
CH Activation by a Heavy Metal Cation: Production of H2 from the Reaction of Acetylene with C4H4-Os(+) in Gas phase



Abstract
:1. Introduction
2. Computational Methods
3. Results
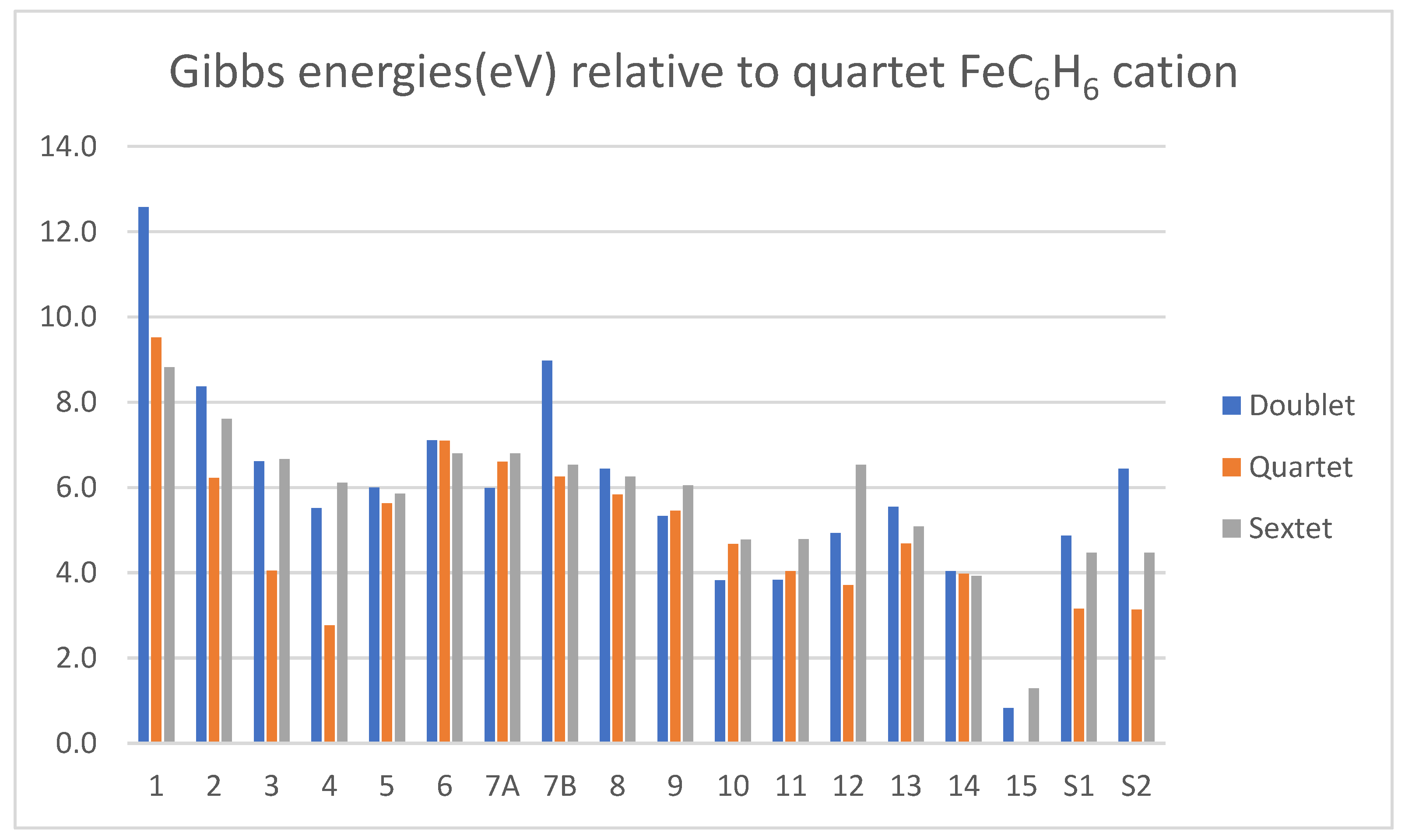
3.1. Energetics and Structures for Fe(+)C6H6 Species
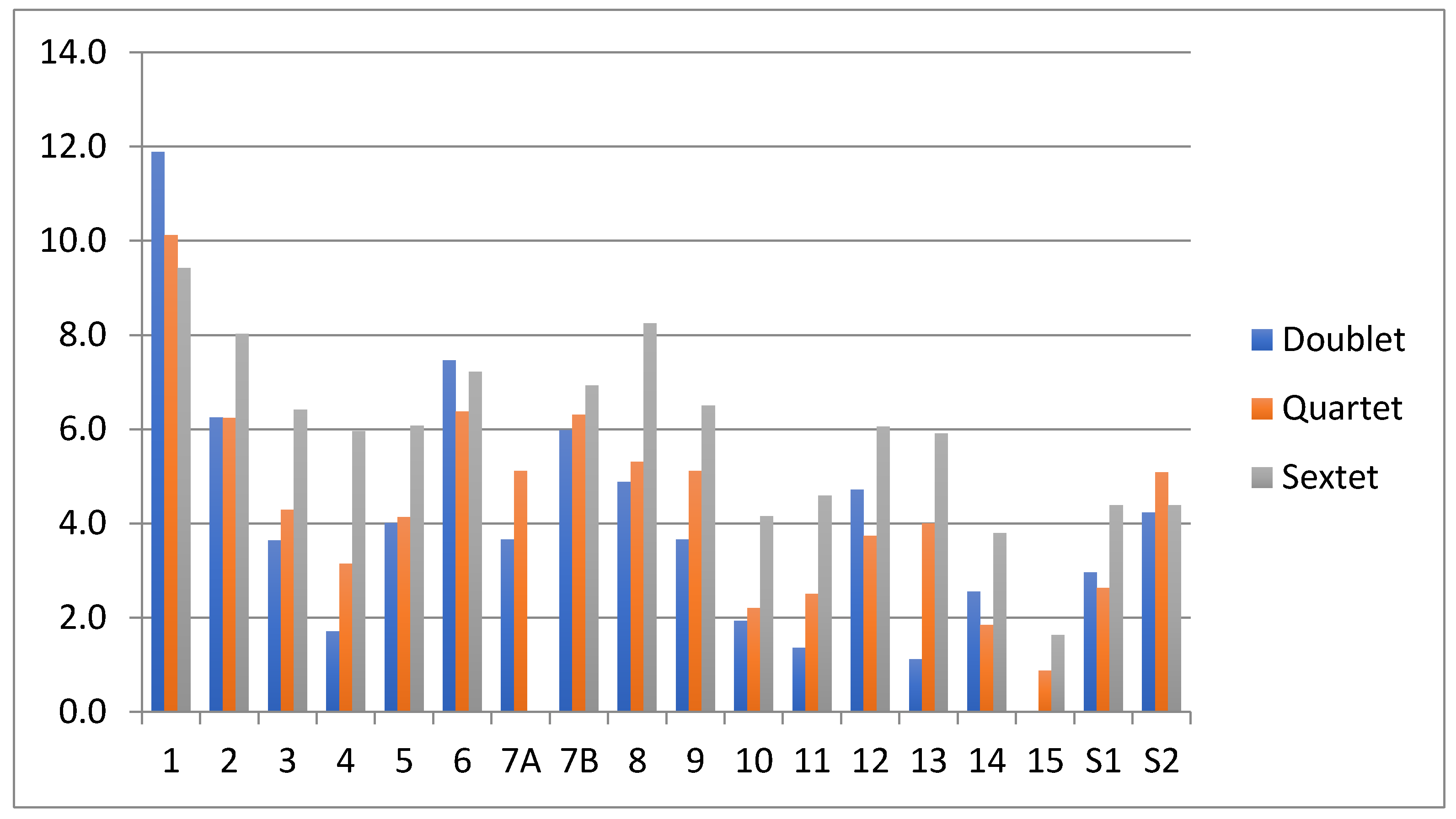
3.2. Energetics and Structures for Os(+)C6H6 Species
3.3. Comparison of MC6H6 Cation Energetics for M = Fe and Os
- A decline is found for both systems in BE as acetylenes are added. However, successive binding energies (BE) of acetylenes are about 50% greater for Os(+) than Fe(+). This favors the production of C–O binding in the Os system.
- The quartet is the ground state of the benzene-Fe(+) complex, consistent with other computational results [20]. The doublet is preferred for the benzene-Os(+) complex.
- The energy requirement for expelling benzene and sextet Os(+) from the ground-state, doublet benzene-Os(+) is 3.67 eV, while producing a sextet Fe(+) from the ground-state quartet, benzene-Fe(+), requires 3.08 eV. This inhibits the benzene production for Os(+) relative to Fe(+).
- Species doublet 14 and doublets S1 and S2 are relatively high in energy for the Fe(+) system, but more accessible in the Os(+) system. This favors H2 production for the heavy metal cation.
- The energy requirement for producing H2 from 14 leaving C6H4-Os(+) is substantial (0.78 eV) but the elimination product at 2.5 eV is lower in energy than the free benzene and separate Os(+), by 3.3 eV.
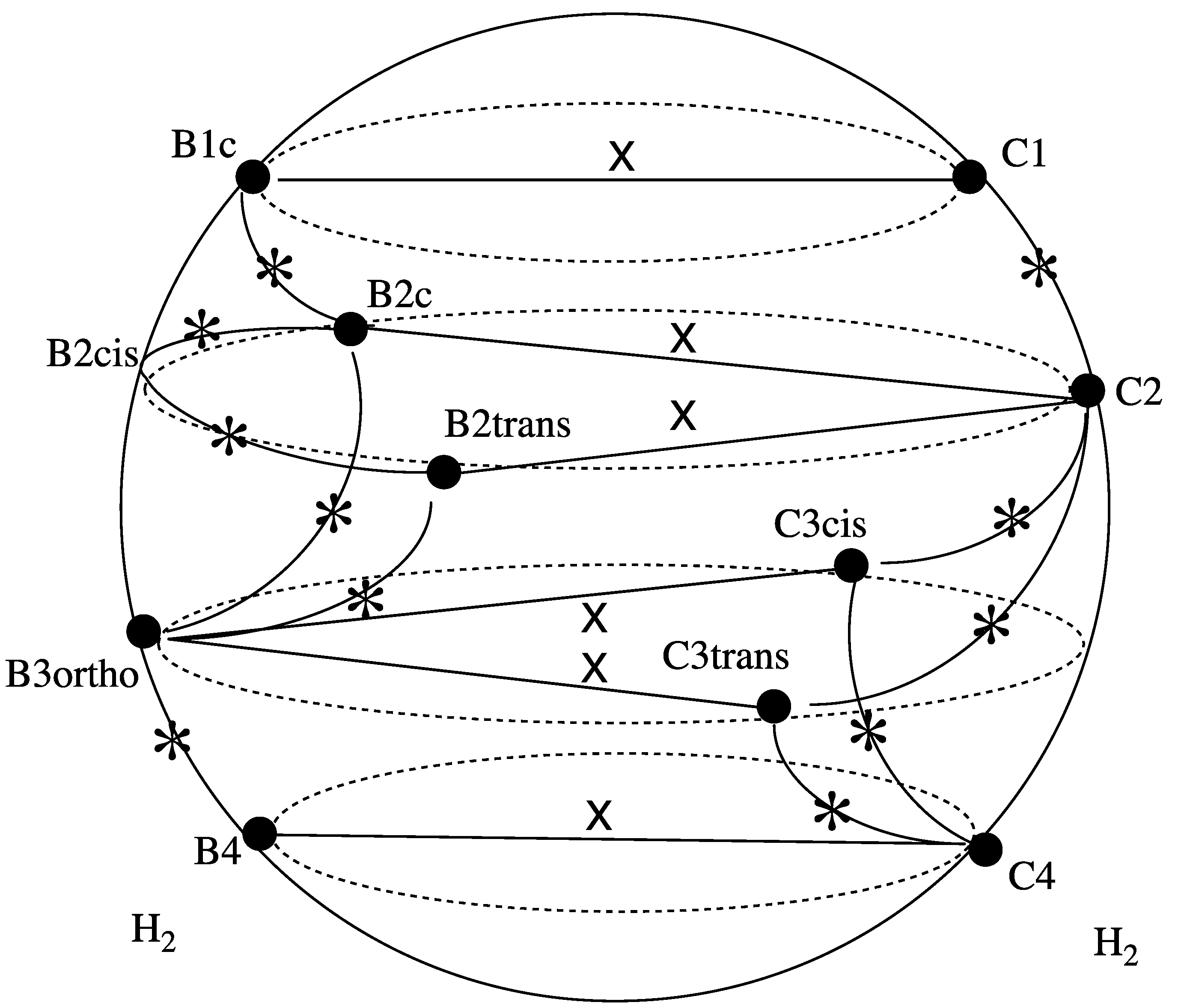
3.4. Reaction Paths for Extrusion of H2 from OsC6H6 Cation
3.4.1. Overview of the Reaction Network for the H2 Extrusion from OsC6H6 Doublet Cation
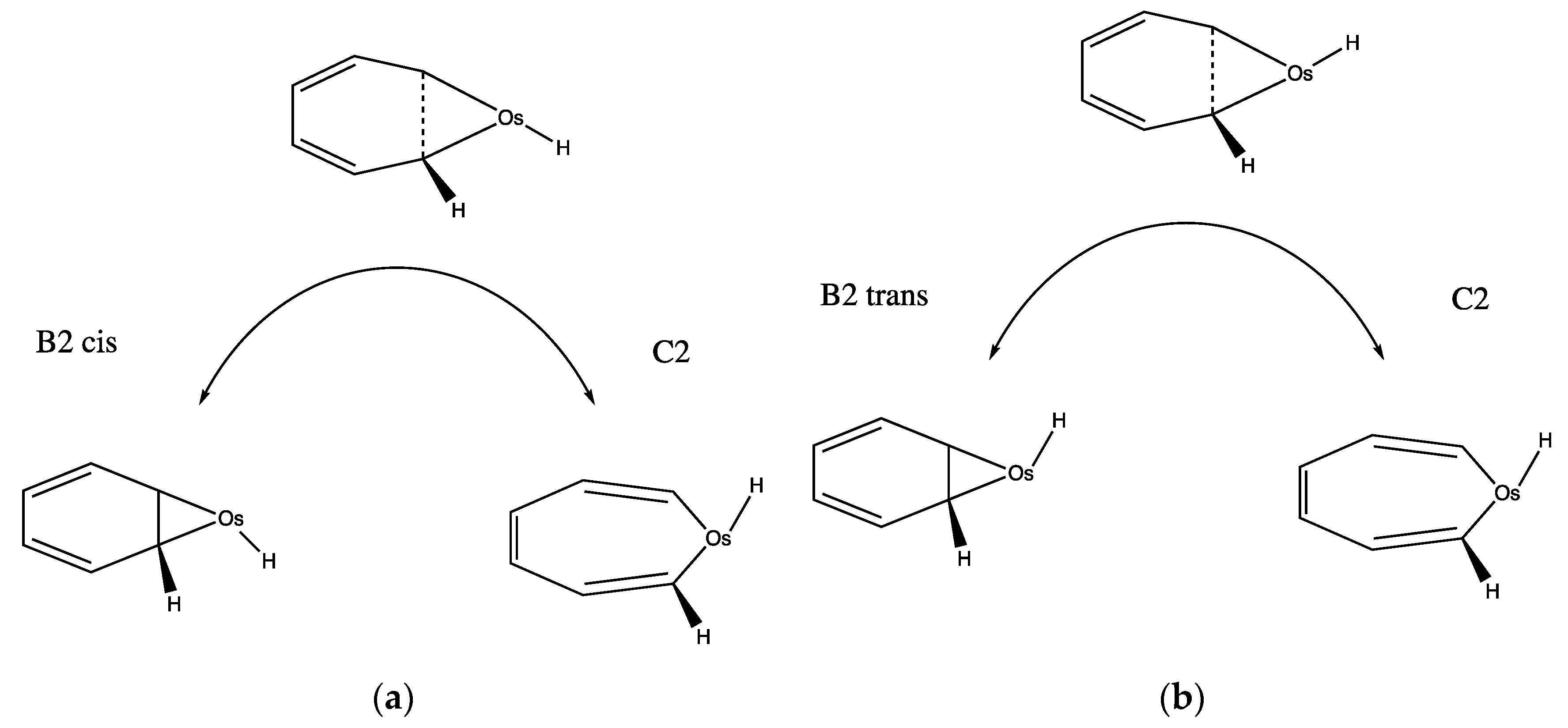
3.4.2. Is the C6v Complex of Os with Benzene OsC6H6 (+) a Sink?
3.4.3. Continuation of the B Path toward H2 Extrusion
3.4.4. Overview of the C Reaction Path for H2 Extrusion from 1-Osma-cyclohepta 2,4,6-triene (C1)
3.4.5. Second H Transfer (C Path)
3.4.6. Can Crossover from C to B Paths Occur?
3.4.7. Extrusion of H2 from OsC6H6 (+)
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, L.; Corma, A. Metal catalysts for heterogeneous catalysis: From single atoms to nanoclusters and nanoparticles. Chem. Rev. 2018, 118, 4981–5079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, H. Ménage á trois: Single atom catalysis, mass spectrometry, and computational chemistry. Catal. Sci. Technol. 2017, 7, 4302–4314. [Google Scholar] [CrossRef]
- Woodham, A.P.; Meijer, G.; Fielicke, A. Activation of molecular oxygen by anionic gold clusters. Angew. Chem. Int. Ed. 2012, 51, 4444. [Google Scholar] [CrossRef] [PubMed]
- Harding, D.J.; Kerpal, C.; Meijer, G.; Fielicke, A. Activated Methane on Small Cationic Platinum Clusters. Angew. Chem. Int. Ed. 2012, 51, 817–819. [Google Scholar] [CrossRef] [PubMed]
- Böhme, D.K.; Schwarz, H. Gas-phase catalysis by atomic and cluster metal ions: The ultimate single-site catalysts. Angew. Chem. Int. Ed. 2005, 44, 2336–2354. [Google Scholar] [CrossRef] [PubMed]
- Schröder, D.; Sulzle, D.; Hrusak, J.; Bohme, D.K.; Schwarz, H. Neutralization-reionization mass spectrometry as a novel probe to structurally characterize organic ligands generated in the Fe(I) mediated oligomerization of acetylene in the gas phase. Int. J. Mass Spectrom. Ion Process. 1991, 110, 145–156. [Google Scholar] [CrossRef]
- Duncan, M.A. Infrared spectroscopy to probe structure and dynamics in metal ion–molecule complexes. Int. Rev. Phys. Chem. 2003, 22, 407–435. [Google Scholar] [CrossRef]
- Schnabel, P.; Irion, M.P.; Weil, K.G. Evidence for low-pressure catalysis in the gas phase by a naked metal cluster: The growth of benzene precursors on iron (Fe4+). J. Phys. Chem. 1991, 95, 9688–9694. [Google Scholar] [CrossRef]
- Schnabel, P.; Weil, K.G.; Irion, M.P. Proof of the Catalytic Activity of a Naked Metal Cluster in the Gas Phase Angew. Chem. Int. Ed. Engl. 1992, 31, 636–638. [Google Scholar] [CrossRef]
- Gehret, O.; Irion, M.P. Reactions of Fe+n clusters (n = 2–11) with C6H6 and C6D6. Ligand isomerization in the benzene precursor ion Fe4(C2H2)3+. Chem. Phys. Lett. 1996, 254, 379–383. [Google Scholar]
- Diefenbach, M.; Schwarz, H. Cationic Transition Metal Arene Interactions. In Encyclopedia of Computational Chemistry; John Wiley & Sons: Chicester, UK, 2004. [Google Scholar]
- Wesendrup, R.; Schwarz, H. Catalytic Benzene Formation in the Gas-Phase Reactions of MC4H4+ (M = Ru, Rh) with C2H2. Organometallics 1997, 16, 461–466. [Google Scholar] [CrossRef]
- Armentrout, P.B.; Kuijpers, S.E.J.; Luschchikova, O.V.; Hightower, R.L.; Boles, G.C.; Bakker, J.M. Spectroscopic Identification of the Carbyne Hydride Structure of the Dehydrogenation Product of Methane Activation by Osmium Cations. J. Am. Soc. Mass Spectrom. 2018, 29, 1781–1790. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-N.; Liu, Z.; Wu, H.; Zhang, D.; Li, W.; Huang, Z.; Wang, G.; Xu, F.; Ding, C.-F.; Zhou, M. Reactions of Transition-Metal Carbyne Cations with Ethylene in the Gas Phas. J. Phys. Chem. A. 2020, 124, 2628–2633. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, X.; Liu, Z.; Wu, H.; Zhang, D.; Ding, X. CC Exchange in Activation/Coupling Reaction of Acetylene and Methane Mediated by Os+: A comparison iith Ir+, Pt+, and Au+. J. Phys. Chem. Lett. 2020, 11, 8346–8351. [Google Scholar] [CrossRef] [PubMed]
- Altun, Z.; Bleda, E.A.; Trindle, C. Single metal catalysis: DFT and CAS modeling of species involved in the Fe cation assisted transformation of acetylene to benzene. Mol. Phys. 2017, 115, 2185–2200. [Google Scholar] [CrossRef]
- Chrétien, S.; Salahub, D.R. Kohn-Sham density-functional study of the formation of benzene on iron clusters Fe/Fen+ (n = 1–4). J. Chem. Phys. 2003, 119, 12291. [Google Scholar] [CrossRef]
- Chai, D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Fox, Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Fox, Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | G | G-G0 (eV) | Barriers (eV) | See |
|---|---|---|---|---|
| B1 closed | −322.593606 | 0 | Figure 5 | |
| B1 closed to B2 closed | −322.496390 | 2.645 | 2.645:1.284 | |
| B2 closed | −322.543587 | 1.361 | ||
| B2 closed to B2 cis | −322.509613 | 2.285 | 0.924: 0.551 | |
| B2cis | −322.5298548 | 1.734 | Figure 6 | |
| B2cis to B2 trans | −322.512661 | 2.202 | 0.468:0.592 | |
| B2trans | −322.534412 | 1.610 | ||
| B3 ortho | −322.533566 | 1.633 | ||
| B2trans to B3 ortho | −322. 526011 | 1.839 | 0.229:0.206 | |
| B2 cis to B3 ortho | −322.522966 | 1.922 | 0.188:0.289: | |
| B3 cis | −322.521829 | 1.953 | ||
| B3cis to B3 trans | −322.505038 | 2.410 | 0.457:0.820 | |
| B3 trans | −322.535153 | 1.590 | ||
| B3ortho to B3 trans | −322.524222 | 1.887 | 0.254:0.297 | |
| B3 ortho to B3′ ortho | −322.532158 | 1.687 | 0.054:0.054 | Figure 7 |
| H2 + B4 | −322.500601 | 2.530 | Figure 11 |
| Species | G | G-G0 (eV) | See |
|---|---|---|---|
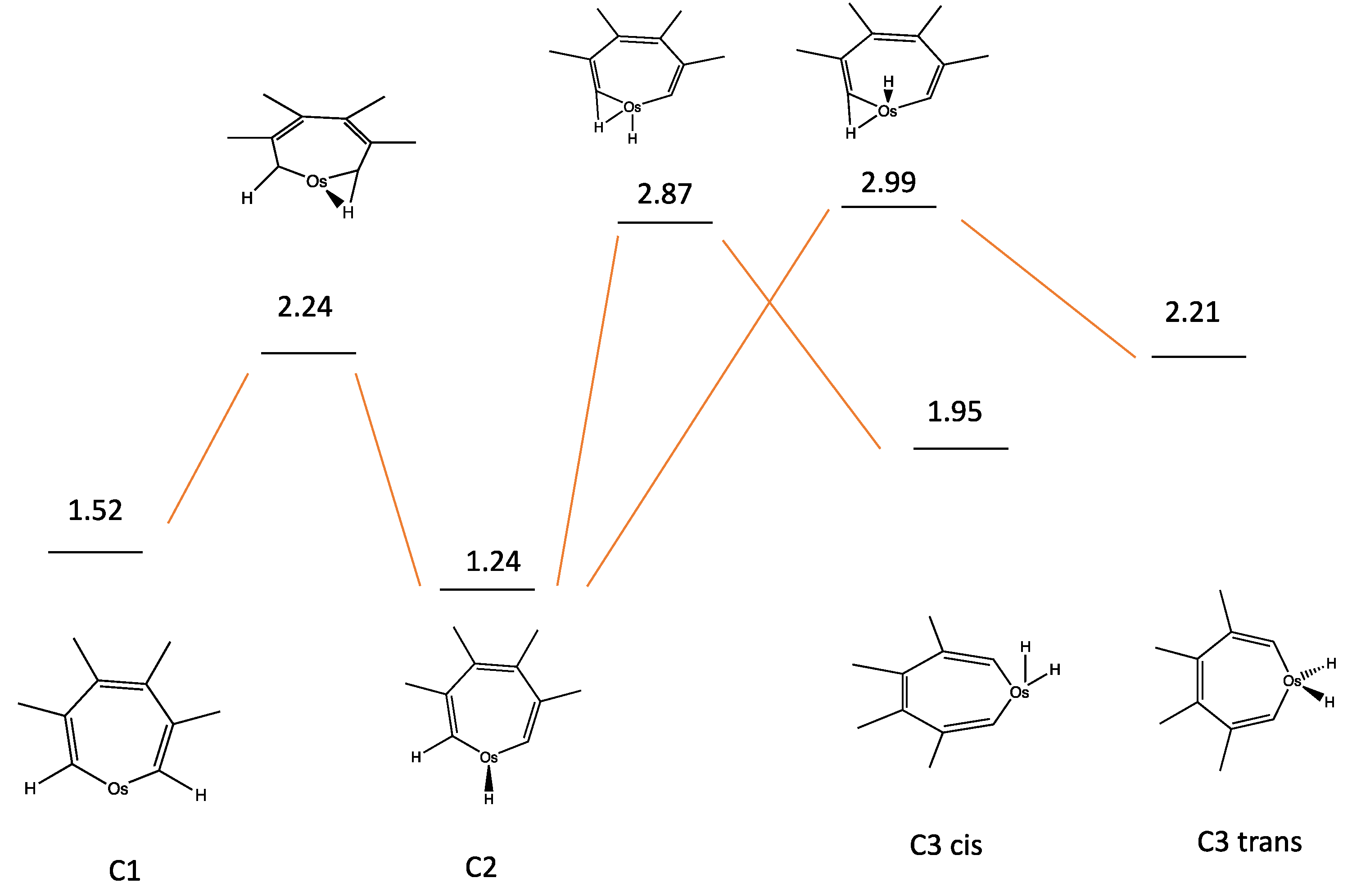
| C1 | −322.537790 | 1.518 | Figure 8 |
| C1 to C2 | −322.511164 | 2.243 | |
| C2 | −322.548169 | 1.236 | |
| C2 to C3 cis | −322.488162 | 2.869 | Figure 9 |
| C3 cis | −322.521893 | 1.951 | |
| C2 to C3 trans | −322.483549 | 2.994 | |
| C3 trans | −322.512357 | 2.210 | |
| C3 cis | −322.521893 | 1.951 | |
| C3 cis to C3 trans | −322.510759 | 2.259 | |
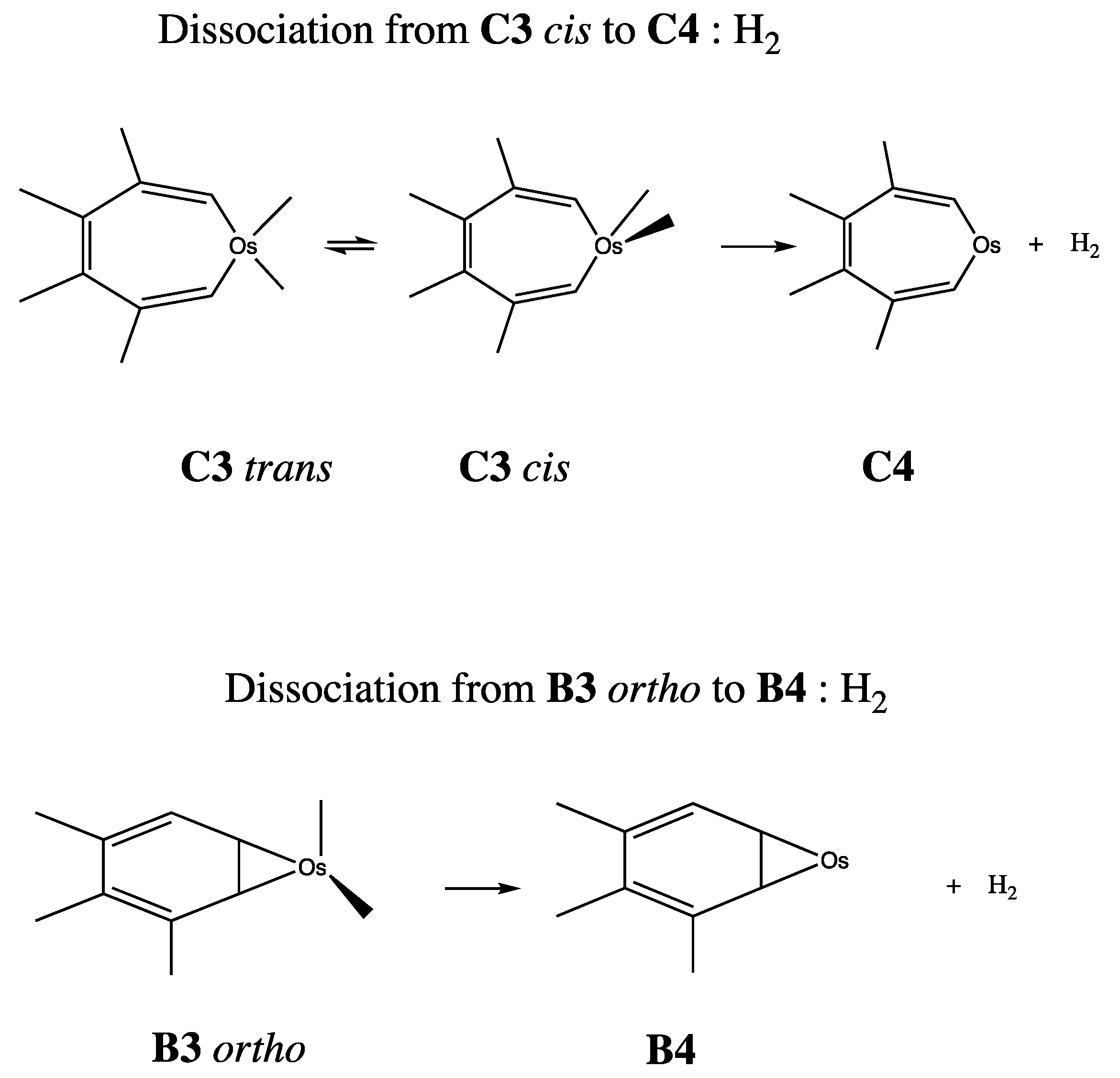
| H2 + C4 | −322.475475 | 3.214 | Figure 11 |
| Species | G | G-G0 (eV) | See |
|---|---|---|---|
| B2 trans to C2 TS | −322.506130 | 2.380 | Figure 10 |
| B2 cis to C2 TS | −322.509119 | 2.298 | |
| H2 + C4 to H2 + B4 | −322.466052 | 3.470 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altun, Z.; Bleda, E.A.; Trindle, C. CH Activation by a Heavy Metal Cation: Production of H2 from the Reaction of Acetylene with C4H4-Os(+) in Gas phase. C 2021, 7, 68. https://doi.org/10.3390/c7040068
Altun Z, Bleda EA, Trindle C. CH Activation by a Heavy Metal Cation: Production of H2 from the Reaction of Acetylene with C4H4-Os(+) in Gas phase. C. 2021; 7(4):68. https://doi.org/10.3390/c7040068
Chicago/Turabian StyleAltun, Zikri, Erdi Ata Bleda, and Carl Trindle. 2021. "CH Activation by a Heavy Metal Cation: Production of H2 from the Reaction of Acetylene with C4H4-Os(+) in Gas phase" C 7, no. 4: 68. https://doi.org/10.3390/c7040068
APA StyleAltun, Z., Bleda, E. A., & Trindle, C. (2021). CH Activation by a Heavy Metal Cation: Production of H2 from the Reaction of Acetylene with C4H4-Os(+) in Gas phase. C, 7(4), 68. https://doi.org/10.3390/c7040068