

Doping of Graphene Nanostructure with Iron, Nickel and Zinc as Selective Detector for the Toxic Gas Removal: A Density Functional Theory Study



Abstract
:1. Introduction
2. Materials, Modeling and Computational Details
2.1. Adsorptive Removal of Toxic Gases
2.2. Langmuir Adsorption Model & Charge Density Analysis
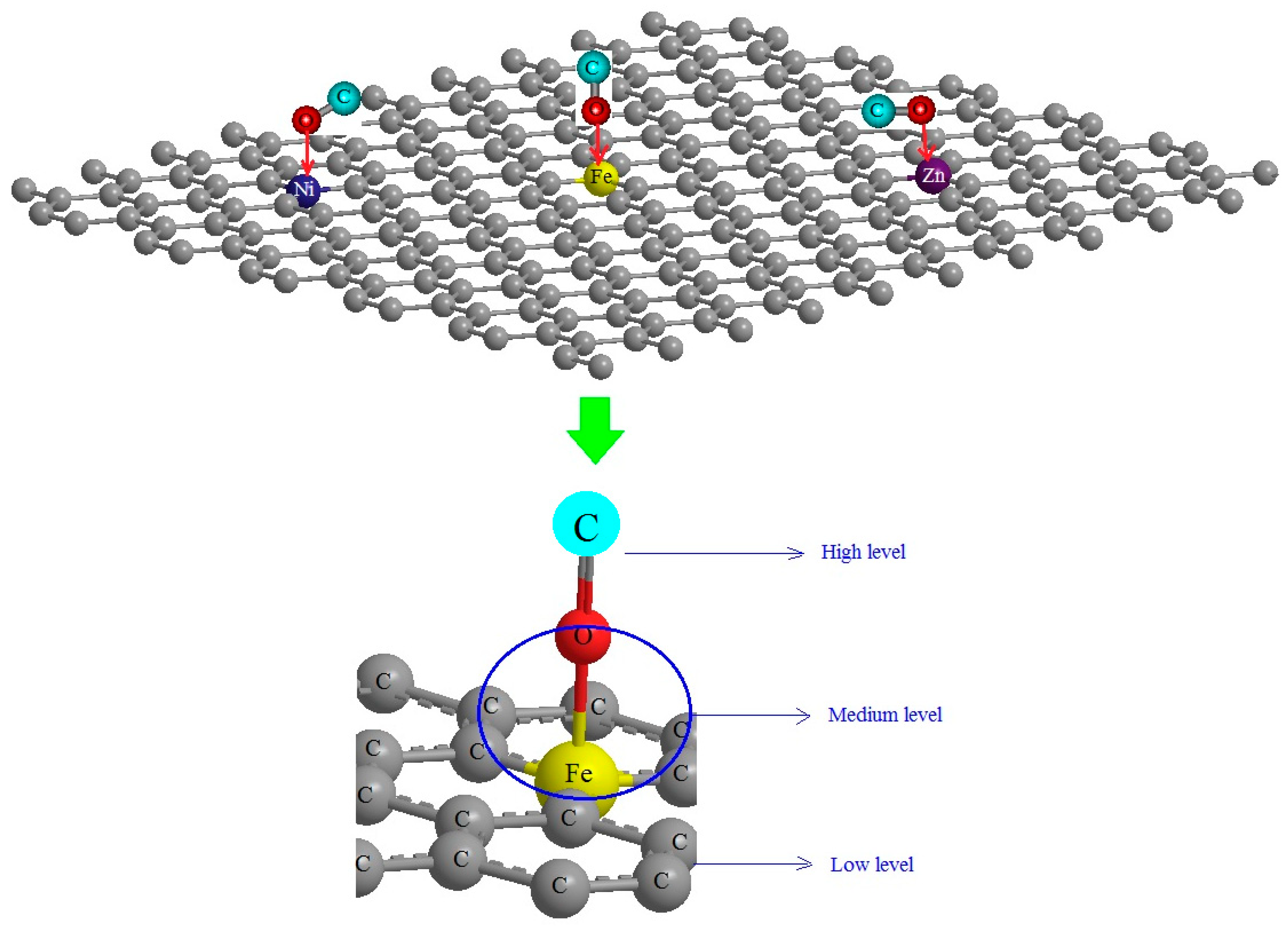
2.3. ONIOM by Density Functional Theory
3. Results
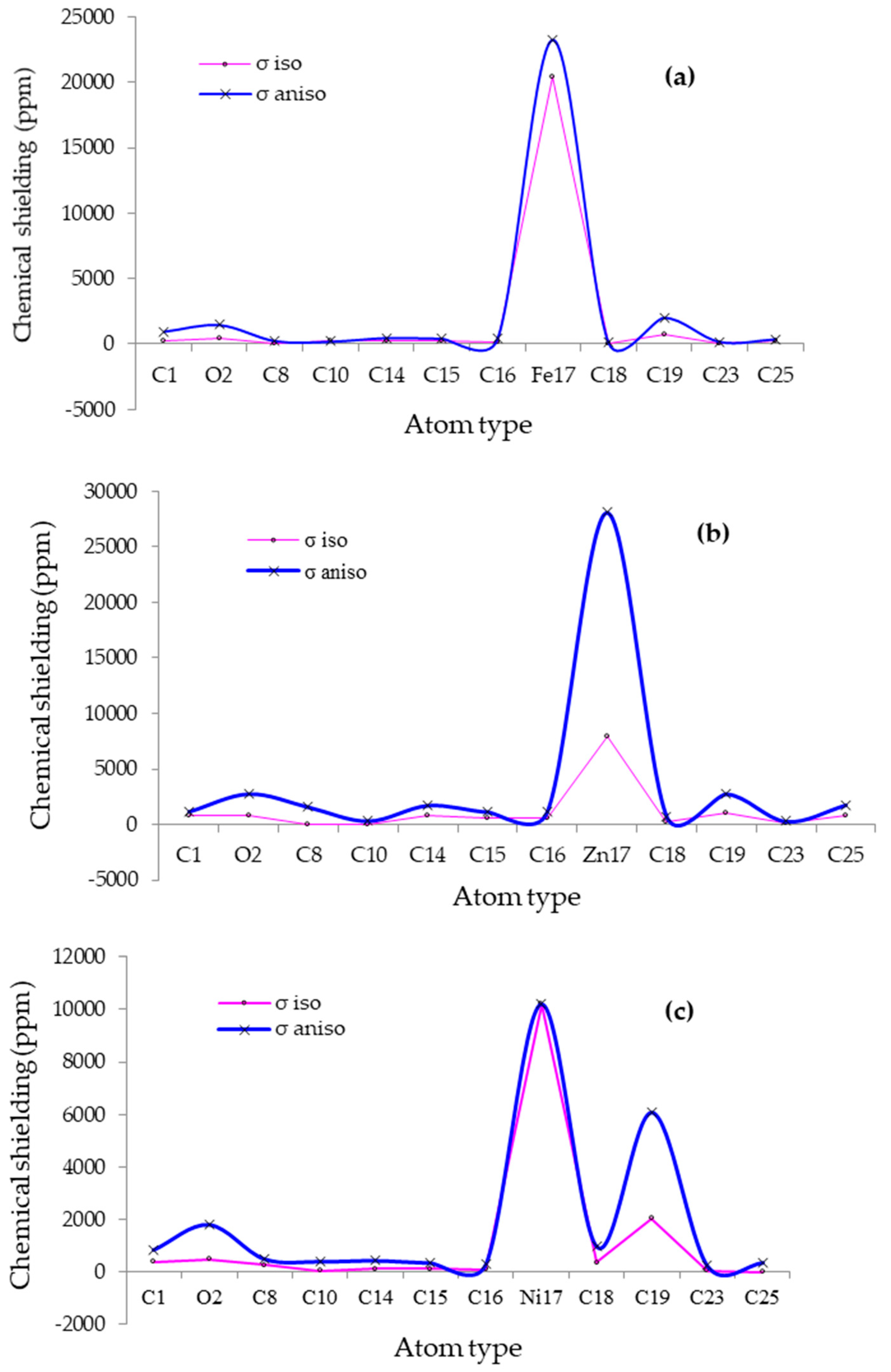
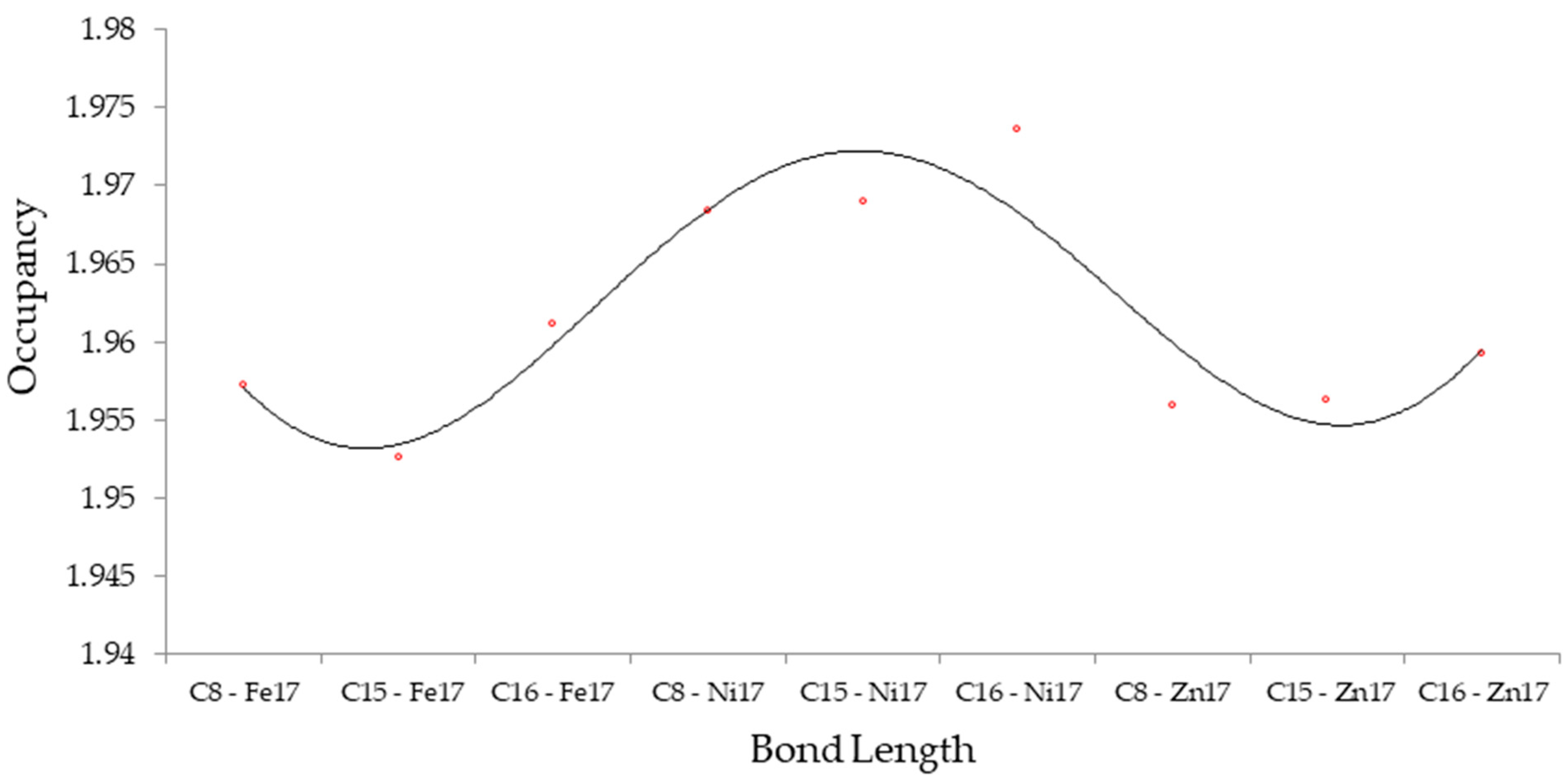
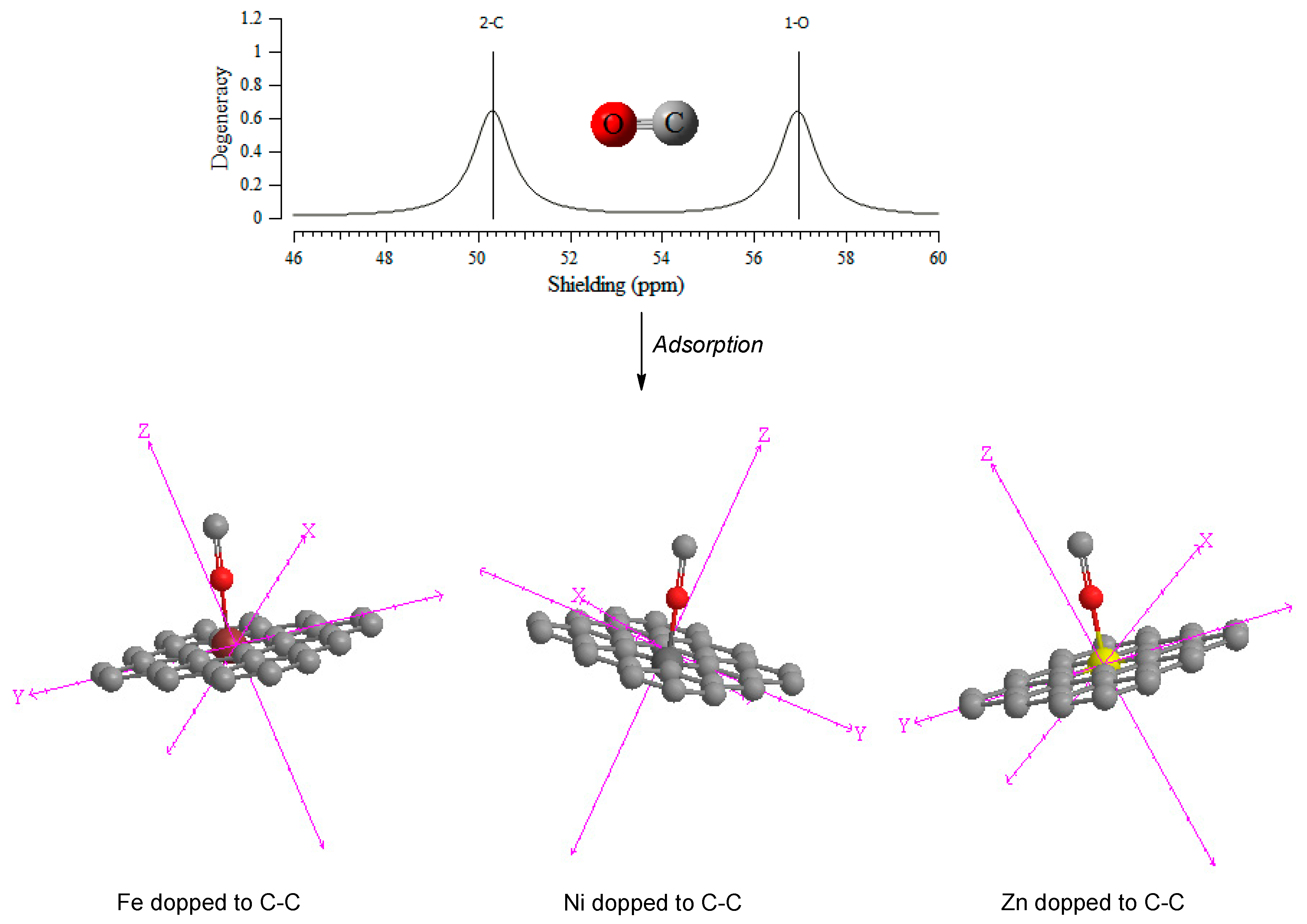
3.1. NMR Spectroscopy & NBO Analysis
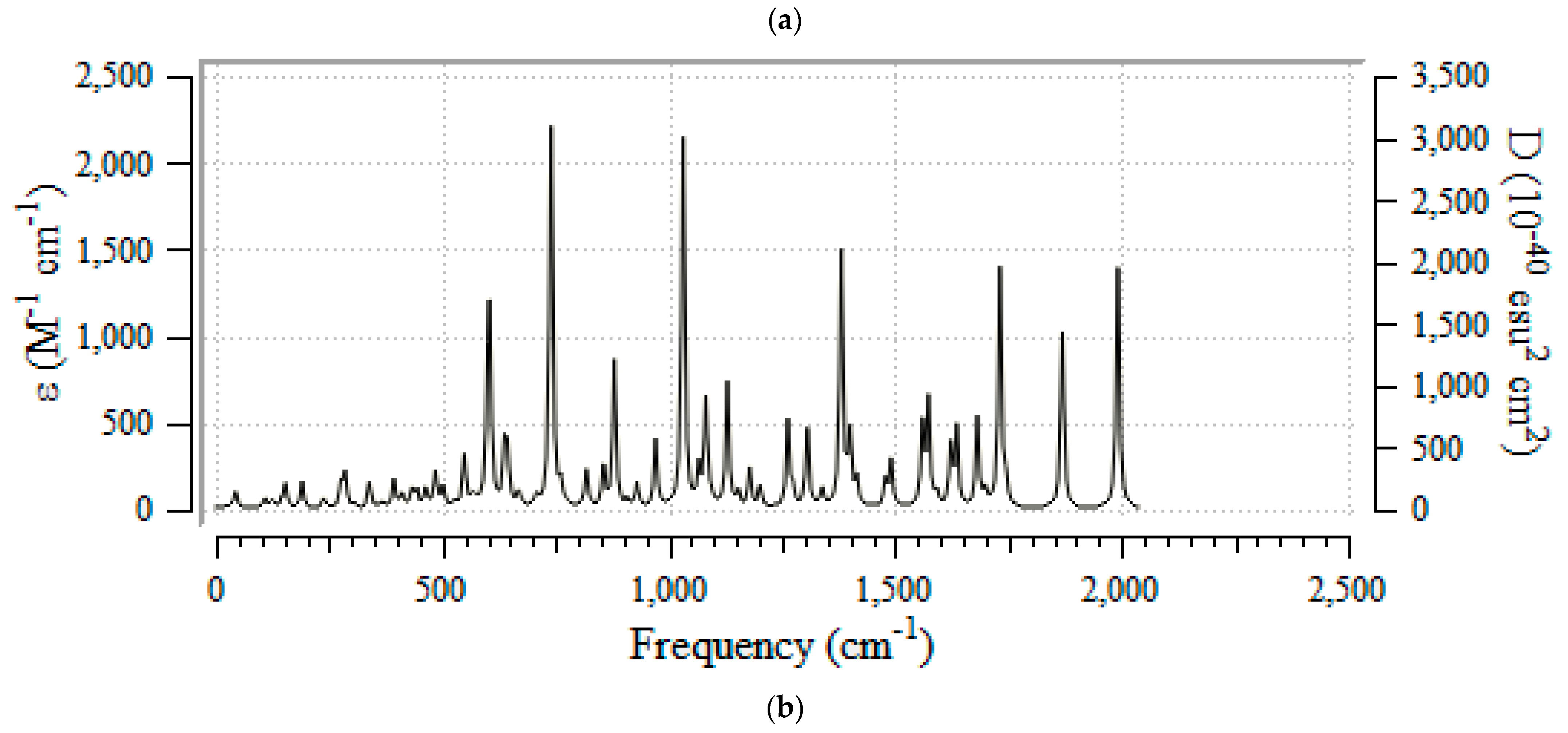
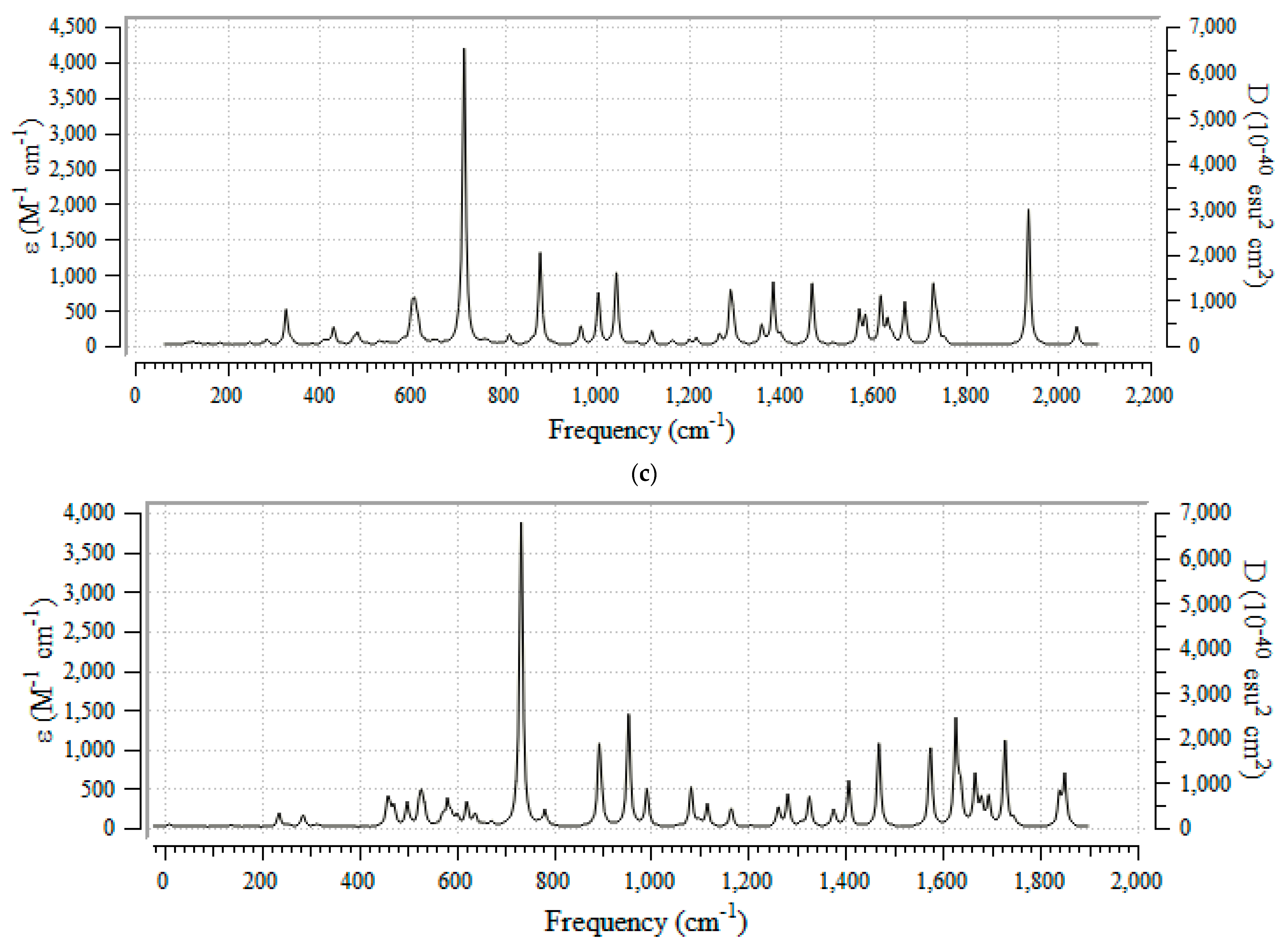
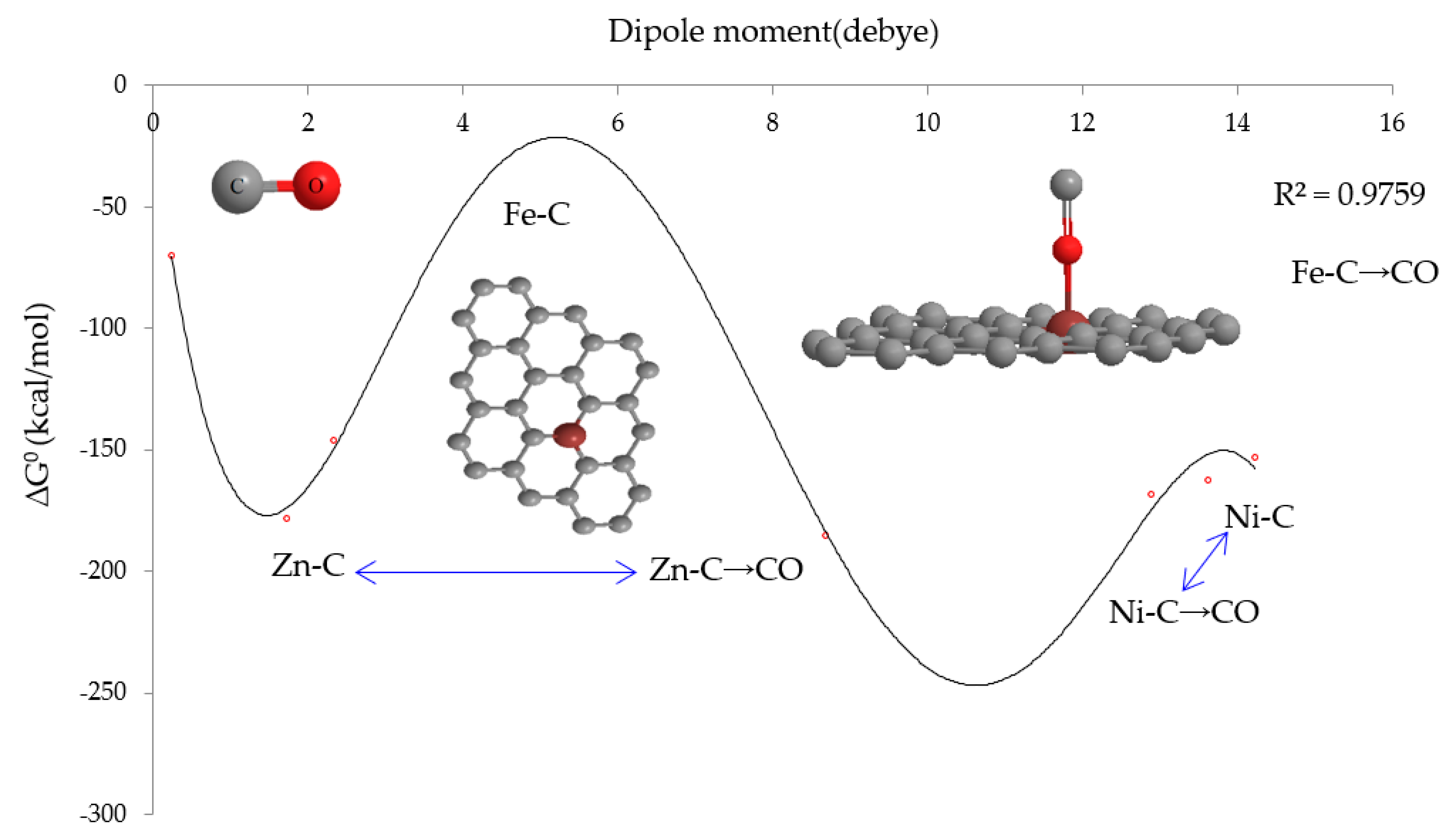
3.2. Thermodynamic Properties & IR Spectroscopy Analysis
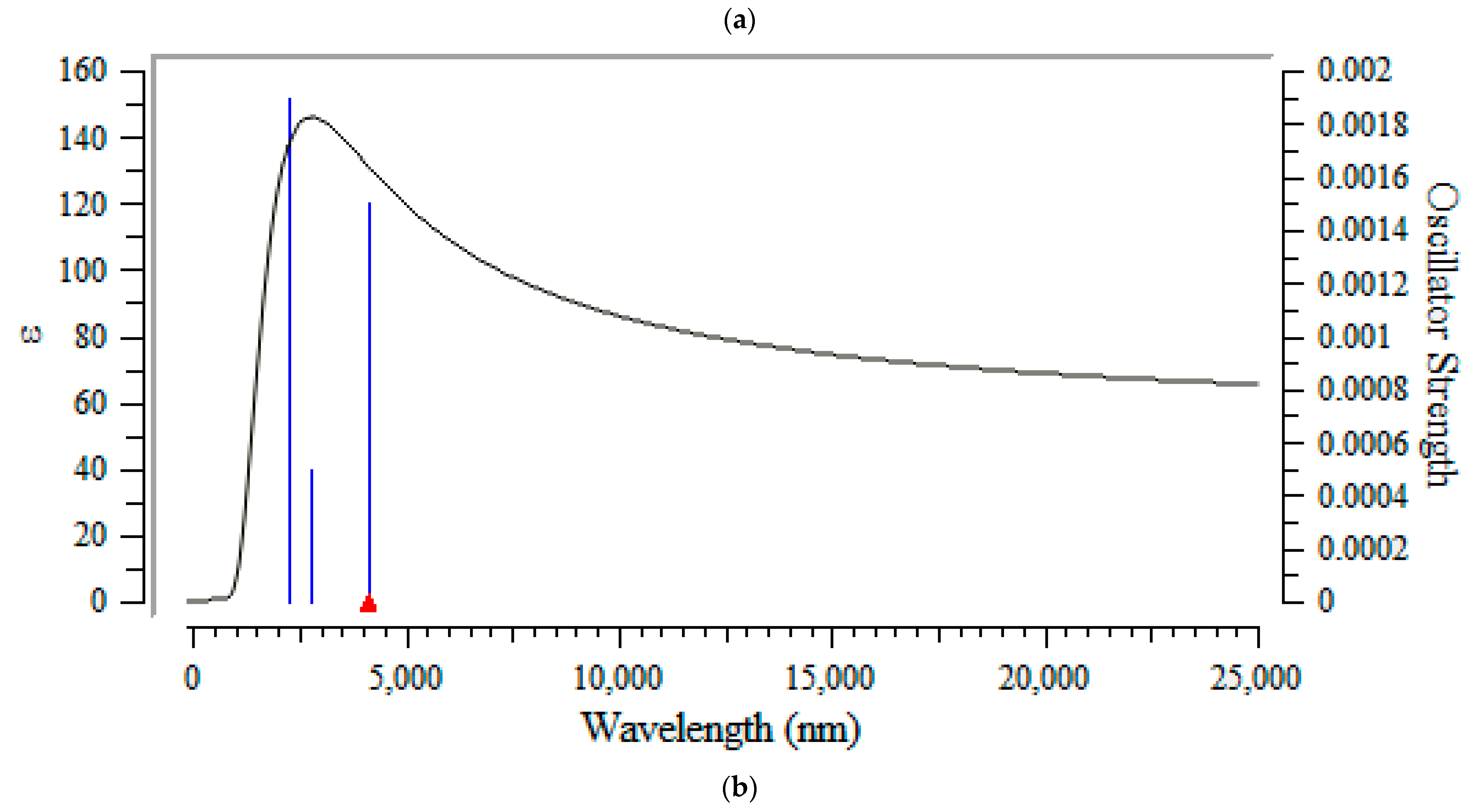
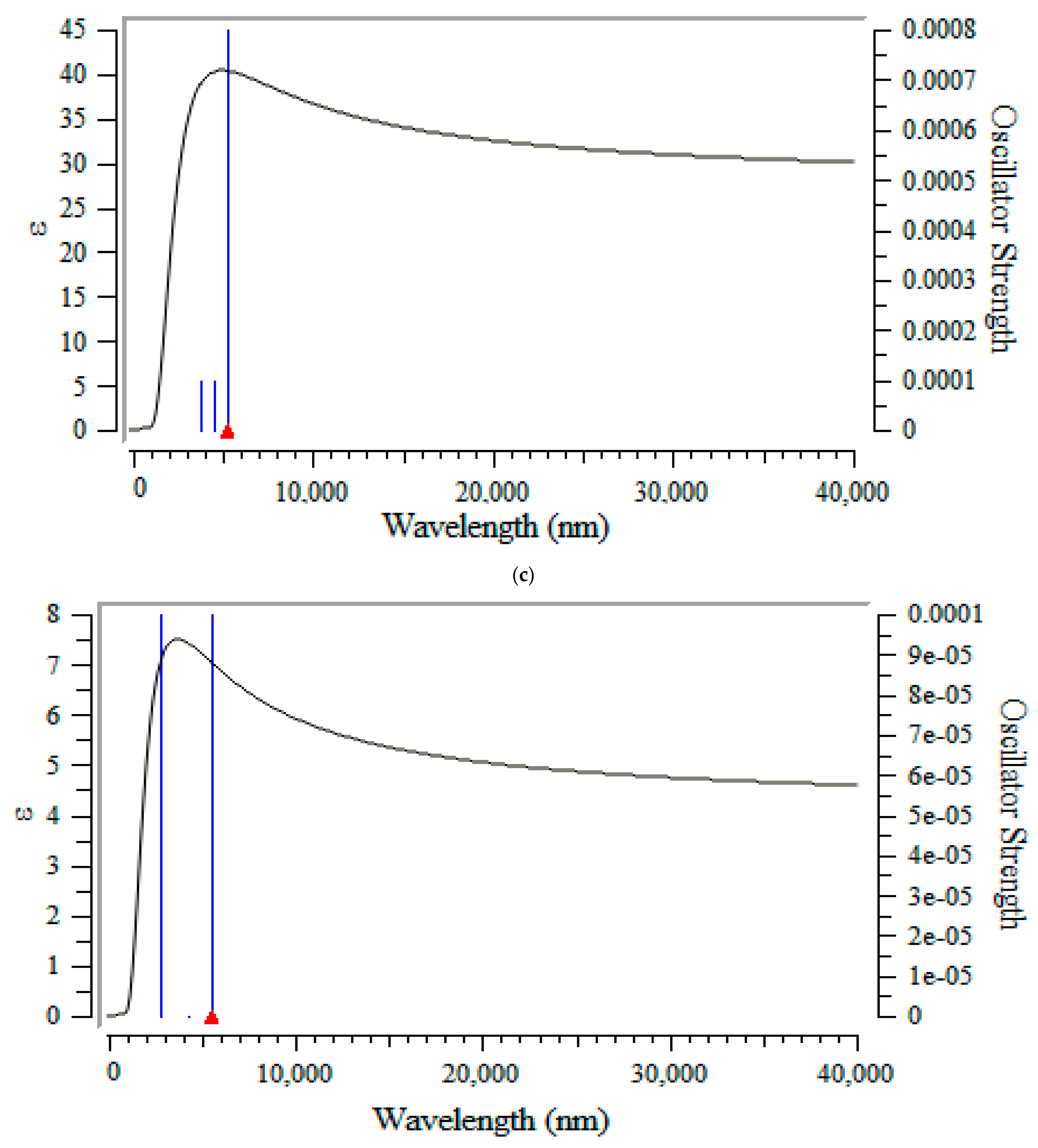
3.3. Frontier Molecular Orbital Analyis and Ultravoilet & Viible Spectroscopy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mendes, P.C.D.; Ocampo-Restrepo, V.K.; Da Silva, J.L.F. Ab initio investigation of quantum size effects on the adsorption of CO2, CO, H2O, and H2 on transition-metal particles. Phys. Chem. Chem. Phys. 2020, 22, 8998–9008. [Google Scholar]
- Wang, M.; Zhang, B.; Ding, J.; Xu, N.; Bernards, M.T.; He, Y.; Shi, Y. Three-Dimensional Nitrogen-Doped Graphene AerogelSupported MnO Nanoparticles as Efficient Electrocatalysts for CO2 Reduction to CO. ACS Sustain. Chem. Eng. 2020, 8, 4983–4994. [Google Scholar]
- Ocampo-Restrepo, V.K.; Zibordi-Besse, L.; Da Silva, J.L.F. Ab initio investigation of the atomistic descriptors in the activation of small molecules on 3 d transition-metal 13-atom clusters: The example of H2, CO, H2O, and CO2. J. Chem. Phys. 2019, 151, 214301. [Google Scholar] [CrossRef] [PubMed]
- Montejo-Alvaro, F.; Martínez-Espinosa, J.A.; Rojas-Chávez, H.; Navarro-Ibarra, D.C.; Cruz-Martínez, H.; Medina, D.I. CO2 Adsorption over 3d Transition-Metal Nanoclusters Supported on Pyridinic N3-Doped Graphene: A DFT Investigation. Materials 2022, 15, 6136. [Google Scholar] [CrossRef]
- Cruz-Martínez, H.; Rojas-Chávez, H.; Montejo-Alvaro, F.; Peña-Castañeda, Y.; Matadamas-Ortiz, P.; Medina, D. Recent Developments in Graphene-Based Toxic Gas Sensors: A Theoretical Overview. Sensors 2021, 21, 1992. [Google Scholar] [PubMed]
- Montejo-Alvaro, F.; González-Quijano, D.; Valmont-Pineda, J.A.; Rojas-Chávez, H.; Juárez-García, J.M.; Medina, D.I.; CruzMartínez, H. CO2 Adsorption on PtCu Sub-Nanoclusters Deposited on Pyridinic N-Doped Graphene: A DFT Investigation. Materials 2021, 14, 7619. [Google Scholar]
- Lisovski, O.; Piskunov, S.; Bocharov, D.; Zhukovskii, Y.F.; Kleperis, J.; Knoks, A.; Lesnicenoks, P. CO2 and CH2 Adsorption on Copper-Decorated Graphene: Predictions from First Principle Calculations. Crystals 2022, 12, 194. [Google Scholar] [CrossRef]
- Ali, M.; Tit, N.; Yamani, Z.H. First principles study on the functionalization of graphene with Fe catalyst for the detection of CO2: Effect of catalyst clustering. Appl. Surf. Sci. 2020, 502, 144153. [Google Scholar] [CrossRef]
- Salih, E.; Ayesh, A.I. Pt-doped armchair graphene nanoribbon as a promising gas sensor for CO and CO2: DFT study. Phys. E Low-Dimens. Syst. Nanostruct. 2021, 125, 114418. [Google Scholar] [CrossRef]
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Nasibulin, A.G.; Pikhitsa, P.V.; Jiang, H.; Brown, D.P.; Krasheninnikov, A.V.; Anisimov, A.S.; Queipo, P.; Moisala, A.; Gonzalez, D.; Lientschnig, G.; et al. A Novel Hybrid Carbon Material. Nat. Nanotechnol. 2007, 2, 156–161. [Google Scholar]
- Moisala, A.; Nasibulin, A.G.; Shandakov, S.D.; Jiang, H.; Kauppinen, E.I. On-Line Detection of Single-Walled Carbon Nanotube Formation during Aerosol Synthesis Methods. Carbon 2005, 43, 2066–2074. [Google Scholar] [CrossRef]
- Delgado, J.L.; Herranz, M.; Martín, N. The Nano-Forms of Carbon. J. Mater. Chem. 2008, 18, 1417. [Google Scholar] [CrossRef]
- Falcao, E.H.; Wudl, F. Carbon Allotropes: Beyond Graphite and Diamond. J. Chem. Technol. Biotechnol. 2007, 82, 524–531. [Google Scholar]
- Langenhorst, F.; Campione, M. Ideal and Real Structures of Different Forms of Carbon, with Some Remarks on Their Geological Significance. J. Geol. Soc. 2019, 176, 337–347. [Google Scholar] [CrossRef]
- Zhou, Q.; Luo, S.; Xue, W.; Liao, N. Highly selective nitrogen dioxide gas sensing of ReS2 nanosheets: A first-principles study. Appl. Surf. Sci. 2023, 609, 155388. [Google Scholar] [CrossRef]
- Chen, M.; Yi, X.; Hu, X.; Zhou, X.; Tian, J.; Li, X. Correlation between the activity of Fe@ (N, S, and P) doped graphene catalysts and the coordination environment: A density functional theory study. Int. J. Hydrog. Energy 2023, 48, 171–179. [Google Scholar] [CrossRef]
- Singla, M.; Sharma, D.; Jaggi, N. Effect of transition metal (Cu and Pt) doping/co-doping on hydrogen gas sensing capability of graphene: A DFT study. Int. J. Hydrog. Energy 2021, 46, 16188–16201. [Google Scholar] [CrossRef]
- Joel, E.F.; Lujaniene, G. Progress in Graphene Oxide Hybrids for Environmental Applications. Environments 2022, 9, 153. [Google Scholar] [CrossRef]
- Bijesh, P.; Selvaraj, V.; Andal, V. A review on synthesis and applications of nano metal Oxide/porous carbon composite. Mater. Today Proc. 2021, 55, 212–219. [Google Scholar]
- Li, F.; Jiang, X.; Zhao, J.; Zhang, S. Graphene oxide: A promising nanomaterial for energy and environmental applications. Nano Energy 2015, 16, 488–515. [Google Scholar] [CrossRef]
- El-Shazly, E.A.A.; Moussa, S.I.; Dakroury, G.A. Recovery of Some Rare-Earth Elements by Sorption Technique onto Graphene Oxide. J. Sustain. Met. 2022, 8, 715–731. [Google Scholar] [CrossRef]
- Su, Y.; Wang, J.; Wang, B.; Yang, T.; Yang, B.; Xie, G.; Zhou, Y.; Zhang, S.; Tai, H.; Cai, Z.; et al. Alveolus-inspired active membrane sensors for self-powered wearable chemical sensing and breath analysis. ACS Nano 2020, 14, 6067–6075. [Google Scholar] [CrossRef]
- Ma, D.; Zhang, J.; Li, X.; He, C.; Lu, Z.; Lu, Z.; Lu, Z.; Yang, Z.; Wang, Y. C3N monolayers as promising candidates for NO2 sensors. Sens. Actuators B Chem. 2018, 266, 664–673. [Google Scholar] [CrossRef]
- Pacheco, M.; Pacheco, J.; Valdivia, R.; Santana, A.; Tu, X.; Mendoza, D.; Frias, H.; Medina, L.; Macias, J. Green Applications of Carbon Nanostructures Produced by Plasma Techniques. MRS Adv. 2017, 2, 2647–2659. [Google Scholar] [CrossRef]
- Leenaerts, O.; Partoens, B.; Peeters, F.M. Adsorption of H2O, NH3, CO, NO2, and NO on graphene: A first-principles study. Phys. Rev. B 2008, 77, 125416. [Google Scholar] [CrossRef] [Green Version]
- Bi, F.; Zhao, Z.; Yang, Y.; Gao, W.; Liu, N.; Huang, Y.; Zhang, X. Chlorine-Coordinated Pd Single Atom Enhanced the Chlorine Resistance for Volatile Organic Compound Degradation: Mechanism Study. Environ. Sci. Technol. 2022, 56, 17321–17330. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, Z.; Zhao, S.; Xiang, S.; Gao, W.; Wang, L.; Xu, J.; Wang, Y. The promoting effect of alkali metal and H2O on Mn-MOF derivatives for toluene oxidation: A combined experimental and theoretical investigation. J. Catal. 2022, 415, 218–235. [Google Scholar] [CrossRef]
- Wen, M.; Dong, F.; Yao, J.; Tang, Z.; Zhang, J. Pt nanoparticles confined in the ordered mesoporous CeO2 as a highly efficient catalyst for the elimination of VOCs. J. Catal. 2022, 412, 42–58. [Google Scholar] [CrossRef]
- Lee, S.W.; Lee, W.; Hong, Y.; Lee, G.; Yoon, D.S. Recent advances in carbon material-based NO2 gas sensors. Sens. Actuators B Chem. 2018, 255, 1788–1804. [Google Scholar] [CrossRef]
- Chatterjee, S.G.; Chatterjee, S.; Ray, A.K.; Chakraborty, A.K. Graphene–metal oxide nanohybrids for toxic gas sensor: A review. Sens. Actuators B Chem. 2015, 221, 1170–1181. [Google Scholar] [CrossRef]
- Xiao, Z.; Kong, L.B.; Ruan, S.; Li, X.; Yu, S.; Li, X.; Jiang, Y.; Yao, Z.; Ye, S.; Wang, C.; et al. Recent development in nanocarbon materials for gas sensor applications. Sens. Actuators B Chem. 2018, 274, 235–267. [Google Scholar] [CrossRef]
- Manna, A.K.; Pati, S.K. Tuning the electronic structure of graphene by molecular charge transfer: A computational study. Chem.–Asian J. 2009, 4, 855–860. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Shen, C.; Chai, Y. Adsorption of CO molecules on doped graphene: A first-principles study. AIP Adv. 2016, 6, 025317. [Google Scholar] [CrossRef]
- Boyd, A.; Dube, I.; Fedorov, G.; Paranjape, M.; Barbara, P. Gas Sensing Mechanism of Carbon Nanotubes: From Single Tubes to High-Density Networks. Carbon 2014, 69, 417–423. [Google Scholar]
- Tabtimsai, C.; Keawwangchai, S.; Nunthaboot, N.; Ruangpornvisuti, V.; Wanno, B. Density Functional Investigation of Hydrogen Gas Adsorption on Fe-doped Pristine and Stone-Wales Defected Single-walled Carbon Nanotubes. J. Mol. Model. 2012, 18, 3941–3949. [Google Scholar] [CrossRef]
- Rather, S. Hydrogen Uptake of Ti-Decorated Multiwalled Carbon Nanotube Composites. Int. J. Hydrogen Energy 2021, 46, 17793–17801. [Google Scholar]
- Zhang, X.; Dai, Z.; Wei, L.; Liang, N.; Wu, X. Theoretical Calculation of the Gas-Sensing Properties of Pt-Decorated Carbon Nanotubes. Sensors 2013, 13, 15159–15171. [Google Scholar] [CrossRef]
- Zhang, X.; Dai, Z.; Chen, Q.; Tang, J. A DFT Study of SO2 and H2S Gas Adsorption on Au-Doped Single-Walled CarbonNanotubes. Phys. Scr. 2014, 89, 065803. [Google Scholar]
- Hanaor, D.A.H.; Ghadiri, M.; Chrzanowski, W.; Gan, Y. Scalable Surface Area Characterization by Electrokinetic Analysis of Complex Anion Adsorption (PDF). Langmuir 2014, 30, 15143–15152. [Google Scholar] [CrossRef]
- Mollaamin, F.; Shahriari, S.; Monajjemi, M.; Khalaj, Z. Nanocluster of Aluminum Lattice via Organic Inhibitors Coating: A Study of Freundlich Adsorption. J. Clust. Sci. 2022, 1–16. [Google Scholar] [CrossRef]
- Zhao, J.; Buldum, A.; Han, J.; Lu, J.P. Gas Molecule Adsorption in Carbon Nanotubes and Nanotube Bundles. Nanotechnology 2002, 13, 195–200. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods I. Method. J. Comput. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Svensson, M.; Humbel, S.; Froese, R.D.J.; Matsubara, T.; Sieber, S.; Morokuma, K. ONIOM: A Multilayered Integrated MO + MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test for Diels–Alder Reactions and Pt(P(t-Bu)3)2 + H2 Oxidative Addition. J. Phys. Chem. 1996, 100, 19357–19363. [Google Scholar] [CrossRef]
- Mollaamin, F. Features of parametric point nuclear magnetic resonance of metals implantation on boron nitride nanotube by density functional theory/electron paramagnetic resonance. J. Comput. Theor. Nanosci. 2014, 11, 2393–2398. [Google Scholar] [CrossRef]
- Argaman, N.; Makov, G. Density functional theory: An introduction. Am. J. Phys. 2000, 68, 69. [Google Scholar] [CrossRef]
- Chermette, H. Density functional theory: A powerful tool for theoretical studies in coordination chemistry. Coord. Chem. Rev. 1998, 180, 699. [Google Scholar]
- Chermette, H.J. Chemical reactivity indexes in density functional theory. Comput. Chem. 1999, 20, 129. [Google Scholar]
- Mollaamin, F.; Monajjemi, M. Molecular modelling framework of metal-organic clusters for conserving surfaces: Langmuir sorption through the TD-DFT/ONIOM approach. Mol. Simul. 2022, 1–12. [Google Scholar] [CrossRef]
- Ladeira, A.C.Q.; Ciminelli, V.S.T.; Duarte, H.A.; Alves, M.C.M.; Ramos, A.Y. Mechanism of anion retention from EXAFS and density functional calculations: Arsenic (V) adsorbed on gibbsite. Geochim. Cosmochim. Acta 2001, 65, 1211. [Google Scholar]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. General performance of density functionals. J. Phys. Chem. A 2007, 111, 10439. [Google Scholar] [CrossRef]
- Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory; Wiley-VCH: New York, NY, USA, 2001. [Google Scholar]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. B 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Kim, K.; Jordan, K.D. Comparison of Density Functional and MP2 Calculations on the Water Monomer and Dimer. J. Phys. Chem. 1994, 98, 10089–10094. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models, 2nd ed.; Wiley: Hoboken, NJ, USA, 2004. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Fry, R.A.; Kwon, K.D.; Komarneni, S.; Kubicki, J.D.; Mueller, K.T. Solid-State NMR and Computational Chemistry Study of Mononucleotides Adsorbed to Alumina. Langmuir 2006, 22, 9281–9286. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute Hardness: Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Politzer, P.; Abu-Awwad, F. A comparative analysis of Hartree-Fock and Kohn-Sham orbital energies. Theor. Chem. Acc. 1998, 99, 83–87. [Google Scholar] [CrossRef]
- Aihara, J. Reduced HOMO–LUMO Gap as an Index of Kinetic Stability for Polycyclic Aromatic Hydrocarbons. J. Phys. Chem. A 1999, 103, 7487–7495. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Bassler, G.C.; Morrill, T.C. Spectrometric Identification of Organic Compounds, 5th ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1981. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CO→Fe-Doped/Gr | CO→Ni-Doped/Gr | CO→Zn-Doped/Gr | ||||||
|---|---|---|---|---|---|---|---|---|
| Atom | σ iso | σ aniso | Atom | σ iso | σ aniso | Atom | σ iso | σ aniso |
| C1 | 233.4257 | 968.2802 | C1 | 382.6002 | 844.5628 | C1 | 817.8193 | 1200.6025 |
| O2 | 394.9638 | 1463.9175 | O2 | 495.3022 | 1809.7355 | O2 | 788.7503 | 2796.0869 |
| C8 | 29.2897 | 240.5192 | C8 | 260.1665 | 501.7185 | C8 | 11.2789 | 1596.7923 |
| C10 | 204.8650 | 198.5053 | C10 | 48.3307 | 404.4537 | C10 | 48.7486 | 342.4682 |
| C14 | 235.1941 | 480.5288 | C14 | 113.4488 | 438.0594 | C14 | 780.6997 | 1754.9402 |
| C15 | 247.1604 | 421.2191 | C15 | 111.3020 | 348.6676 | C15 | 562.6576 | 1162.1428 |
| C16 | 166.2217 | 406.5082 | C16 | 104.6419 | 310.2320 | C16 | 576.8997 | 1196.7425 |
| Fe17 | 20,464.6572 | 23,260.8181 | Ni17 | 10,207.7265 | 10,207.7265 | Zn17 | 7974.7632 | 28,112.8322 |
| C18 | 38.1089 | 134.3989 | C18 | 362.1582 | 967.9775 | C18 | 244.7286 | 749.2167 |
| C19 | 752.6628 | 2028.2108 | C19 | 2042.4580 | 6083.9999 | C19 | 1100.1399 | 2780.5764 |
| C23 | 89.6758 | 183.7640 | C23 | 58.0015 | 232.5619 | C23 | 177.0834 | 302.6073 |
| C25 | 269.0681 | 379.3128 | C25 | 3.1888 | 359.3453 | C25 | 834.3115 | 1785.4044 |
| Chemical shielding (CS) tensors in principal axes system evaluate the isotropic chemical-shielding (σiso), anisotropic chemical-shielding (σaniso) [64]: σisoσaniso | ||||||||
| CO → TM-Doped/Gr Nanosheet | Bond Orbital | Occupancy | Hybrids |
|---|---|---|---|
| CO → Fe-doped/Gr | BD (1) C8–Fe17 | 1.95721 | 0.8066 (sp1.69) C + 0.5910 (sp0.31 d3.07) Fe |
| BD (1) C15–Fe17 | 1.95267 | 0.8154 (sp1.40) C + 0.5789 (sp0.34 d3.23) Fe | |
| BD (1) C16–Fe17 | 1.96117 | 0.8180 (sp1.46) C + 0.5753 (sp0.40 d4.30) Fe | |
| CO → Ni-doped/Gr | BD (1) C8–Ni17 | 1.96844 | 0.8015 (sp1.59) C + 0.5980 (sp0.34 d2.00) Ni |
| BD (1) C15–Ni17 | 1.96893 | 0.8094 (sp1.39) C + 0.5872 (sp0.38 d2.24) Ni | |
| BD (1) C16–Ni17 | 1.97360 | 0.8191 (sp1.04) C + 0.5737 (sp0.58 d4.28) Ni | |
| CO → Zn-doped/Gr | BD (1) C8–Zn17 | 1.95595 | 0.8224 (sp1.45) C + 0.5689 (sp2.02 d0.43) Zn |
| BD (1) C15–Zn17 | 1.95624 | 0.8055 (sp1.33) C + 0.5926 (sp1.39 d1.08) Zn | |
| BD (1) C16–Zn17 | 1.95929 | 0.8052 (sp1.35) C + 0.5930 (sp1.44 d1.27) Zn |
| Compound | ∆Ho × 10−4 (kcal/mol) | ∆Go × 10−4 (kcal/mol) | So (Cal/K.mol) | Dipole Moment (Debye) |
|---|---|---|---|---|
| Fe-C | −146.2782 | −146.2816 | 111.175 | 2.3199 |
| Ni-C | −162.4793 | −162.4828 | 116.150 | 13.6226 |
| Zn-C | −178.2030 | −178.2066 | 120.533 | 1.7301 |
| :C≡O: | −69.784 | −69.798 | 47.100 | 0.2373 |
| :C≡O:→Fe-C | −153.2461 | −153.2501 | 131.502 | 14.2253 |
| :C≡O:→Ni-C | −168.3894 | −168.3930 | 119.303 | 12.8804 |
| :C≡O:→Zn-C | −185.1696 | −185.1733 | 123.534 | 8.6863 |
| Gas→TM-Doped Gr@NS | LUMO | HOMO | ∆E | µ | χ | η | ζ | ψ |
|---|---|---|---|---|---|---|---|---|
| CO→ Fe-C |  −0.00480 |  −0.12195 | 3.1878 | −1.7245 | 1.7245 | 1.5939 | 0.3137 | 0.9329 |
| CO→ Ni-C |  −0.00314 |  −0.13644 | 3.6272 | −1.8991 | 1.8991 | 1.8136 | 0.2757 | 0.9943 |
| CO→ Zn-C |  −0.00907 |  −0.15289 | 3.9135 | −2.2035 | 2.2035 | 1.9567 | 0.2555 | 1.2407 |
| ∆E = ELUMO − EHOMO; µ = (EHOMO + ELUMO)/2; χ = −(EHOMO + ELUMO)/2; η = (ELUMO − EHOMO)/2; ζ = 1/(2η); ψ = µ2/(2η) | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollaamin, F.; Monajjemi, M. Doping of Graphene Nanostructure with Iron, Nickel and Zinc as Selective Detector for the Toxic Gas Removal: A Density Functional Theory Study. C 2023, 9, 20. https://doi.org/10.3390/c9010020
Mollaamin F, Monajjemi M. Doping of Graphene Nanostructure with Iron, Nickel and Zinc as Selective Detector for the Toxic Gas Removal: A Density Functional Theory Study. C. 2023; 9(1):20. https://doi.org/10.3390/c9010020
Chicago/Turabian StyleMollaamin, Fatemeh, and Majid Monajjemi. 2023. "Doping of Graphene Nanostructure with Iron, Nickel and Zinc as Selective Detector for the Toxic Gas Removal: A Density Functional Theory Study" C 9, no. 1: 20. https://doi.org/10.3390/c9010020
APA StyleMollaamin, F., & Monajjemi, M. (2023). Doping of Graphene Nanostructure with Iron, Nickel and Zinc as Selective Detector for the Toxic Gas Removal: A Density Functional Theory Study. C, 9(1), 20. https://doi.org/10.3390/c9010020