[60]PCBM, as its trivial name suggests, may be considered as consisting of three parts: a phenyl group, a C61 cyclopropafullerenyl group and a butyric acid methyl ester group. The conformational structure of the conjoined phenyl-cyclopropafullerenyl substituent to the ester is trivial. This is because there is only one conformation of that grouping that is consistent with the molecule’s Cs symmetry. This conformer has phenyl carbons C1 and C4 and cyclopropafullerene carbons C20, C30, C31, C40 and C61 on the mirror plane while all other atoms exist as mirrored pairs bisecting the mirror plane.
3.1. Proposed Conformer Nomenclature
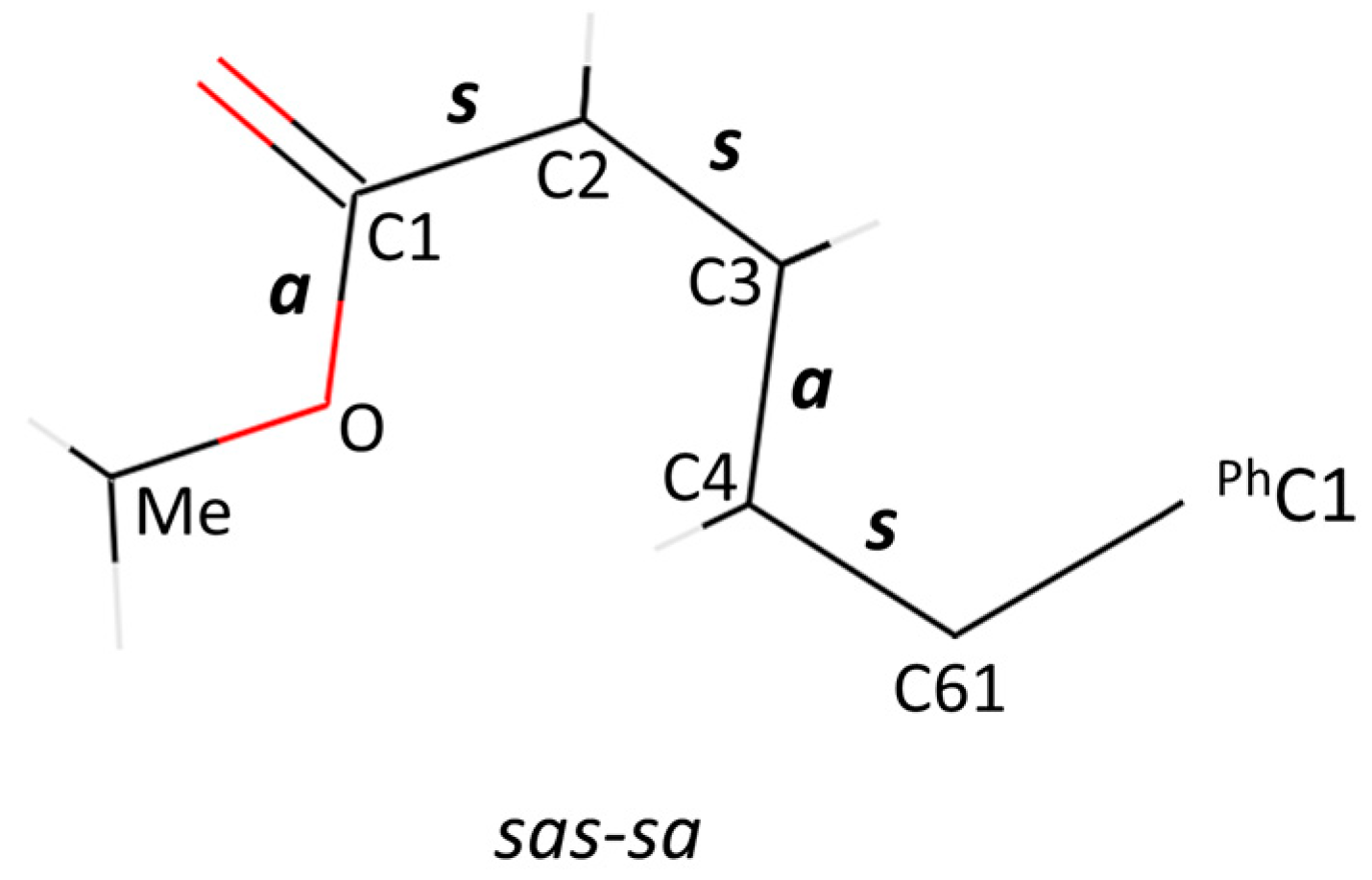
To differentiate the 24 remaining conformers while keeping a reasonably short trivial name, we propose the following nomenclature. In parentheses, prefix [60]PCBM with the five relative orientations about the bonds in the ester group, which are designated “s” for syn and “a” for anti with respect to the relevant single bonded heavy elements of the addend that lie on the mirror plane. We present them in the following order: C61-C4, C4-C3, C3–C2, C2–C1 and C1–O, and place a hyphen after the third bond to differentiate the conformational behaviour of the alkyl chain from that of the ester end.
An example is (
sas-sa)[60]PCBM, which has C61-C4
syn, C4-C3
anti and C3-C2
syn for the alkyl chain part, and C2-C1
syn and C1-O
anti for the COOMe part. A diagram of (
sas-sa)[60]PCBM is given in
Figure 1, which was randomly chosen to illustrate this naming system. For example, the C61-C4 bond has the attached carbons C3 of the ester and C1 of the phenyl ring in a
syn conformation on the mirror plane; hence, the conformation about this bond is denoted as
s. On the other hand, the arrangement of the attached carbons to the C3-C4 bond (C2 and C61, respectively) are in an
anti a conformation. It must be kept in mind that the
s or
a designation refers to the heavy elements (C and O).
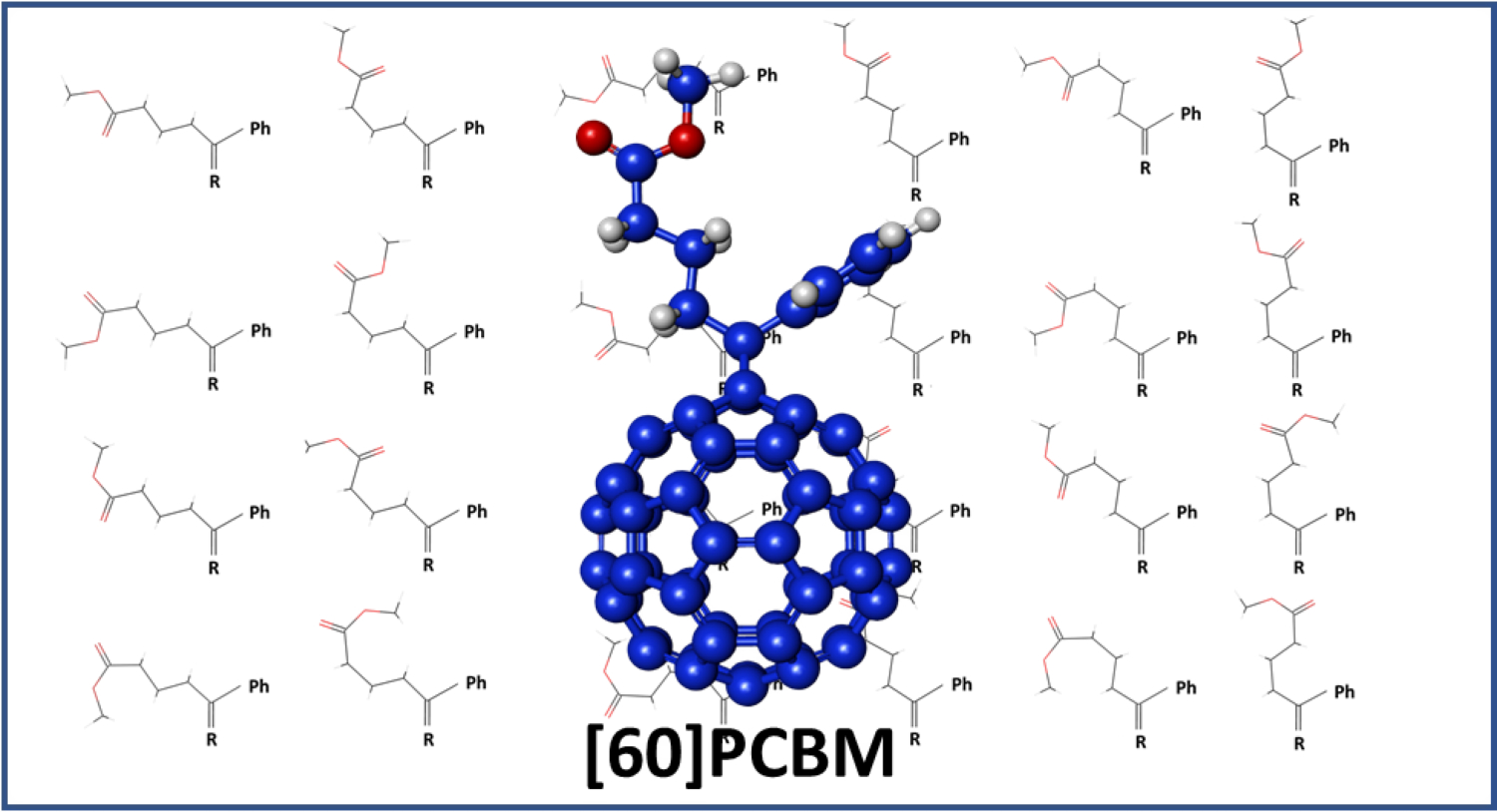
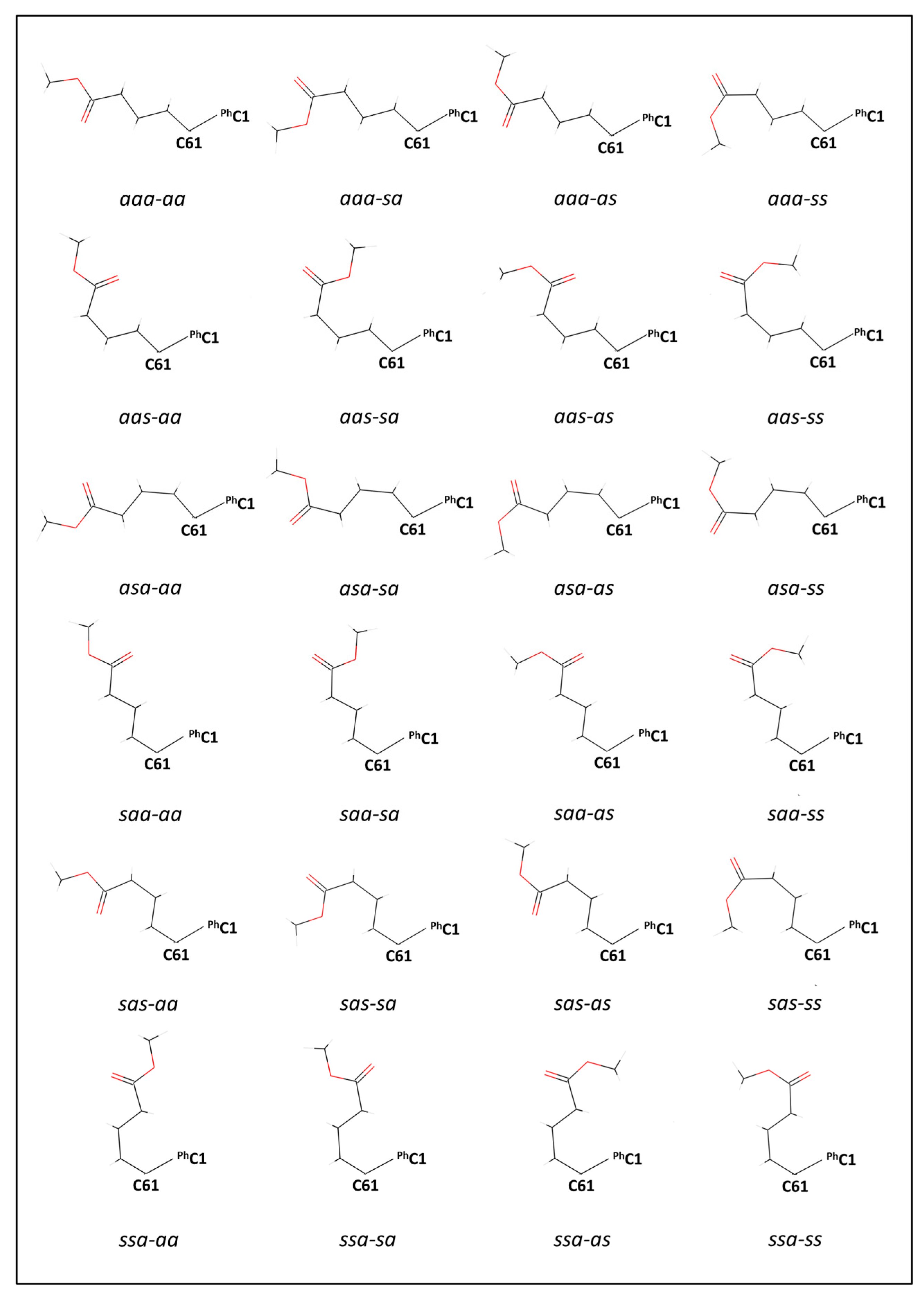
All 24 possible ester conformations are shown in
Figure 2 at their B3LYP/cc-pVTZ optimised geometries. For clarity, as with
Figure 1, the conformationally invariant phenyl and cyclopropafullerenyl groups are omitted and only the relevant atoms are shown.
3.2. Bond Strain and Electronic Energy Analysis
In this part of the work, the initial freely optimised geometry (as detailed in
Section 2) had its five relevant dihedral set to angles to either 0° (
syn) or 180° (
anti) to generate the 24
CS symmetric conformers. The geometries of the 24 conformers were then re-optimised subject to the five dihedral angles, each being constrained as appropriate, while all bond lengths, bond angles and other dihedral angles could freely vary. The initial geometry from which the 24 conformers were generated was at a minimum on the B3LYP/cc-pVTZ potential energy surface. As such, with all 24 conformers being generated from this one optimised structure by varying it along only one coordinate (one dihedral angle), the 24 conformers should be near the minima.
It is apparent from inspecting
Figure 2 that many of the bond angles are unrealistic. For example, the C1-C2-C3 bond angle in (
aas-ss)[60]PCBM at the end of the second row is exceptionally wide at 138° where about 112° is expected. As further examples of the unrealistic bond angles, the six
xxx-ss conformers have their C1-O-Me bond angles ranging from 126.6° to 129.7°. These are all very wide compared to experimentally known values from powder x-ray diffraction studies, −116.0° [
16]. Hence, even without simulating the
13C NMR spectra of the six
xxx-ss conformers, these may be discounted as candidates for the proper input geometry. Similarly, all 12
axx-xx and four
ssa-xx conformers have extremely large C61-C4-C3 bond angles, ranging from 120.7°–135.9° (
cf. experiment [
16] at 114.4°); conversely, the remaining eight
sxx-xx conformers (
saa-xx and
sas-xx) have more realistic angles (ranging from 113.9°–115.4°). This suggests that the 12
axx-xx and four
ssa-xx conformers may be also discounted. A full table of every relevant bond angle is given as
supporting information (Table S1).
To give a quantitative measure of the bond strain for each of the 24 conformers, all the relevant bond angles for each conformer were measured using Gaussview software [
17]. Following this, the absolute differences from the experimental solid state [
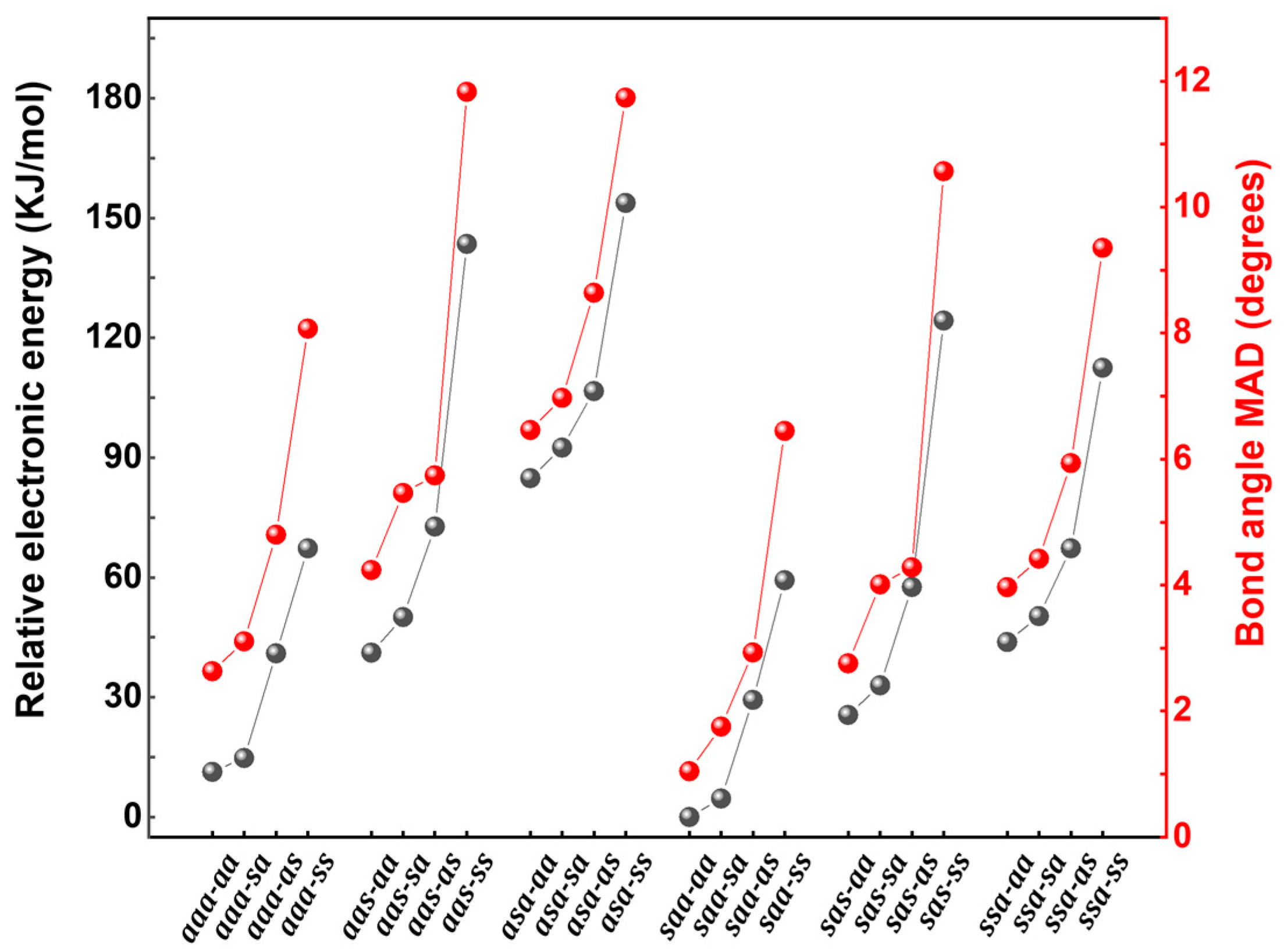
16] bond angle were obtained, and finally, from these, the mean absolute deviation (MAD) was obtained for each conformer. These 24 MADs are plotted against their relevant conformer in
Figure 3. The MADs range from 0.6 to 12.3 degrees per bond angle. Within each of the six
xxx- combinations, it is the
xxx-ss that have the highest MADs (which were qualitatively rejected earlier in the discussion), validating this method.
The degree of bond strain in the conformers is also likely to be reflected in their total electronic energies. As such, the total electronic energy of each of the 24 conformers is also given in
Figure 3. These are presented as relative to the lowest energy conformer, which is set to zero. These energies range up to 153 kJ/mol (1.59 eV) above that of the most stable conformer.
Two initial deductions can be drawn from
Figure 3. The first is that there is a very close correlation between MAD of the bond angles and the relative total electronic energy, with remarkably similar trends in both. The second is that entire pattern breaks into two separate series of three separate four-membered groups. One series involves the 12
axx-xx conformers and another involves the 12
sxx-xx conformers, within each series, there are three groups of four conformers (with each group consisting of an
xxx-aa, -
sa, -
as, -
ss set).
The members of each of the six groups all follow the same trend. That is, the xxx-aa member is the most stable (having the lowest total electronic energies and bond strain MADs), closely followed by the -sa member. There is then a much larger step to the -as member followed by an even larger step to the -ss member. This indicates that the -COOMe part is quite unstable having the C1-O bond syn and is even more unstable if the C2-C1 bond is also syn. It also indicated that provided the C1-O bond is anti, there is little difference in terms of energy or bond strain on whether the C2-C1 bond is anti or syn.
Just as the members of the groups all follow the same trend, the groups themselves within their respective series also follow a matching trend in that there is an increase in instability and/or bond strain in the order xaa-xx < xas-xx < xsa-xx.
The final trend compares the two series and indicates that members of the
sxx-xx series are shifted downwards by about 20 kJ/mol (200 meV) in electronic energy and about 2° in bond angle MAD relative to their respective analogs in the
axx-xx series. If the C61-C4 bond is
anti, then C3C4 bond is oriented slightly towards the fullerene. Therefore, to alleviate steric issues with the ester group becoming too close to the fullerene, the
PhC1-C61-C4 bond angle becomes strained in that it decreases from the known value of 115.5° [
16] (
PhC1 is used to avoid ambiguity as C1 of the phenyl and C1 of the C
60 are both bonded to C61). It decreases to 107.6° for all
aaa-xx conformers, 106.9–106.4° for the
aas-xx conformers and 103.5–102.7° for the
asa-xx conformers. Conversely, if the C61-C4 bond is
syn, then the C3-C4 bond is oriented almost vertically away from the fullerene. However, this then brings it closer to the phenyl group. To alleviate similar steric issues with the ester group becoming too close to the phenyl, the
PhC1-C61-C4 bond angle opens relatively slightly to 115.9–116.3° for the
saa-xx conformers, 117.1–117.3° for the
aas-xx conformers and 120.0–120.4° for the
asa-xx conformers. The average deviation for each group is slightly larger for the
axx series than it is for the
sxx series, which may explain why the members of the former series have slightly decreasing electronic energies and bond strain MADs. It is also noticed that both these trends in increasing bond strain follow the same
xaa-xx <
xas-xx <
xsa-xx trend for series members, as identified above. Hence, it seems likely that the C61-C4 bond plays a significant role in the conformational behaviour of [60]PCBM, with a strong preference for it being carbon-
syn.
3.3. Simulation of the 13C NMR Spectra (sp3 Hybridised Carbons)
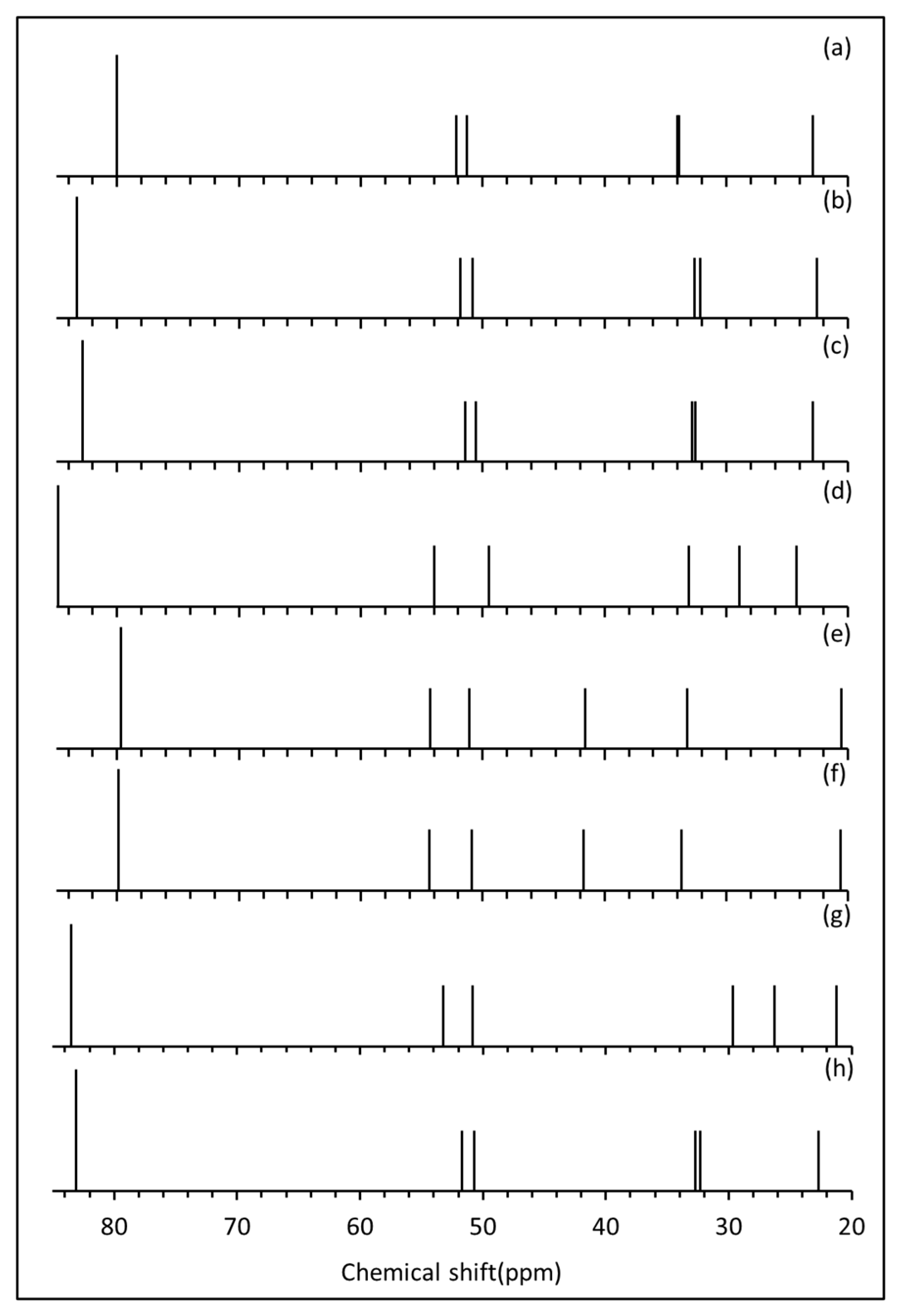
Based on these analyses, the conformers with the six lowest bond angle strains, as measured by their mean absolute deviations of the relevant bond angles from experiment, were shortlisted to have their 13C NMR spectra simulated. These six conformers also had the six lowest electronic energies, and are in order of increasing bond angle MAD, saa-aa, saa-sa, aaa-aa, sas-aa, aaa-as and aaa-sa.
13C NMR spectra of the six candidate conformers were simulated. These are presented for the
sp3 region, together with the experimental spectrum, in
Figure 4. Inspection of these spectra gives a clear indication that only the (b)
saa-aa and (c)
saa-sa conformations have simulated spectra that can be compared favourably to that of the experimental spectrum. Both follow the experimental line pattern very closely. It is also indicated that
saa-sa is the better of the two on groups, having the spacings closer to the experiment. Conversely, both (d)
saa-as and (g)
sas-aa have the two lines near 50 ppm, much wider spacing than the experiment, and the other three lines are quite evenly spaced rather than having the experimental groupings. Finally, (e)
aaa-aa and (f)
aaa-sa are the worst of the six shortlisted conformers, with each having the first five lines with reasonably equal spacings.
The observation from the six simulated sp3 NMR spectra that saa-aa and saa-sa are very similar to each other and are the two closest to experiment agrees with the conclusion made earlier from the bond strain and electronic energy analysis. This is because these two conformers have the lowest two bond strains and electronic energies of all the conformers, but their values are very similar. Conversely, of the six simulated spectra, aaa-aa and aaa-sa also appear very similar but give the worst agreement with the experiment. This also accords with the previous analysis, as these have the highest two bond strains and electronic energies of the six shortlisted conformers but are very close to each other in value. That the bond angle MADs (average bond strains), the total electronic energies and the quality of the simulated 13C NMR spectra all follow the same trends provides compelling evidence that saa-aa and saa-sa are the two most stable structures for the conformational arrangements of the ester group and are thus the two most occupied conformers.
The discussion above is limited to the
sp3 region for two reasons: (1) it is a simple area of the spectrum where comparisons between the different conformers are easy to make, and (2) it involves the very carbons that are relevant to the conformations of the ester (nb., the
sp2 fullerene and phenyl carbons do not conformationally vary in any of the 24 conformation). Analysis of the far more complicated
sp2-hybridised carbons also indicates that
saa-sa performed slightly better than
saa-aa; for this reason, the
saa-as conformer was used in our recent publication on the full spectrum [
14].
3.4. Conformational Analysis of the Ester Group
Insights into the dynamical behaviour of the ester group may be obtained with reference to our recent experimental study of second-order spin–spin couplings in the
1H NMR spectrum. The observed very strong second-order spin–spin couplings at the C4 carbon resulted from restricted rotation about the ester C61-C4 and C4-C3 bonds [
12]. For these two bonds, there is
gauche-syn/anti-gauche′ and back again restricted rotation. That is, for these two bonds, there is no continued rotation from
gauche to
gauche′ for steric reasons [
12]. Hence, it seems that
axx-xx conformers cannot be obtained owing to this restricted rotation. For the same reasons, the
xsx-xx conformers also cannot be obtained. This restricted rotation explanation is consistent with the NMR simulations in
Figure 4, as these give relatively poor results when the ester is symmetry-constrained into these unfavourable conformations. The poor agreement of the
sas-aa conformer’s NMR spectrum with the experiment may also be explained in terms of dynamical behaviour. Although these conformers may be obtained as there is some degree of full rotation about the C2–C3 bond [
12], occupation of the
syn conformation in this bond should be relatively short lived.
To further investigate these conclusions and extend them to all heavy atom bonds of the ester groups, we conducted a DFT investigation. To this end, we considered the optimised conformer with the lowest total electronic energy and bond strain, (
saa-aa)[60]PCBM, to be a “standard” to compare other conformers. This involved re-optimising the geometry of the standard (
saa-aa)[60]PCBM so that all bond lengths, bond angles and dihedral angles were fixed, but then the following dihedral angles were each in turn varied from 0° to 180° in 10° steps: (a)
PhC1-C61-C4-C3, (b) C61-C4-C3-C2, (c) C4-C3-C2-C1, (d) C3-C2-C1-O and (e) C2-C1-O-Me. In this way, plots of relative energy against dihedral angle may be obtained for each of the five heavy-atom dihedral angles of the ester group (
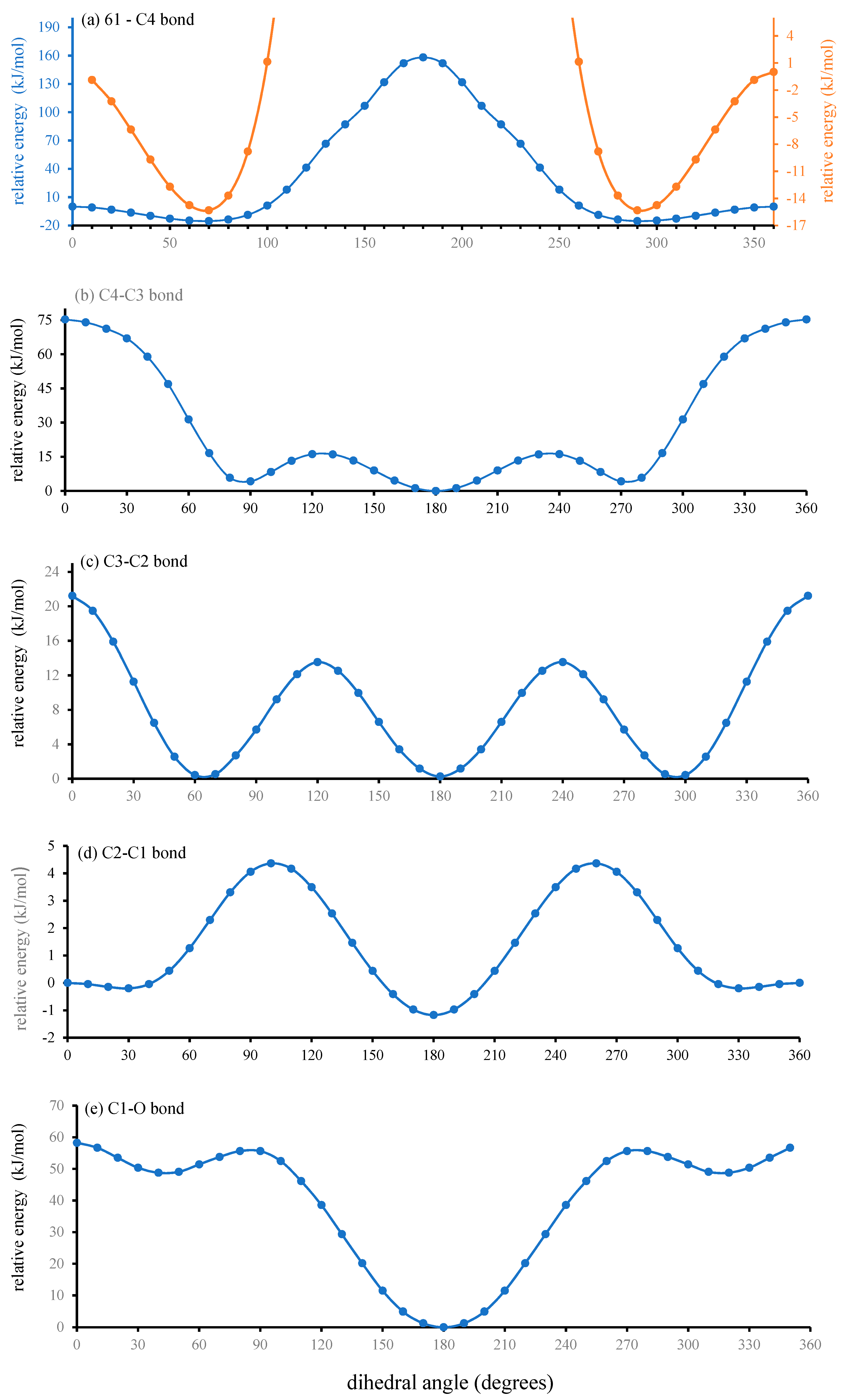
Figure 5). The fixing of the bond lengths and angles may be justified under the Born–Oppenheimer approximation. This is because bond length/angle variations are vibrational motion, and these are extremely slow relative to the rotational motion of dihedral angle variation. Hence, bond lengths and bond angles may, to a good approximation, be considered fixed on the time scale of the rotations. These five plots of relative energy against dihedral angle are considered, in turn, below in the abovementioned order.
C61-C4 Bond
A plot of the
PhC1-C61-C4-C3 dihedral angle (which represents rotation about the C61-C4 bond) against relative energy is presented in
Figure 5a. The blue graph is shown at full scale, whereas for clarity, the orange graph is an expansion in energy. This figure reveals two minima at near-gauche and near-gauche’ dihedral angles of +68° and 292° (−68°). The slight variation away from the ideal
gauche dihedral angle of 60° is likely to be due to the large asymmetry in the barriers on either side of these minima. The barrier to rotation from
gauche to
gauche’
via the
syn conformer (0°) is 15.3 kJ/mol (176.7 meV), which corresponds to 6.18 ×
kT at room temperature (RT). A Boltzmann population analysis indicates that about 1 in 500 molecules (0.2%) have enough thermal energy to overcome this barrier. As the barrier is the same height in both directions, the
gauche and
gauche’ conformers are equally populated.
On the other hand, rotation from
gauche to
gauche’
via the
anti (180°) conformer is impossible, as the barrier is 177 kJ/mol (1.84 eV) or 71.5 ×
kT RT. A Boltsmann analysis suggests that only 10
−29% of molecules could overcome this barrier. However, the energy difference between (
aaa-aa)[60]PCBM and (
saa-aa)[60]PCBM from
Figure 3, at 11.6 kJ/mol (120 meV), is much less that that from
Figure 5a. A Boltzmann/Arrhenius analysis based on this energy difference suggests that population of the
aaa-aa conformer would be slightly less than 1% of that of the
saa-aa conformer, and as will be shown for the remaining dihrdral angles, the populations would be far below 1% for the other 11
axx-xx conformers. However, even a 1% population is unlikely to be achieved. This is because the 11.6 kJ/mol energy difference is that of a vibrationally relaxed conformer. As mentioned earlier, with vibrational motions of the bonds (length and angle changes) being about 2 orders of magnitude slower than rotations about the bonds, it can be concluded that the relative population of the
aaa-aa conformer is effectively zero. This is because with rotational rapid ascent up the barrier towards
aaa-aa along the coordinate of
Figure 5a, the configuration would be rapidly repelled from further assent long before it had any opportunity to even start vibrationally relaxing.
Within the gauche and gauche’ conformers about the C61-C4 bond, the ester group undergoes librational behaviour whereby the PhC1-C61-C4-C3 dihedral varies between −17° and +14° (gauche) and −14° and +17° (gauche’) either side of the minima, the extremes of which correspond to kT at room temperature (2.48 kJ/mol or 25.7 meV) above the minima in energy.
There are two shoulders on the central peak near 130 and 230 degrees. These are at elevated instability as they occur at dihedral angles about C61-C4 where one or the other of the two hydrogens on C4 is eclipsed with the hydrogen-less PhC1 carbon.
It should be noted that the
aaa-aa conformer and the
saa-aa conformer are related to each other by a rotation of 180° about the C61-C4 bond. As such, they lie on a line through the potential energy surface (PES). Considering the PES along this line while retaining
CS symmetry should show that the
aaa-aa conformer is a maximum and
saa-sa is a minimum. The
saa-aa conformer does not, however, represent the global minimum on the B3LYP/cc-pVTZ PES of (
xaa-aa)[60]PCBM. Inspection of
Figure 5a shows that the global minimum regarding the relative orientation about the C61-C4 bond has a dihedral angle of about 70°. Confirmation of this comes from an OPT-FREQ calculation that reveals this dihedral angle to be 68.14° with all other relevant dihedrals angles being within a degree of 180°. This structure is lower in energy than the similarly optimised (
saa-aa)[60]PCBM structure. In addition, it has no imaginary vibrational frequencies (indicative of a minimum on the PES). However, this structure has
C1 point group symmetry rather than the experimentally observed
CS symmetry. As such, simulations based on this structure in solution would not, in general, resemble the experiment. For example, simulated
13C NMR spectra of PCBM would split into 72 resonances instead of the experimentally seen 42 resonances; degenerate energy levels would also be split. As such, it seems likely that the barrier for
gauche(’) to
syn may be lower than that calculated, and that in reality, the librational motion goes back and forth between 90° and −90° (270°), with the
syn conformation (0°) being the centre of the wide motion.
From the analysis of the orientational behaviour about the C61-C4 bond, it can be concluded that none of the 12 axx-xx conformers of [60]PCBM has any significant population. As such, (aaa-aa)[60]PCBM, the most commonly used representation of [60]PCBM in the literature, is essentially energetically forbidden, which suggests that at least some of the many ab initio calculations based on it may have unreliable conclusions.
C4-C3 Bond
The plot of dihedral angle rotation about the C4-C3 bond against relative energy, shown in
Figure 5b, indicates that there are three relatively stable conformers,
gauche (92°),
anti (180°) and
gauche’ (272°), and that the
syn conformer (0°) at about 75 kJ/mol (~800 meV) represents an unsurmountable barrier (over 30 ×
kT at RT) to rotation from
gauche’ to
syn to
gauche. This barrier would be much higher (at about 4.2 eV) if the neighbouring C3–C2 dihedral angle was not able to change to partially alleviate the bond strain.
The greater variation away from the ideal gauche angle than that seen for the C61–C4 bond above is due to the even greater asymmetry in the barriers. On this occasion, the barrier for rotation from gauche(’) to anti is 12.18 kJ/mol (126.3 meV, 4.91 × kT at RT), and that to rotate from anti to gauche(’) is 16.32 kJ/mol (169.2 meV, 6.58 × kT at RT).
A Boltzmann analysis based on these barriers indicates that 0.734% of molecules have sufficient thermal energy to overcome the barrier and rotate from the
gauche(’) conformers to the
anti conformer, and that 0.138% of molecules may similarly rotate from
anti to
gauche(’). An Arrhenius analysis indicates that there is a 16:84 population ratio of the two conformers, with the
anti (180°) conformer being the more favoured. As the barrier to rotation from
anti to
gauche and that
anti to
gauche’ are identical, the
gauche and
gauche’ conformers would be equally populated (at
ca. 8% each). These findings are consistent with the conclusion we recently drew on the populations of these three conformers based on well-resolved second-order spin–spin coupling splitting of the hydrogens attached to Carbon C4 by those attached to C3. Based on a CJG conformer analysis, a 12½:75:12½ population ratio for the
gauche,
anti and
gauche’ conformers was estimated [
12]. The slight discrepancy might come from imperfect assumptions made in the CJG analysis, in this work, or both. Nevertheless, both this work and our recent CJG estimate from second-order
1H NMR spin–spin couplings have the general conclusion that the
syn conformer is extremely unstable, the
anti conformer is the most stable with
gauche(’) islands of stability either side of the
anti conformer, and that the population ratio of the
gauche,
anti and
gauche’ conformers is about 1:8:1. With the
syn conformation being unstable, all
xsx-xx conformers have essentially zero population.
Like the PhC1-C61-C4-C3 dihedral angle discussed above, there is librational motion about the minima of the gauche, anti and gauche’ conformers. In this case, the librations occur over a 30° range, ±15° either side of the anti conformer (i.e., between C61-C4-C3-C2 dihedral angles of 165°–195°), and over an 18° range, ±9° either side of the gauche(’) conformers (i.e., between dihedral angles of 79°–97° and 263°–181°, respectively).
C3-C2 Bond
A plot of relative electronic energy vs the C4-C3-C2-C1 dihedral angle is given in
Figure 5c. Three equal minima are seen at the
gauche dihedral angles of −68°, −68° (292°) and at the
anti dihedral angle of 180°, with equal
gauche-anti and
gauche’-anti barriers of 13.4 kJ/mol (140 meV, 5.47 ×
kT at RT). The more stable 68°, 180° and 292° dihedrals have the hydrogens of C3 and C4 staggered, whereas the unstable 0°, 120° and 240° have those hydrogens eclipsed.
The barrier to rotation through the
syn conformer is higher at 21 kJ/mol (220 meV). Like the previously discussed bond, this barrier would be much higher, 155 kJ/mol (1.6 eV) this time, if it wasn’t for the ability of the neighbouring C2–C1 dihedral angle to change and thereby partially alleviate the bond strain. Again, like the previous bond, the
syn conformation is unstable compared to the
anti conformation, meaning that all
xxs-xx conformers also have essentially zero population. An Arrhenius analysis of these barriers suggests that the
gauche-
anti-gauche’ transition occurs 20 times more often than that of
gauche-
syn-gauche’. This situation is like that for the C4-C3 bond, where the rest of the ester group “wags” back and forth from
gauche to
gauche’ via the stable middle conformer (
anti in this case) and very rarely by the unstable conformer. However, unlike the
1H NMR resonances from the hydrogen on carbon C4 being split by those of C3, the NMR spectrum does not indicate significant second-order spin–spin couplings for the hydrogens of carbon C2 being split by those of C3, which we recently interpreted as resulting from near free rotation about the C3-C2 bond [
12]. However, this interpretation is not correct. This is because the
gauche-
anti-gauche’ transition occurring 20 times more often than the
gauche-
syn-gauche’ transition is inconsistent with free rotation. Therefore, a new explanation is needed of the experimental
1H NMR observation of a first-order 1:2:1 triplet for the two symmetrically equivalent hydrogens of carbon C2 by those of carbon C3. Instead of “free rotation”, the lack of second-order effects is better explained by the near equal energy barriers for the
gauche(’) to
anti and the
anti to
gauche(’) transitions, which would result in near equal populations of the three conformers. This is because at second order, the usual first-order 1:2:1 triplet occurs when the populations of the three staggered conformers about this bond are equal. Hence, is it the effectively equal
gauche(’)-
anti and
anti-
gauche(’) barriers, rather than free rotation about the C3-C2 bond, that gives the near equal populations that account for the lack of second-order NMR effects ordinarily expected from non-free rotation.
C2-C1 Bond
From
Figure 5d, it is apparent that there are two stable conformers about this bond,
syn at 0° and
anti at 180°, with the
anti conformer being slightly more stable by 0.974 kJ/mol (10.1 meV). The two wells near +30° and −30° (330°) are so shallow, at only 0.19 kJ/mol (2 meV, 0.078 ×
kT at RT), that they are insignificant in relation to ester dynamics at room temperature.
About the
syn conformer, there is librational motion over a very wide range of about 150°. That is, between +75° and −75° (285°). Meanwhile, about the more stable
anti conformer, the librational range is only 70°, at ±35° (i.e., between C3-C2-C1-O dihedral angles of 155° and 215°). However, the energy barrier for rotation from
anti to
syn is far lower than those discussed so far at only 2.2
kT at RT (5.53 kJ/mol, 57.3 meV), and that for
syn to
anti is even lower at 1.8
kT at RT (4.56 kJ/mol, 47.3 meV). As such, these barriers suggest that there is almost free rotation about this bond. Indeed, a Boltzmann indicates that about 16% of molecules have sufficient thermal energy to rotate from
syn to
anti at room temperature, and that about 11% of molecules have sufficient energy to rotate the other way. An Arrhenius analysis based on these barriers indicates that there is an approximate 60:40 population ratio of the two conformers with the
anti conformer being the more favoured. The much low barriers to full rotation about this bond account for the observation made from
Figure 3 that the
xxx-
aa and
xxx-
sa conformers, which involve this bond, have the lowest bond strain differences. With this, in simulating molecular spectra of [60]PCBM, it may be advantageous to use a 60:40 weighted average of relevant spectra of (saa-aa)[60]PCBM and (saa-sa)[60]PCBM. For example,
Figure 4 h shows a weighted
13C NMR spectrum of the
sp3-hybridised carbons of [60]PCBM, which is remarkably close to the experimental spectrum (
Figure 4a).
C1-O Bond
The final ester bond to have a dihedral angle for heavy elements is the C1-O single bond. The plot of relative electronic energy as a function of the C2-C1-O–Me dihedral angle (
Figure 5e) shows three minima near
gauche (45°),
anti (180°) and
gauche’ (−45° or 315°) dihedral angles. However, only the
anti conformer is stable, and thereby has any significant population. This is because there is a relatively small barrier of 2.8
kT at RT (6.9 kJ/mol, 72 meV) to
gauche(’) to anti rotation, but an essentially unsurmountable barrier of 22.5
kT at RT (53.7 kJ/mol, 577 meV) for the reverse rotation, giving an approximate 1:10
8:1 population ratio of the
gauche,
anti and
gauche’ conformers. Within the stable
anti conformer, there is librational motion about the C1-O bond between dihedral angles of 166° and 194°; i.e., ±14° either side of
anti.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




