette, IN, USA, 17–20 July 1990. [Google Scholar]
- Parks, G.; Boyd, R.; Cornish, J.; Remick, R. Hydrogen Station Compression, Storage, and Dispensing Technical Status and Costs; National Renewable Energy Lab: Golden, CO, USA, 2014.
- Sdanghi, G.; Maranzana, G.; Celzard, A.; Fierro, V. Review of the current technologies and performances of hydrogen compression for stationary and automotive applications. Renew. Sustain. Energy Rev. 2019, 102, 150–170. [Google Scholar] [CrossRef]
- Sdanghi, G.; Nicolas, V.; Mozet, K.; Schaefer, S.; Maranzana, G.; Celzard, A.; Fierro, V. A 70 MPa hydrogen thermally driven compressor based on cyclic adsorption-desorption on activated carbon. Carbon 2020, 161, 466–478. [Google Scholar] [CrossRef]
- Sdanghi, G.; Nicolas, V.; Mozet, K.; Maranzana, G.; Celzard, A.; Fierro, V. Modelling of a hydrogen thermally driven compressor based on cyclic adsorption-desorption on activated carbon. Int. J. Hydrogen Energy 2019, 44, 16811–16823. [Google Scholar] [CrossRef]












© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sdanghi, G.; Canevesi, R.L.S.; Celzard, A.; Thommes, M.; Fierro, V. Characterization of Carbon Materials for Hydrogen Storage and Compression. C 2020, 6, 46. https://doi.org/10.3390/c6030046
Sdanghi G, Canevesi RLS, Celzard A, Thommes M, Fierro V. Characterization of Carbon Materials for Hydrogen Storage and Compression. C. 2020; 6(3):46. https://doi.org/10.3390/c6030046
Chicago/Turabian StyleSdanghi, Giuseppe, Rafael L. S. Canevesi, Alain Celzard, Matthias Thommes, and Vanessa Fierro. 2020. "Characterization of Carbon Materials for Hydrogen Storage and Compression" C 6, no. 3: 46. https://doi.org/10.3390/c6030046
APA StyleSdanghi, G., Canevesi, R. L. S., Celzard, A., Thommes, M., & Fierro, V. (2020). Characterization of Carbon Materials for Hydrogen Storage and Compression. C, 6(3), 46. https://doi.org/10.3390/c6030046