3.1. Difference in Thermal Expansion Coefficient between Diamond and Catalyst as a Reason for Stresses Generated in Diamond
An evident and, in a sense, trivial cause of diamond cracking is a difference in the thermal expansion coefficient (TEC) between the diamond α
diam and the solid metallic catalyst α
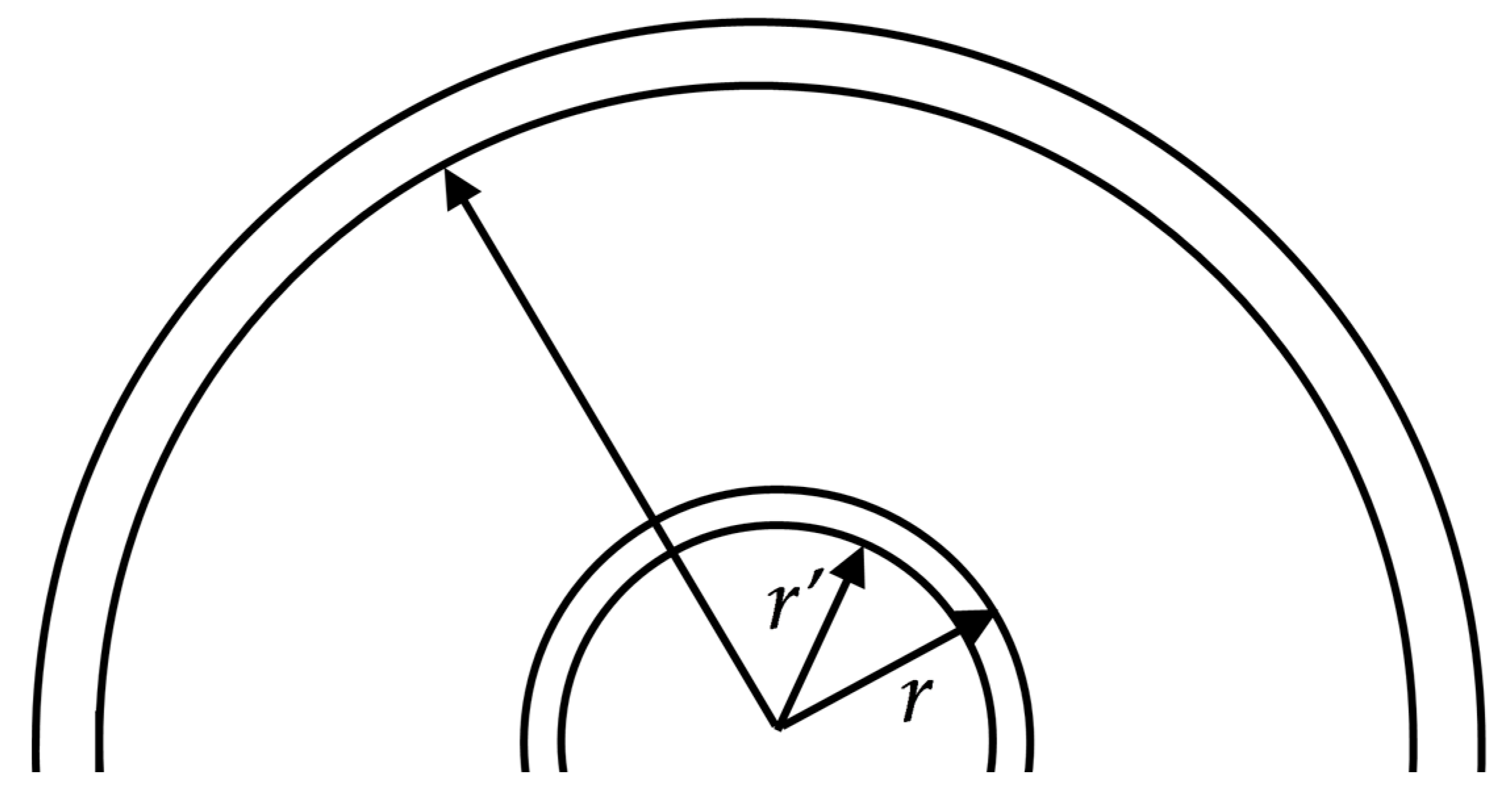
cat. It is easily accounted for in a simple model of a rigid spherical diamond core and a spherical catalyst shell enclosing it. In the absence of such a core, the cooled hollow-ball-shaped catalyst, initially without mechanical stresses and external pressure, reduces its inner radius from a value of
r (equal to the original diamond radius) to a smaller value
r′ =
r∙(1 − α
cat∙δT), where δT is the temperature decrease (
Figure 2).
As the rigid diamond core has a smaller TEC, this value is even smaller: , such that the stress tensor component σrr in the catalyst along the shell–diamond contact contour is approximately , where is the effective Young’s modulus of the catalyst. This takes into account the simultaneous tangential stretch of the catalytic shell and includes the Poisson factor.
By equating the radial component of the catalyst stress tensor σ
ρρ with a similar value for the diamond, we obtain
. Finding
r″ from here, one comes to the relationship
(The last equality in this relationship takes into account that E
diam = 1100 GPa >> E
cat = 200–300 GPa). Since the difference between the TEC of the diamond and of the catalyst has the order of the TECs themselves, i.e., 10
−6, the radial stress should not exceed 1–10 MPa if the temperature decreases even by 10
3 K. The same applies to the tangential stress. Therefore, the difference between the TECs of the diamond and of the shell can hardly be considered a probable cause of the cracking of the diamonds and other causes should be analyzed.
3.2. Basic Concepts of Plasticity Theory in the Model under Consideration
Based on the loss of mass of the graphite substance, which is the carbon source in diamond growth, it is easy to estimate the carbon content in the substance initially forming a pure metallic catalyst. It is not less than 5–10%. Therefore, we conventionally correlate this catalyst with cast iron, especially since it completely loses its catalytic role at the stage of reducing the parameters p and T.
The diamond growth conditions (the pressure of 5.5 GPa and the temperature of 1750–1800 K) are close to the liquid–solid-phase equilibrium curve for iron and transition metals of the iron group (nickel–cobalt, following the book [
14]). Because of this circumstance and the fact that the composition and morphology of the catalyst are not reliably known, we consider the initial point of the descent trajectory in the
p,T diagram as a solid phase of the catalyst and compare it with the phase diagram in the
p,T axes constructed for pure iron.
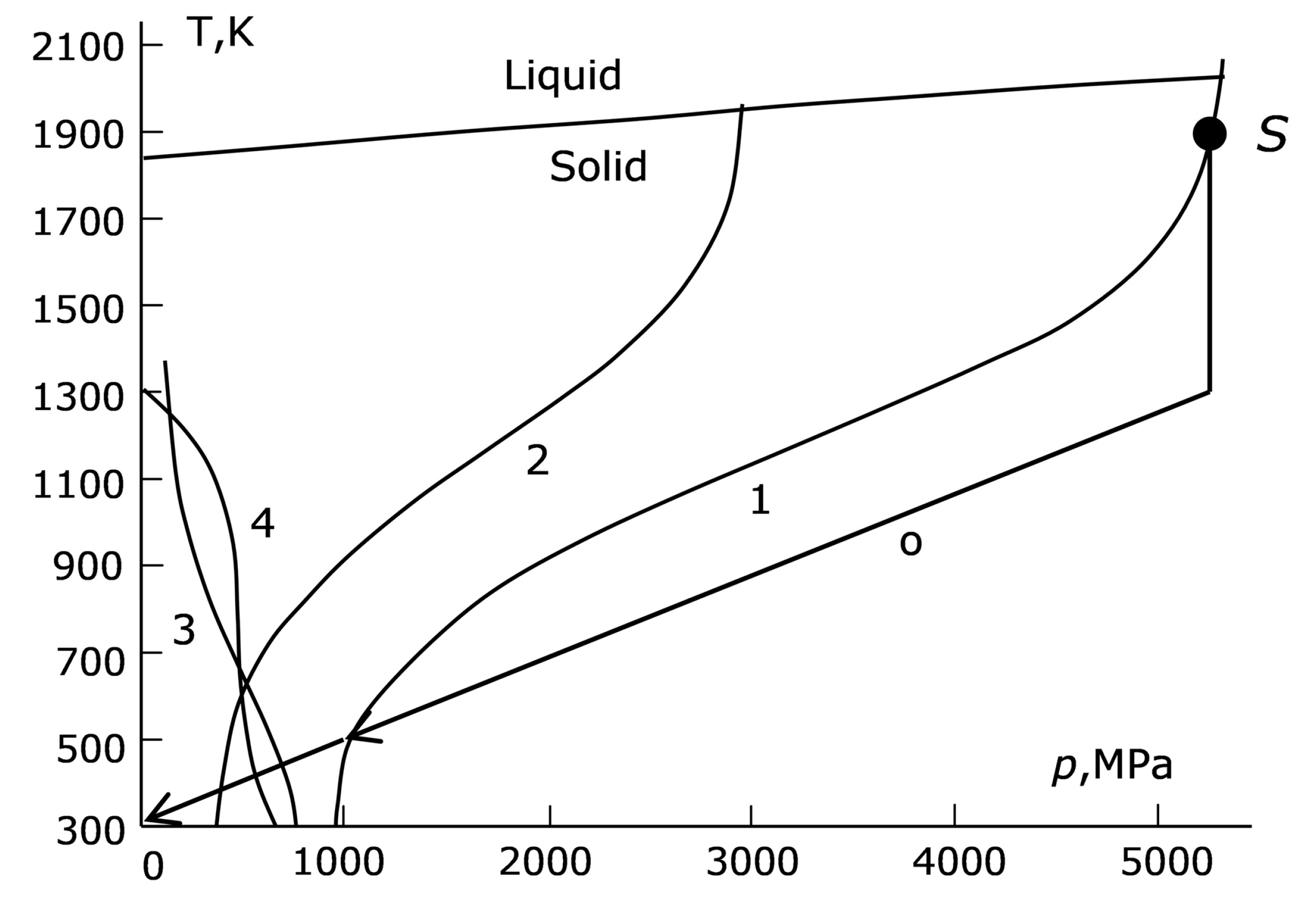
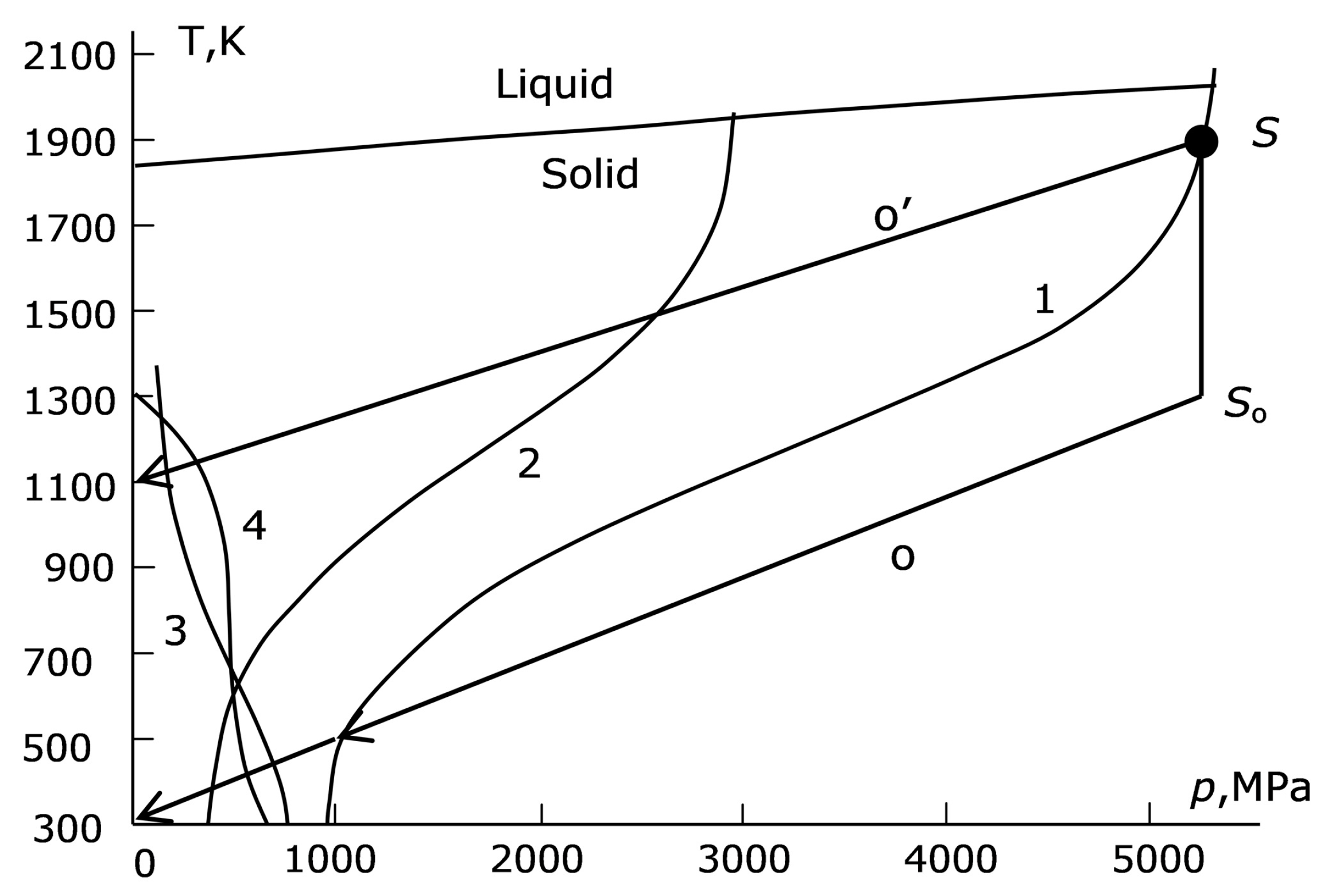
As the pressure and the temperature decrease from the diamond growth conditions (the bold black dot S in the upper right corner of
Figure 3) to the normal conditions, we enter the region corresponding to a strongly plastic state. To evaluate this region, we can proceed from S.N. Zhurkov’s theory determining the effective time τ
relax of plastic deformation or plastic fracture, or crack formation [
15], as
where σ is some generalized stress in the system; (
U0, γ) is a pair of parameters corresponding to one of the above processes; and γ is called the activation volume. When describing plastic deformation, it is considered that the energy parameter
U0 is close to 2/3 of the vaporization energy, and “bare” time τ
0 is close to the inverse frequency of atomic vibrations in the lattice, i.e., 10
−13 s.
The activation barrier
in (1) is proportional to the stress
. At the Zhurkov stress limit
, this barrier turns to zero. If the value σ
Zh is identified with the plasticity onset limit of the catalyst material σ
T(
p,T) (considered to be known), one can draw a curve on the (
p,T) plane (curve 1,
Figure 3). To the right of this curve, the plastic state is established instantly, and the material is as plastic as it can be. Let us discuss the values of σ
Zh and σ
T which can be used when drawing curve 1.
The value of σ
Zh is known with sufficient accuracy only for a small number of metals. So, in [
15], the values of
U0 = 3.8, 4.4, 3.6, and 5.7 eV are suggested for Ni, Fe, Cu, and Pt, respectively. The values of γ for Ni, Cu, and Pt are 0.7, 1.6, and 5.2 nm
3, such that σ
Zh is 851.8, 354, and 1726 MPa for these metals, respectively. From indirect data for iron in the same paper, σ
Zh = 865–880 MPa.
The initial information on the dependence σ
T(
p,T), essential for drawing curve 1 in
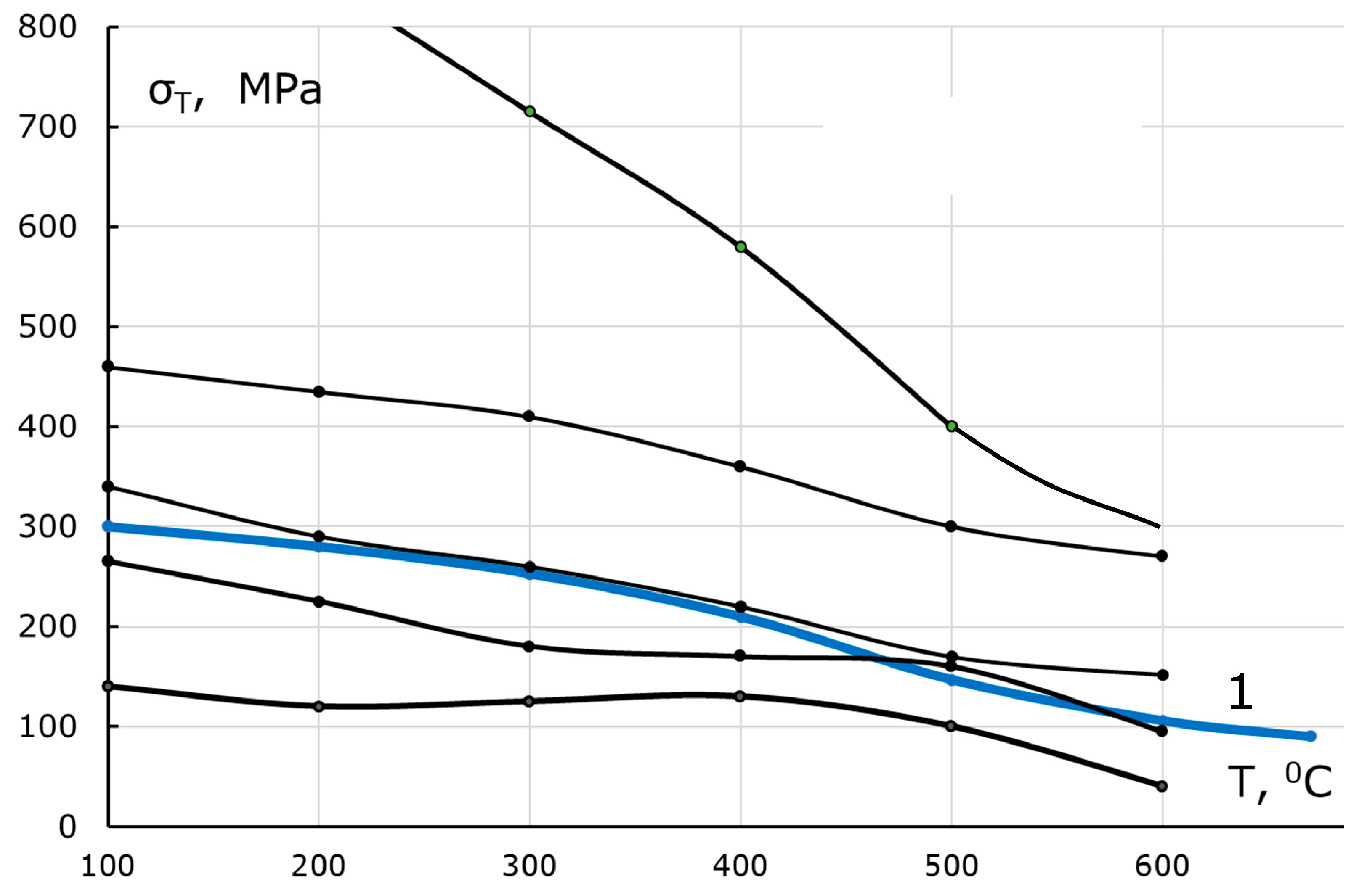
Figure 3, is rather scarce, though the parameter σ
T(T) at normal pressure is a quite standard characteristic both for cast irons and steels (
Figure 4).
The dependences σ
T(T) in
Figure 4 monotonically decrease. As a rule, they are known at temperatures up to 1000 K, which are practically interesting for structural materials. Focusing on obtaining qualitative conclusions and considering that the dependence σ
T(T) tends toward some constant value σ
T (
p =
pn = 1 atm, T
∞) at temperatures up to 2000 K, we assume that
where T
∞ is a conditional “infinitely high” temperature.
Formula (2) suggests that the plasticity onset limit decreases with the temperature growth to decrease “e” times at T
∞. The fitting temperature T
1 is assumed to be T
1 = 980 K. As can be seen from a typical s-s diagram (strain ε-stress diagram,
Figure 5), this formula does not contain all necessary information. Simultaneously to decreasing σ
T with temperature, there is an increase in the “plasticity plateau” of the strain ε, such that the dependence of plasticity on temperature is ambiguous.
Unfortunately, the known diagrams of cast irons similar to those shown in
Figure 5 have been constructed only for tensile experiments with bars. It is difficult to predict on their basis the plastic medium behavior under the all-round compression of the growth cell complicated by rigid inclusions in the form of a diamond or a diamond group. It can be assumed then that the horizontal sections of the σ(ε) dependences are transformed into sloping ones (
Figure 5b).
Therefore, we assumed that at temperatures considerably higher than room temperature Tn, the plasticity onset limit σT is sufficiently representative of the same value in all-round compression.
Other available information on the dependence σ
T(
p,T) can be found in the dependence of σ
T (or of some proportional value such as activity or fracture limit) on pressure at normal temperature T
n. Such dependences have been plotted for some cast irons and steels in a number of works, information on which is collected in [
16].
These curves look like straight (or nearly straight) lines brought to a pressure not exceeding 1 GPa. Their expression at normal temperature has the form
where the quantities σ and ω are accepted equal to σ
T(
pn,T
n) = 300 MPa, ω
n = 0.6 at T = T
n, and ω
∞ = 0.6 at T = T
∞; the values of the both pressures and σ
T(
p,T
n) are expressed in MPa. The limit of 1 GPa up to which the dependences (3) are known is obviously insufficient. However, it is known that P.W. Bridgman’s investigations of the mechanical properties of metals, glasses, and minerals [
17,
18], carried out in the 1930s–1940s, reached external pressures up to 30 thousand atm. A sharp increase in the plasticity of the materials was noted. Yet, the same source [
16] suggests that all-round uniform pressure cannot increase the strength and the plasticity onset limit indefinitely. More specific data are not available.
On the other hand, the industrial hardening of steels in industry is conducted at pressures > 2.5 GPa. Ferrous iron, phosphor bronzes, beryllium, and marble are also hardened under such conditions. The pressure extrusion technology also uses pressures above 1 GPa and there is no information to suggest that after this processing the materials are easier to destroy.
The possibility of reducing plastic properties at ultrahigh pressures could be related to approaching the critical point (
pcrit, T
crit) of the melt-to-solid metal phase transition. The problem of whether such a point exists, above which the melt could not be hardened at any pressure (by analogy with liquid–vapor transitions), is relatively poorly investigated [
19,
20]. In the most recent work on this problem [
21], the estimated critical pressure was not lower than 10
5–10
6 atm for alkali metals and lay in the range of 10
6–10
7 atm for transition metals of the iron group. This pressure is connected with approaching the region of relativistic electronic statistics when the Fermi energy of metal is comparable by the order of magnitude with mc
2.
This pressure is much higher than the conditions of the conventional diamond anvil cell (DAS), not to mention the conditions of diamond synthesis.
Based on the above, the plasticity onset limit at “infinitely high temperature” (conventionally T = T
∞) was chosen by analogy with that at room temperature in (3):
The relationship between the values of σ
T at room temperature and at T
∞ was assumed by
Taken together, the relationship
was given by combining Formulas (3)–(5):
where the function E is introduced in (2).
3.3. Characteristic Curves of the Catalytic Metal on (p,T) Plane, along Which the Catalyst Properties Change Drastically
Let us now return to a qualitative discussion of the dependence
i.e., of curve 1 (
Figure 3). Note that the intersection of this curve with the liquid–solid interface curve at a pressure close to 5.5 GPa, i.e., at the diamond growth operating pressure in a particular technology, is rather random. In general, the start of the trajectory of reducing the
p,T parameters does not necessarily lie at curve 1.
As mentioned above, the material is “superplastic” to the right of curve 1. To the left of curve 1, there is a region of activated plasticity, where the attainment of plasticity requires some finite time. If the plasticity properties are weakened on some trajectory of reducing the p,T parameters within this region, the excess stress over the plasticity onset limit at an earlier point in time may exceed the strength capabilities of the catalyst. In this case, the excess stress is transferred to the diamond, increasing the probability of its fracture.
To the left of the activated plasticity region, there is the elastic region. Its natural right boundary can be considered as the linearity limit σLin on the tensile stress curve. Given the lack of information, this limit was set in two ways.
The first way assumes a constancy of the ratio σ
Lin/σ
T (e.g., σ
Lin/σ
T = 0.6). For many cast irons, this assumption corresponds to reality and allows us to draw an analogue of curve 1 (
Figure 3) with the substitution of σ
Zh with 0.6σ
Zh (curve 2,
Figure 3):
Another way of setting the elastic region boundary assumes that in the elastic section of the s-s curve, the stress varies in proportion to the pressure and does not differ too much from it at the end of this section (curve 3 in
Figure 3):
In addition to curves 2 and 3, the region of strongly activated plasticity was evaluated in yet another way. The parameters
p,T were determined for the point at which the plasticity delay time τ
relax in Formula (1) for the given dependence σ
T(
p,T) reaches a preset value, e.g., one second (curve 4,
Figure 3).
Curves 2 and 3 form a “gorge” with curve 1 in the region of low temperature and of comparatively low pressure (
Figure 3), the crossing of this “gorge” being inevitable when the parameters
p,T decrease. As the plastic properties of the catalyst reduce when approaching the linear region on the left edge of the “gorge”, the conditions here can be considered as extreme ones from the viewpoint of diamond preservation.
Until the “gorge” is reached, the optimal reduction trajectory should leave curve 1 to the left and top of itself. In this case, the catalyst instantly reaches the plastic state at each time moment and the diamond is subjected to the lowest probability of fracture.
So, the condition of not entering the activated plasticity region looks the simplest for the optimal trajectory of reducing the (p,T) parameters, but this condition may contradict other desirable conditions for diamond preservation. It is therefore necessary to understand which trajectories within this region expose the diamond to the greatest danger.
As it is most correct to compare the stress value not with the plasticity onset limit σ
T but with the fracture limit when assessing this danger, we considered that the fracture limit is related to σ
T by an unknown proportional multiplier. As a parameter characterizing the probability of the diamond fracture, we used initially the ratio
of the instantaneous value of pressure and the plasticity onset limit at the earlier time
t − τ
relax, where θ(
t) is the Heaviside theta function.
The introduction of the parameter
D partially compensates for the paucity of information on the plastic properties of the catalytic material under complex deformation and allows one not to construct a numerical field of deformations and stresses for each point of the trajectory (
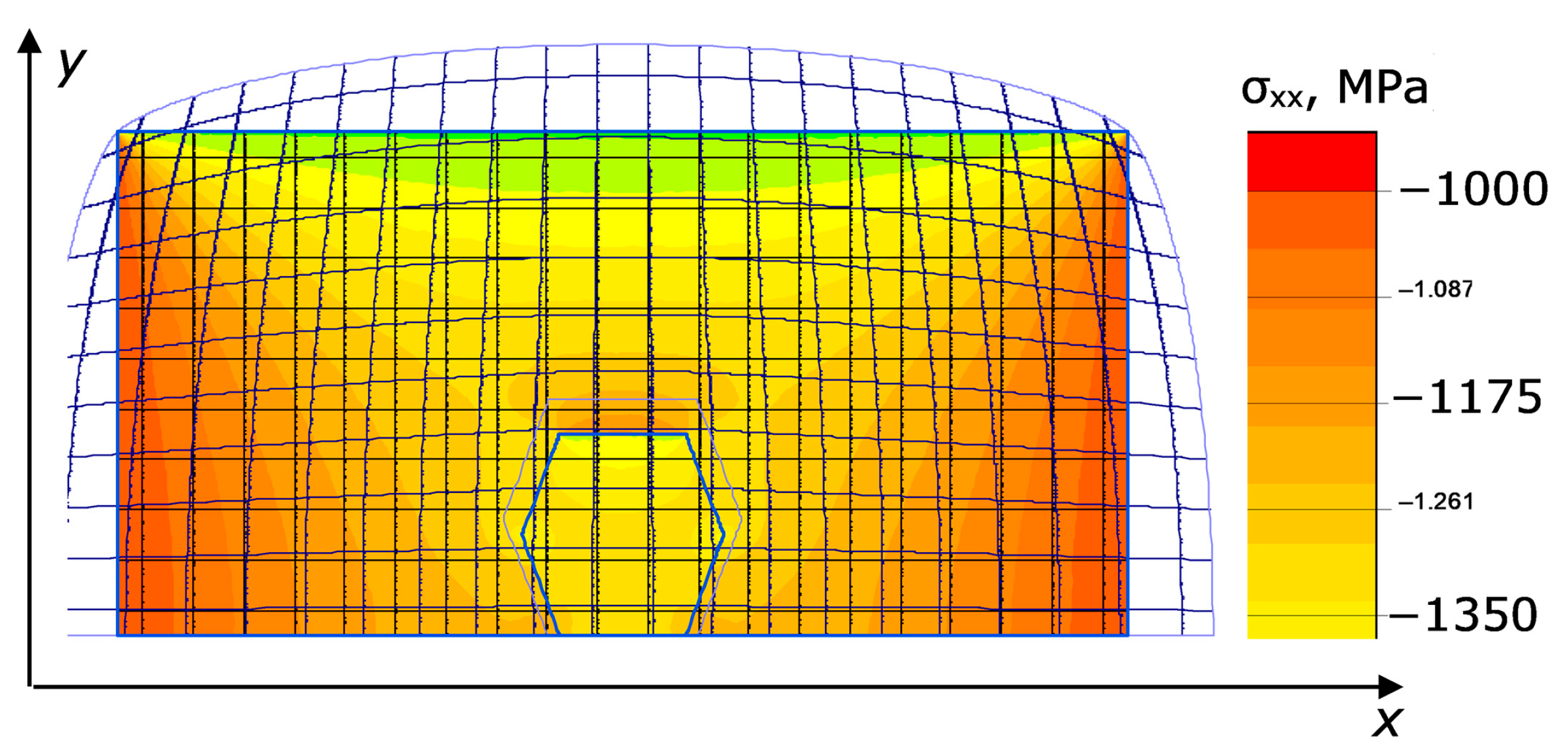
p,T) (such a field always describes an already established plasticity). An example of calculation of the stress field σ
xx in ANSYS 13 software (Static Structure) [
22] in the 2D simulation framework is shown in
Figure 6. The catalytic medium in the cell at an applied pressure of 1 GPa was assumed to be close to a perfectly plastic medium [
23].
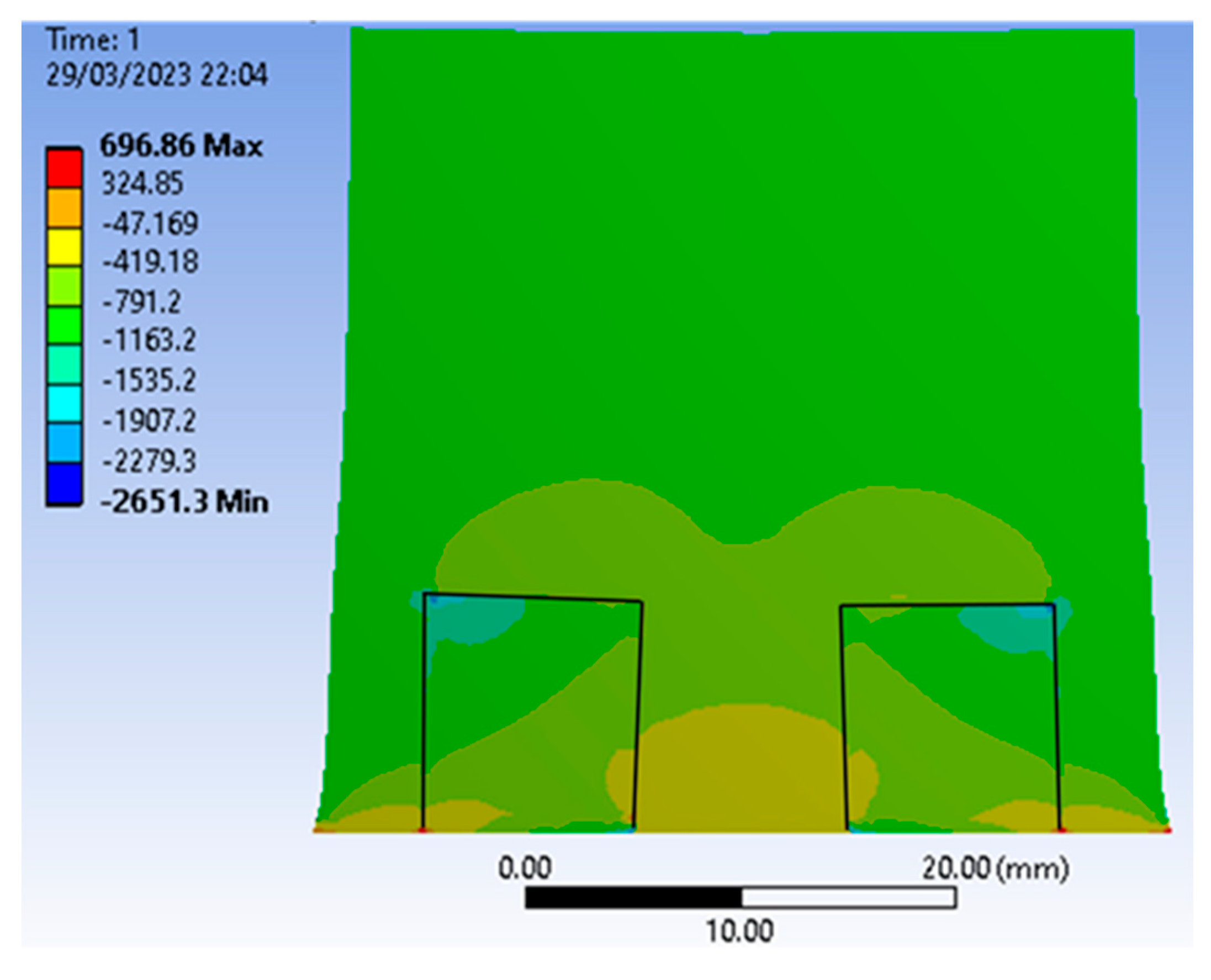
It is clearly seen that the stress level is notably reduced in the area between the diamonds, partially shielded from external pressure. On the contrary, inside the diamonds the stress increases on the sides facing the sides of the container. In general, this stress is distributed rather unevenly across the diamond.
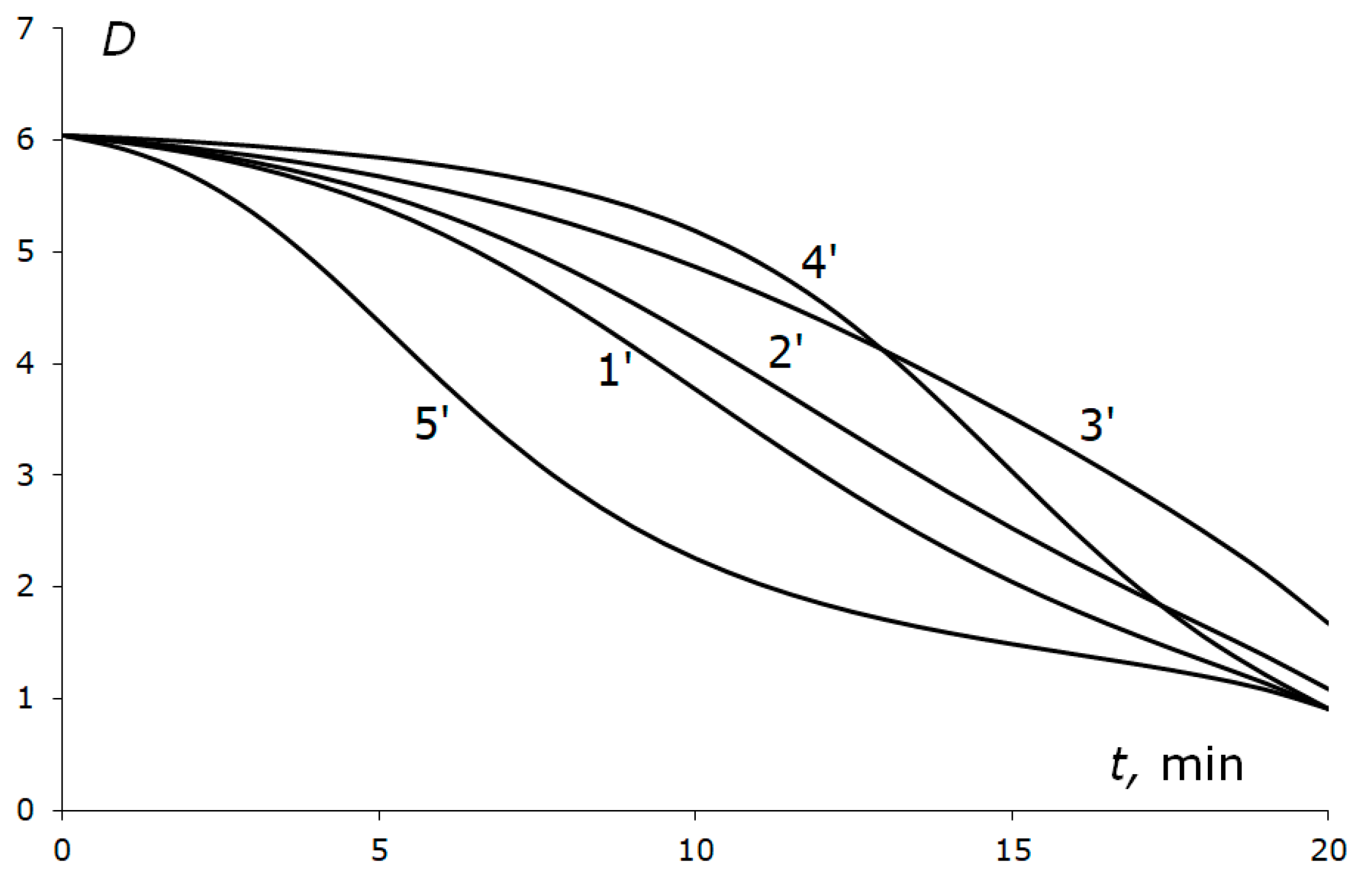
The variations in the parameter
D(
t) for several (
p,T) reduction trajectories on the
p,T plane from the starting point S on curve 1 (
Figure 3) until reaching the elastic region of the catalyst were calculated for the same reduction time of 20 min. The reduction trajectories and the variation in parameter
D are shown in
Figure 7 and
Figure 8, respectively. For curves 1′–3′ in
Figure 7, the pressure reduces uniformly over time from the starting value of
ps = 5250 MPa to
pf = 514 MPa, which approximately corresponds to the elastic region boundary (curve 3,
Figure 3).
The starting temperature T
s was 1888.2 K and the final temperature varied: T
f = 516.5 K for curve 1′, 700 K for 2′, and 1000 K for 3′. On these three curves, the temperature reduced uniformly over time, like pressure. On curve 5′, the temperature reduced to T
f = 516.5 K, initially with acceleration with respect to
p, then slowed down. On curve 4′, it was vice versa. The curves of
Figure 3 are shown as well in
Figure 5 as dashed curves with the same numbers as in
Figure 3.
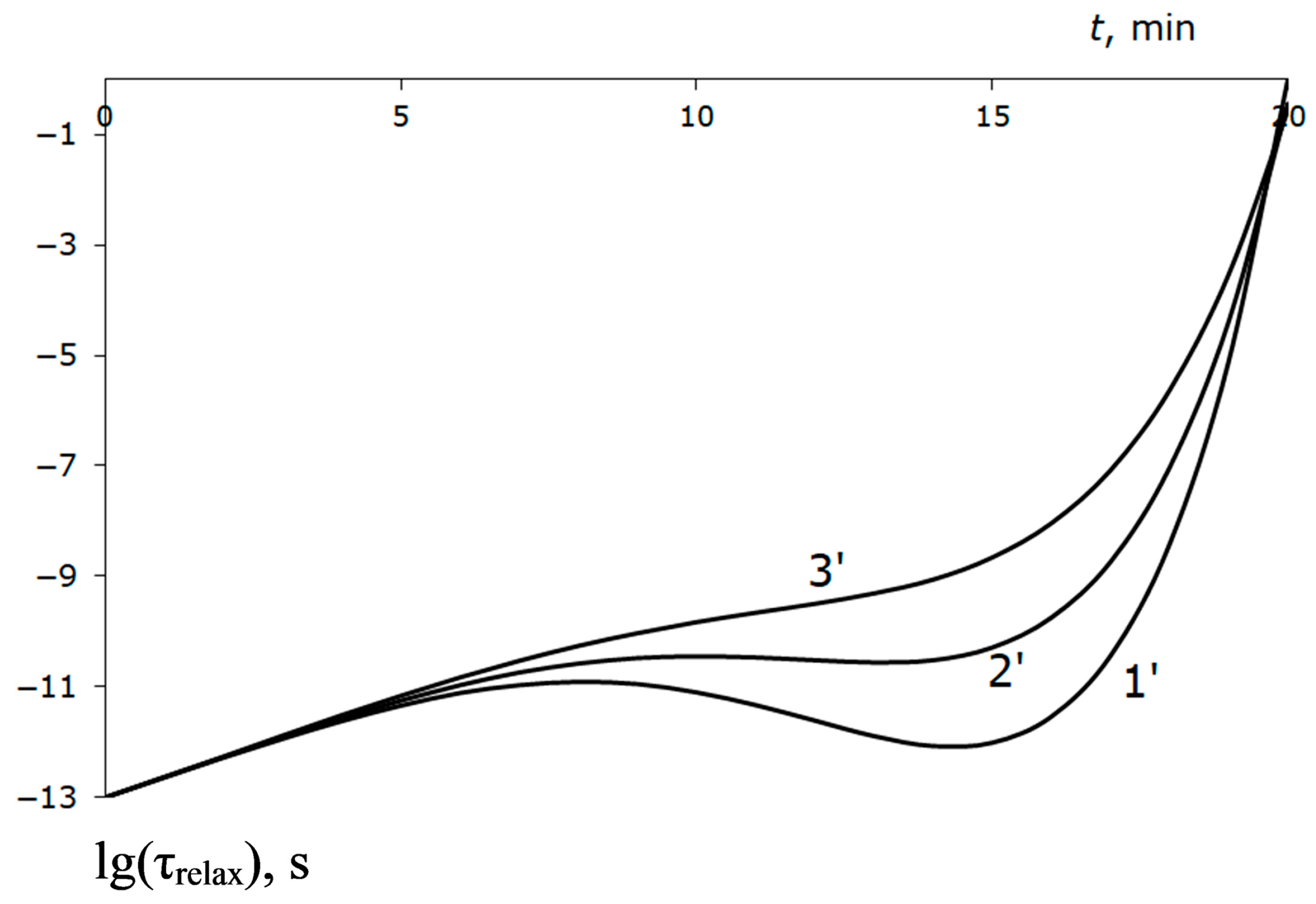
Figure 9 plots the relaxation time change for curves 1′, 2′, and 3′ (
Figure 7). It can be seen that almost to the elastic region boundary, the delay in the development of the plastic state is small. It becomes significant only at the end of the trajectory, when the absolute value of external pressure reduces by 5–6 times.
The variation in static plastic properties is more important. The most dangerous trajectories are the trajectories of slow temperature descent (curves 3′ and 4′ in
Figure 7 and
Figure 8), where the growth of the plastic properties due to the temperature decrease does not manage to compensate for the reduction of these properties due to the pressure reduction.
The parameter
D is maximal at the beginning of the trajectories (i.e., in early moments of time) and it is in this area that the pressure must first be reduced slowly (curve 5′,
Figure 7 and
Figure 8) and the temperature must be reduced much more rapidly. However, the area approaching the elastic region border can also be dangerous.
First of all, the position of curves 2, 3, and 4 in
Figure 3 is known very approximately. In addition, the delay in the plastic state onset can be much higher if the pre-exponent factor τ
0 of the relaxation time is longer than the approximate value of 10
−13 s. Finally, if curves 1′–5′ in
Figure 8 are reconstructed as ratios
D’ of the pressure to the fracture limit σ
fract (instead of plasticity onset limit), they will quantitatively coincide with curve
D only if the plasticity onset limit σ
T and fracture limit σ
fract change proportionally.
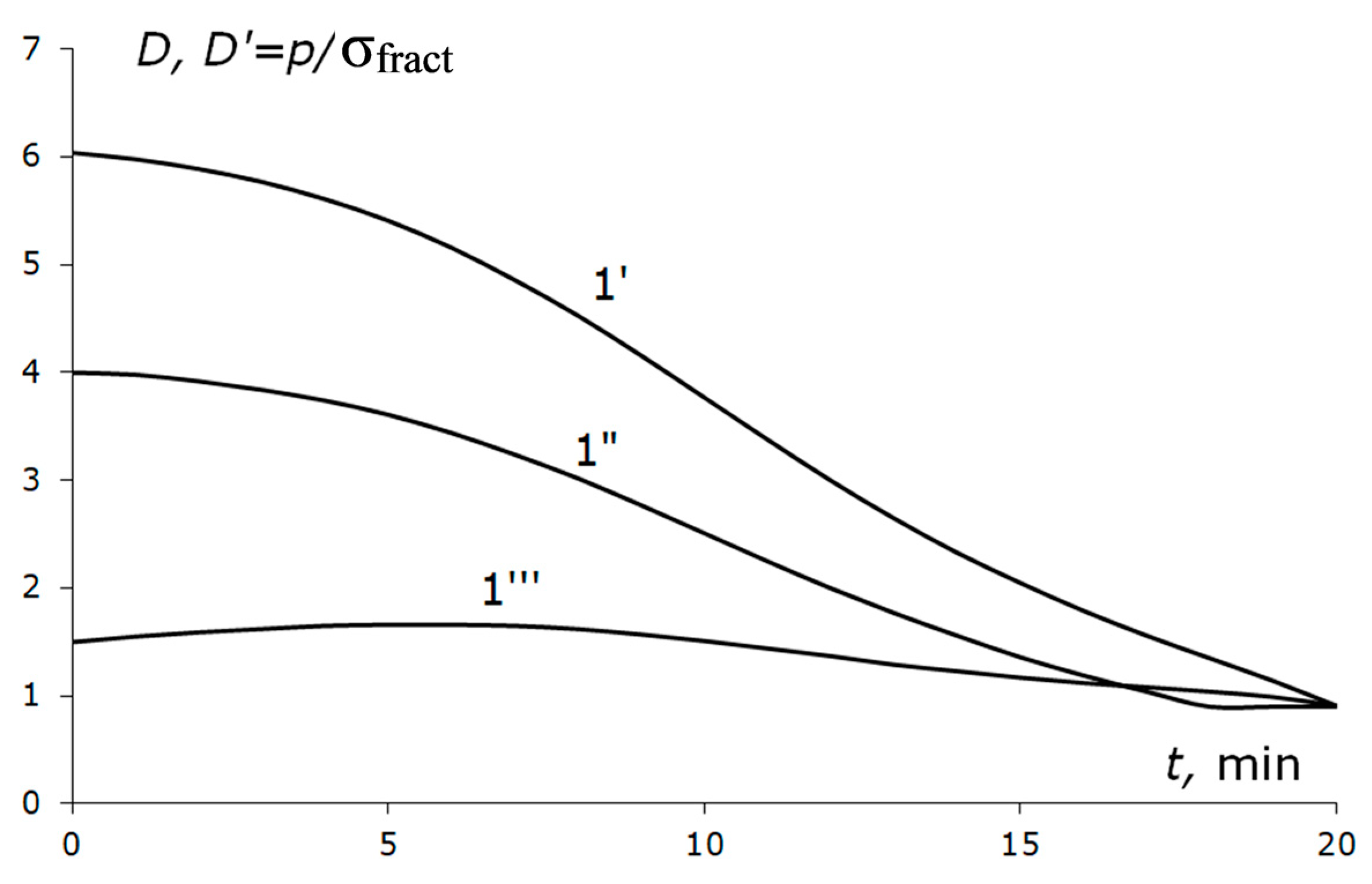
If the fracture limit decreases faster than the plasticity onset limit (as in
Figure 5b) with the temperature decrease, the shape of curve
D′(
t) is much more gentle compared to curve
D (curve 1′′′,
Figure 10). Then, the risk of fracturing the solid catalytic medium is maintained throughout the cooling and pressure descent trajectory. We return to the question of how fitting the parameter σ
fract/σ
T allows us to describe the main body of experimental data in
Section 4.
In practice, it is desirable to significantly decrease the reduction rate of both the pressure and the temperature at pressures in the range of p = 0.6–1 GPa, where the delay in the onset of plasticity becomes large (in the order of minutes). For example, one can take one hour to go through this range.
At lower pressure, the system enters the elastic behavior region of the catalyst. As the next section suggests, only unsteady temperature gradients can influence the stress level in this region. Let us estimate the possible temperature gradients associated with a sharp decrease in the average temperature in the growth cell.
3.4. Unsteady Temperature Gradients and Elastic and Weakly Plastic Behavior of Catalytic Medium
As shown in this section, unsteady temperature gradients can be made small by choosing a reasonable temperature reduction rate. Therefore, we have not conducted a detailed analysis of the temperature distribution in the growth cell. As far as we know, analysis of this sort for different designed anvils with self-consistent consideration of the position of the heating circuit and insulating gaskets was first carried out in [
24]. For our particular problem, we either limited ourselves to a constant temperature in the growth cell (as above) or set a constant temperature gradient down and up the cell.
The elements of the electric circuit for heating the growth cell are a graphite heater and anvils (not shown in
Figure 1), and the elements of the thermal problem are the insulating MgO gasket and the growth cell itself. The graphite heater can be immediately excluded from the consideration of the thermal problem, as well as the current-carrying circuit through the anvils of enormous cross-section. Then, the power supply to the pyrophyllite container is allocated almost entirely in the growth cell and the MgO gasket, the MgO being thin and having lower thermal conductivity than the growth cell. Therefore, the delay time τ
delay in establishing a steady-state temperature distribution in the cell is determined by the heat capacity and thermal conductivity of the catalyst alone. By order of magnitude
where χ
cat is the temperature conductivity of the catalyst medium. The unsteady temperature difference between the center of the cell and the heat deposition point (graphite heater at a periphery of the cell) is
The thermal conductivity , the density , and the heat capacity of the iron catalyst under normal conditions are 0.50 W/(cm∙K), 7.5 g/m3, and 0.9 J/(g∙K), respectively; the half side rcell of the 2 rcell side was assumed to be rcell = 3 cm. Taking into account the considerable variation in these parameters depending on p and T, τdelay~1 min and δT~50 K at uniform temperature reduction from 2000 to 500 K in 40 min. This reduction rate approximately corresponds to the steady-state bottom-top temperature difference in the cell during the diamond growth. At a lower temperature reduction rate of 10 K/min (1000 K for ~1.5 h), the unsteady temperature effects are unimportant and cannot cause significant stresses in the diamond.
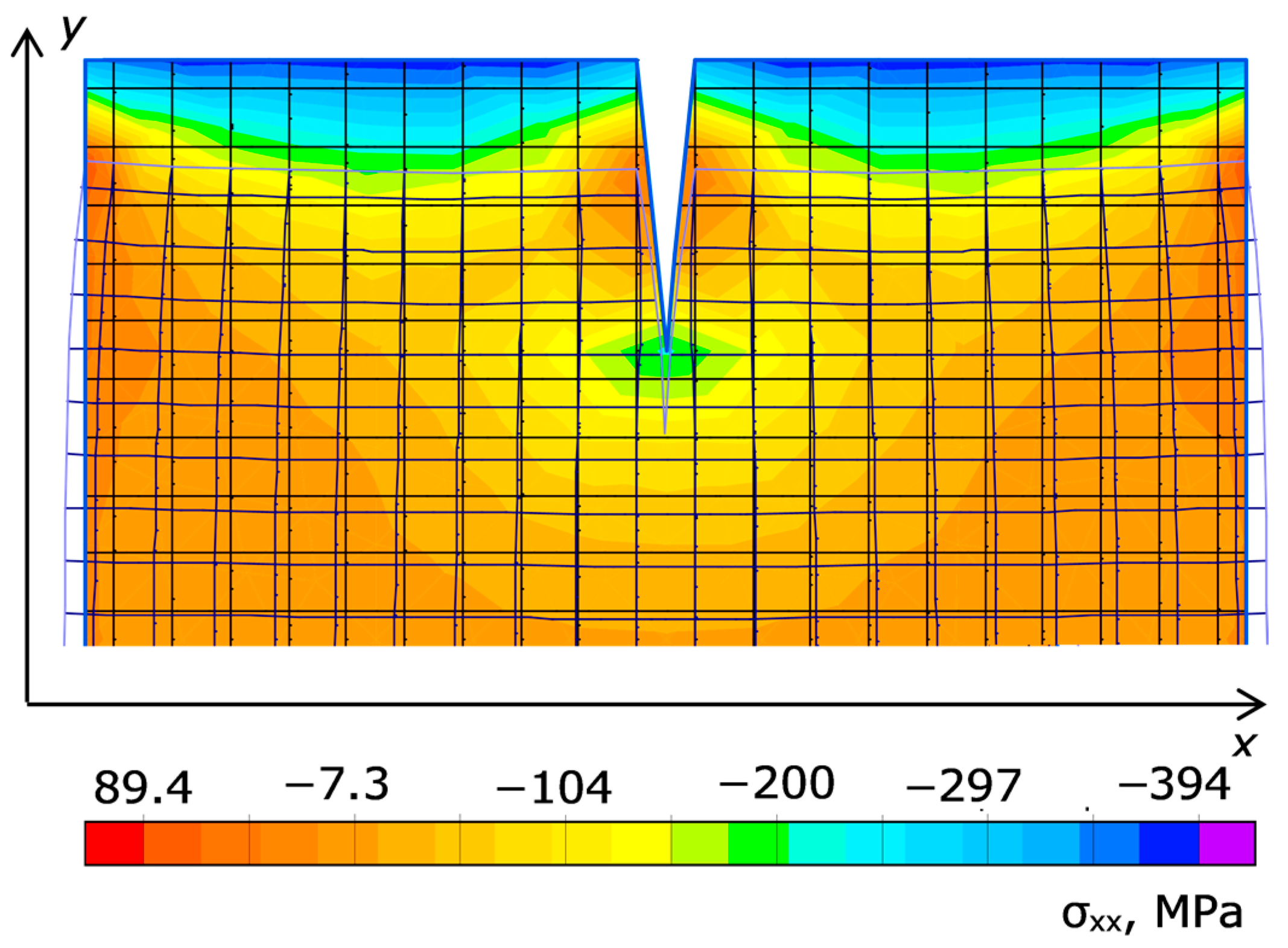
Figure 11 shows the stress fields σ
xx in the diamond and in the catalyst due to the difference in Young’s moduli and TECs in the elastic problem (ELCUT 5.2 package [
25]) at a temperature difference of about 50 K across the cell height. With the 2D simulation, the external pressure was 1 GPa, and the temperature varied from the bottom to the top as 330 + 2.5y (K) up to a height of y = 20 mm. It can be seen that the stress is very slightly higher than the external pressure, although it is accentuated on the diamond first of all.
3.5. Influence of Plastic Properties of the Container and Limitations on the Descent Trajectory of the p,T Parameters Associated with It
Thus, neither the “delayed plasticity” nor the temperature gradients should result in extreme stresses on the diamond at the “dangerous” region below 1 GPa and cause its fracture during the relatively slow reduction in the p,T parameters.
However, the above consideration does not address the strength and plastic properties of the pyrophyllite container enclosing the growth cell. The integrity of the container is practically irrelevant as long as the catalyst material retains its plasticity. This is true up to pressures of ~1 GPa, as shown in
Figure 3. However, in the elastic region of the catalyst, it completely translates superstresses arising in the container.
If the container loses its plasticity and has “pre-acquired” cracks at pressures above 0.7–1 GPa, the loads translated to the diamond can exceed this value many times over.
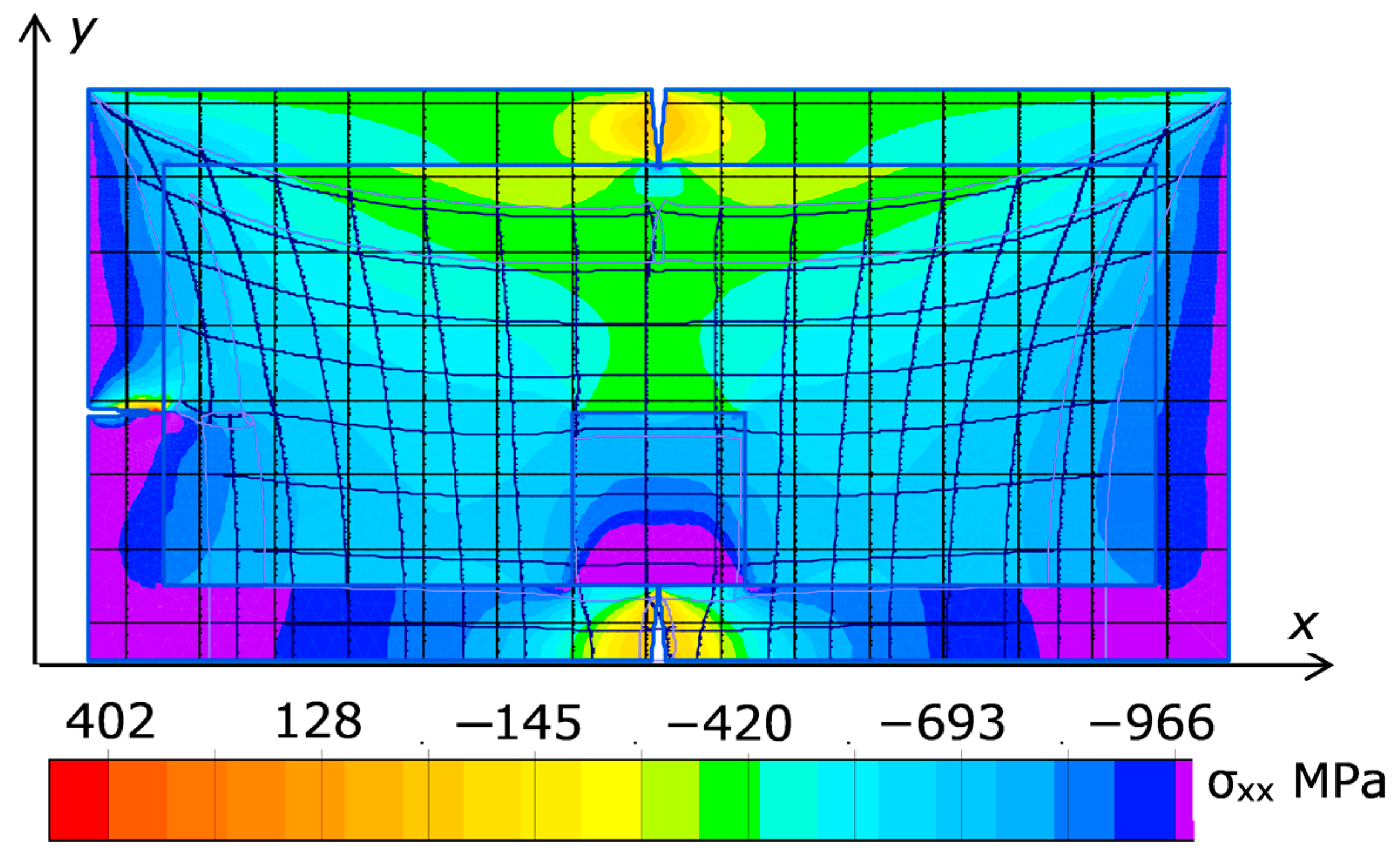
A typical picture of the stress field σ
xx created in an elastic material by a 5 mm-deep and 3 mm-wide triangular crack at the top of the material is shown in
Figure 12.
It can be seen that the maximum rupturing stress occurs at the bottom of the crack and effectively extends deep into the catalyst at least to the depth of the crack. The effective width of the stress exceeds the transverse crack width by about an order of magnitude.
Figure 13 plots the stress field σ
xx initiated in the cell by the three cracks in the pyrophyllite container—top, left, and bottom under the diamond under an all-round load of 1 GPa. The Young’s modulus and Poisson’s ratio of the container are chosen rather arbitrarily as 300 GPa and 0.25, respectively. The dimensions of the diamond and the growth cell are 7 × 7 mm and 40 × 20 mm, respectively; the thickness of the container is 3 mm.
It can be seen that the maximum rupturing stress acting on the diamond is concentrated in its lower part, immediately above the crack (dark blue color).
Let us set a target to minimize the probability of cracks in the container during the reduction of the extreme parameters p,T, assuming that no cracks have yet occurred in the container at the growth times of the diamond.
Since almost nothing is known about the strength properties of pyrophyllite, wholly dehydrated and turned into stone after the diamond growth period is completed, the most stringent requirement for reducing the p and T parameters was as follows: the volume of the pyrophyllite container should not change. Thus, the reduction in the catalyst volume inside the pyrophyllite automatically leads to lower pressure on it from inside. Accordingly, in the parameter dynamics optimal for pyrophyllite container integrity, the external pressure should decrease at the same rate. Then, the external and internal pressures on the container remain approximately equal and the probability of cracking is minimal.
As follows from conventional thermodynamics, the decrease in the internal pressure pint due to the temperature reduction is approximately dpint/dT ≈ (∂p/∂T)V = −(∂p/∂V)T(∂V/∂T)p = 3αcat/ζ, where ζ is the isothermal compressibility of the catalyst material. For solid iron under normal conditions, αcat = 12 × 10−6, ζ = 0.6 × 10−6 1/atm. That gives dpint/dT~50 atm/K for the internal pressure reduction rate, i.e., about 105 atm for the total temperature reduction of ~2000 K during the entire reduction time of the p,T parameters. This value is only twice the ratio of the total drops in the pressure and temperature over the reduction period.
Therefore, the reduction trajectory optimal for the pyrophyllite container preservation lies almost entirely in the activated plasticity region (trajectory o′ in
Figure 14) and has a small slope. Herewith the load on the catalyst is maximum. Reaching the catalyst-safe trajectory requires passing through the initial cooling section without the pressure reduction (S-S
o section), when the risk of the pyrophyllite container failure is maximal, at least from the point in time where the curve o′ in
Figure 14 intersects curve 2.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
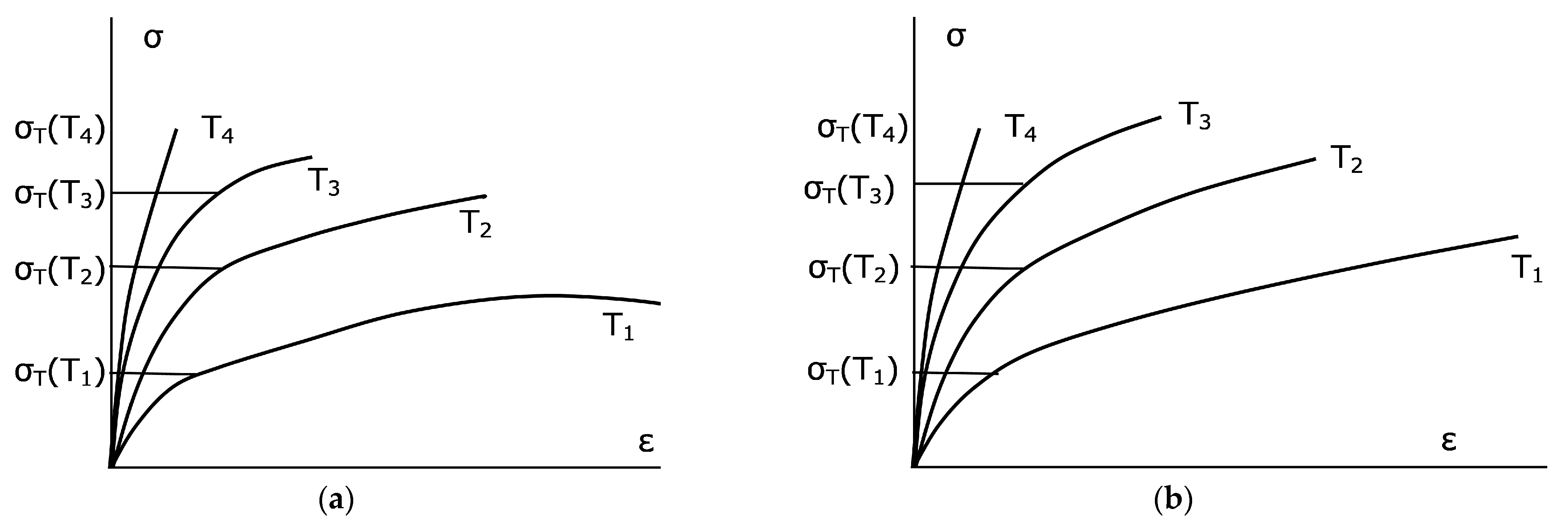{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














