
Cofactor Self-Sufficient Whole-Cell Biocatalysts for the Relay-Race Synthesis of Shikimic Acid

 ,
,
Abstract
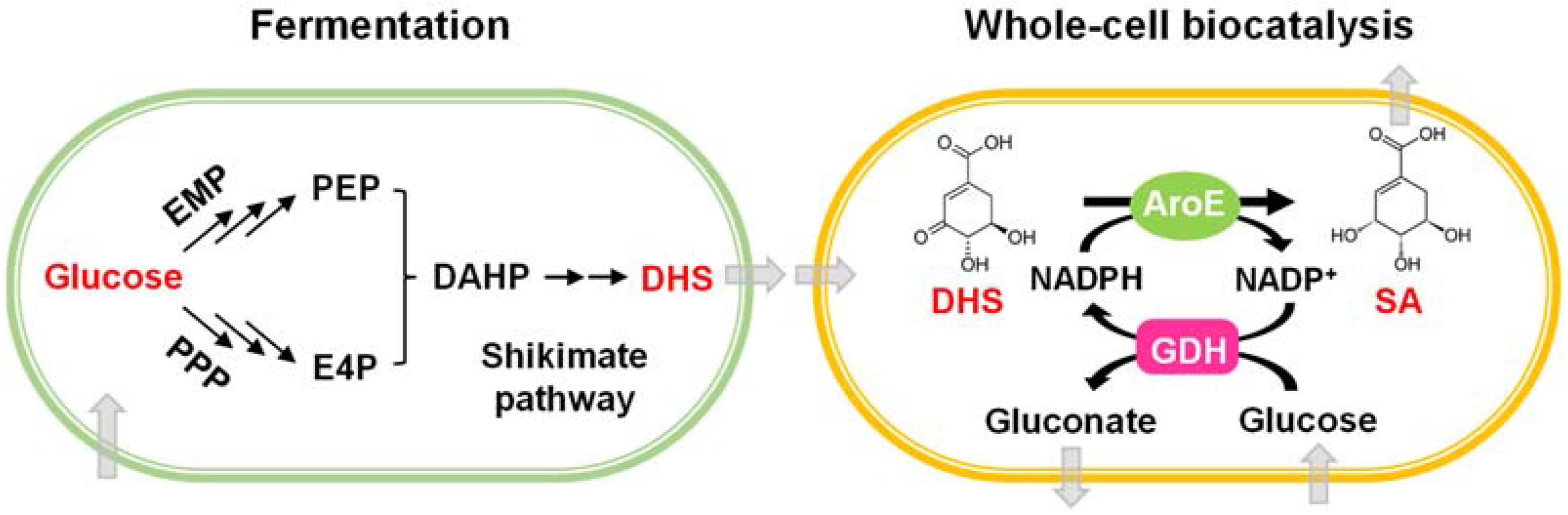
:1. Introduction
2. Materials and Methods
2.1. Strains and Plasmids
2.2. Plasmid Construction
2.3. Deletion of Chromosomal serA
2.4. Media and Growth Conditions
2.5. Enzyme Assays of GDH and AroE
2.6. Whole-Cell Biocatalytic Conditions
2.7. Determination of DHS, SA, and Glycerol Concentrations in Fermentative Broth
3. Results and Discussion
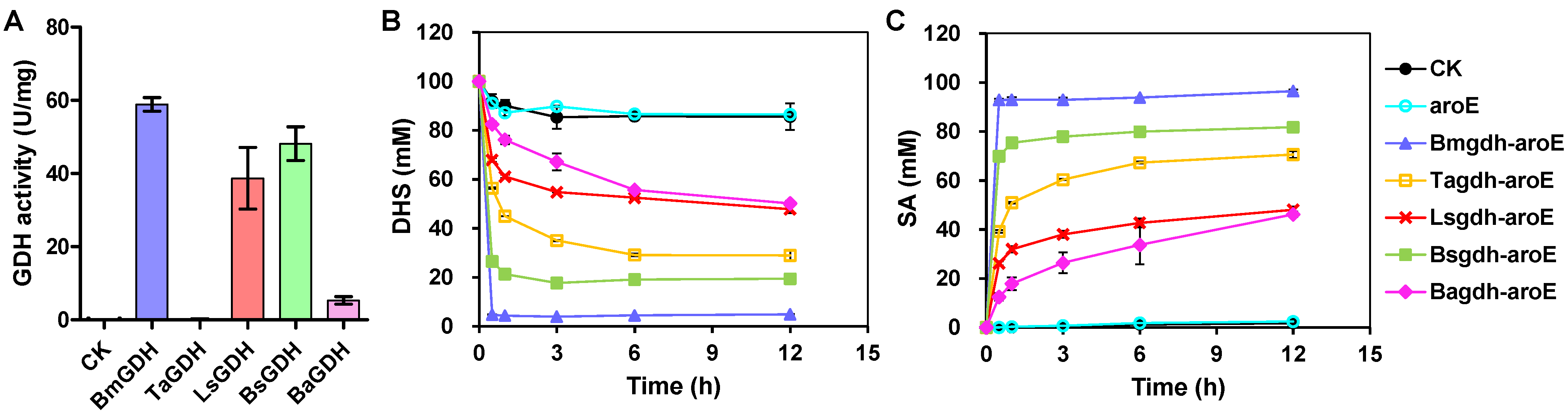
3.1. Screening GDHs to Construct NADPH Self-Sufficient Whole-Cell Biocatalysts
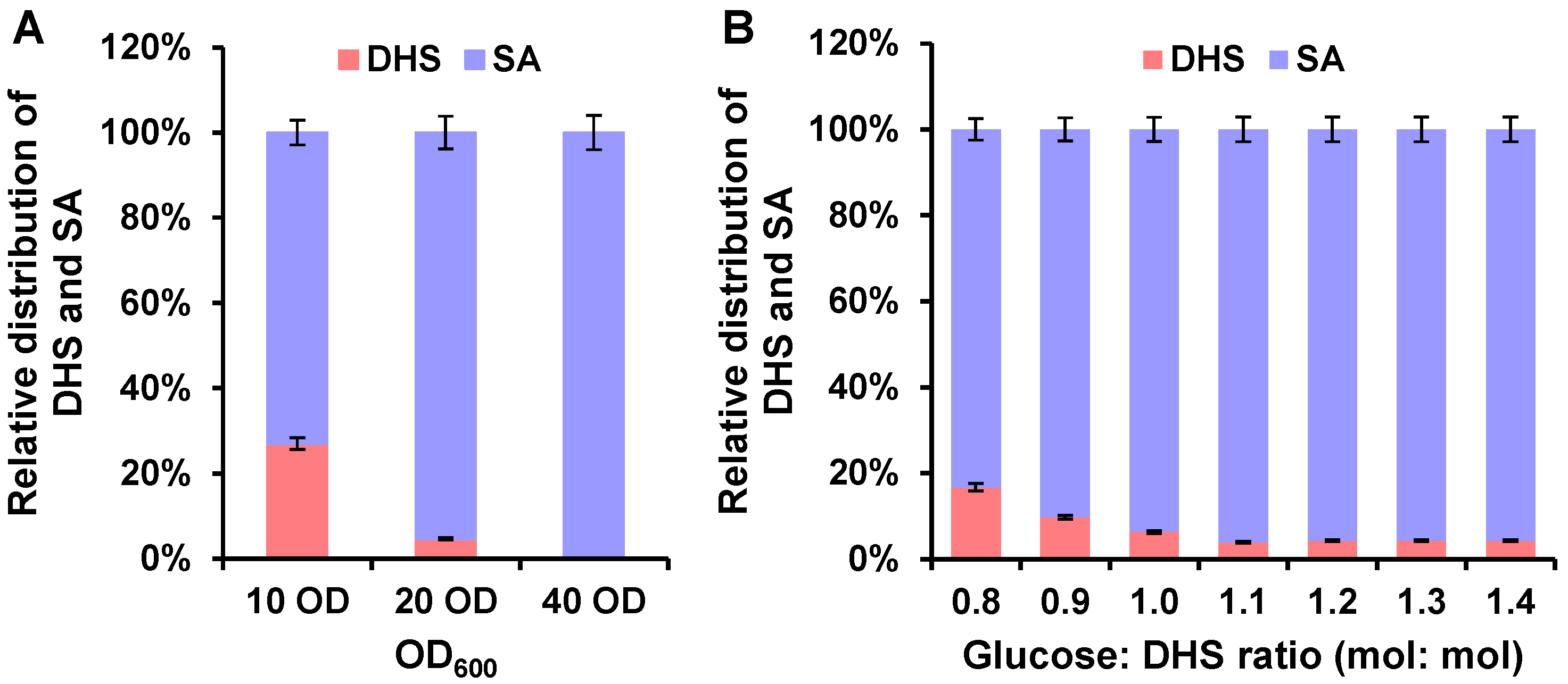
3.2. Optimizing Catalytic Conditions for Efficient Whole-Cell Biocatalysis
3.3. Effect of the Expression Order of Bmgdh and aroE on Whole-Cell Biocatalytic Efficiency
3.4. Optimizing Co-Expression System for Efficient Whole-Cell Biocatalysis
3.5. Optimizing Catalytic Conditions for Efficient Whole-Cell Biocatalysis on a Large Scale
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kogure, T.; Kubota, T.; Suda, M.; Hiraga, K.; Inui, M. Metabolic engineering of Corynebacterium glutamicum for shikimate overproduction by growth-arrested cell reaction. Metab. Eng. 2016, 38, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Candeias, N.R.; Assoah, B.; Simeonov, S.P. Production and synthetic modifications of shikimic acid. Chem. Rev. 2018, 118, 10458–10550. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, D.V.; Sysolyatin, S.V.; Kalashnikov, A.I.; Surmacheva, I.A. Shikimic acid: Review of its analytical, isolation, and purification techniques from plant and microbial sources. J. Chem. Biol. 2012, 5, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Zhang, H. Engineering the shikimate pathway for biosynthesis of molecules with pharmaceutical activities in E. coli. Curr. Opin. Biotechnol. 2016, 42, 1–6. [Google Scholar] [CrossRef]
- Shen, Y.P.; Niu, F.X.; Yan, Z.B.; Fong, L.S.; Huang, Y.B.; Liu, J.Z. Recent advances in metabolically engineered microorganisms for the production of aromatic chemicals derived from aromatic amino acids. Front. Bioeng. Biotechnol. 2020, 8, 407. [Google Scholar] [CrossRef]
- Wu, F.; Cao, P.; Song, G.; Chen, W.; Wang, Q. Expanding the repertoire of aromatic chemicals by microbial production. J. Chem. Technol. Biot. 2018, 93, 2804–2816. [Google Scholar] [CrossRef]
- Chandran, S.S.; Yi, J.; Draths, K.M.; von Daeniken, R.; Weber, W.; Frost, J.W. Phosphoenolpyruvate availability and the biosynthesis of shikimic acid. Biotechnol. Prog. 2003, 19, 808–814. [Google Scholar] [CrossRef]
- Rodriguez, A.; Martinez, J.A.; Baez-Viveros, J.L.; Flores, N.; Hernandez-Chavez, G.; Ramirez, O.T.; Gosset, G.; Bolivar, F. Constitutive expression of selected genes from the pentose phosphate and aromatic pathways increases the shikimic acid yield in high-glucose batch cultures of an Escherichia coli strain lacking PTS and pykF. Microb. Cell. Fact. 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, M.; Zhou, L.; Shen, W.; Algasan, G.; Fan, Y.; Wang, Z. Metabolic engineering of Escherichia coli for improving shikimate synthesis from glucose. Bioresour. Technol. 2014, 166, 64–71. [Google Scholar] [CrossRef]
- Gao, C.; Hou, J.; Xu, P.; Guo, L.; Chen, X.; Hu, G.; Ye, C.; Edwards, H.; Chen, J.; Chen, W.; et al. Programmable biomolecular switches for rewiring flux in Escherichia coli. Nat. Commun. 2019, 10, 3751. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.N.; Seo, S.Y.; Kim, H.J.; Park, J.H.; Park, E.; Choi, S.S.; Lee, S.J.; Kim, E.S. Artificial cell factory design for shikimate production in Escherichia coli. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab043. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Gao, M.; Suastegui, M.; Mei, Y.; Shao, Z. Building microbial factories for the production of aromatic amino acid pathway derivatives: From commodity chemicals to plant-sourced natural products. Metab. Eng. 2020, 58, 94–132. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.; Wang, S.; Iqbal, H.M.N.; Zhao, Y.; Hu, H.; Wang, W.; Zhang, X. Metabolic engineering strategies for enhanced shikimate biosynthesis: Current scenario and future developments. Appl. Microbiol. Biotechnol. 2018, 102, 7759–7773. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.A.; Bolivar, F.; Escalante, A. Shikimic acid production in Escherichia coli: From classical metabolic engineering strategies to omics applied to improve its production. Front. Bioeng. Biotechnol. 2015, 3, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowsett, J.R.; Corbett, J.R. The purification and properties of shikimate dehydrogenase. Biochem. J. 1971, 123, 23P. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, O.; Ano, Y.; Toyama, H.; Matsushita, K. High shikimate production from quinate with two enzymatic systems of acetic acid bacteria. Biosci. Biotechnol. Biochem. 2006, 70, 2579–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.; Li, M.; Liu, Y.; Zhu, M.; Zhao, G.; Zhou, Y.; Zhang, L.; Wu, Y.; Dai, X.; Xia, T.; et al. Functional analysis of 3-dehydroquinate dehydratase/shikimate dehydrogenases involved in shikimate pathway in Camellia sinensis. Front. Plant Sci. 2019, 10, 1268. [Google Scholar] [CrossRef]
- Cui, Y.Y.; Ling, C.; Zhang, Y.Y.; Huang, J.; Liu, J.Z. Production of shikimic acid from Escherichia coli through chemically inducible chromosomal evolution and cofactor metabolic engineering. Microb. Cell Fact. 2014, 13, 21. [Google Scholar] [CrossRef] [Green Version]
- Adachi, O.; Ano, Y.; Shinagawa, E.; Yakushi, T.; Matsushita, K. Conversion of quinate to 3-dehydroshikimate by Ca-alginate-immobilized membrane of Gluconobacter oxydans IFO 3244 and subsequent asymmetric reduction of 3-dehydroshikimate to shikimate by immobilized cytoplasmic NADP-shikimate dehydrogenase. Biosci. Biotechnol. Biochem. 2010, 74, 2438–2444. [Google Scholar] [CrossRef]
- Ghosh, S.; Pawar, H.; Pai, O.; Banerjee, U.C. Microbial transformation of quinic acid to shikimic acid by Bacillus megaterium. Bioresour. Bioprocess. 2014, 1, 7. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Hady, G.N.; Ikeda, T.; Ishida, T.; Funabashi, H.; Kuroda, A.; Hirota, R. Engineering cofactor specificity of a thermostable phosphite dehydrogenase for a highly efficient and robust NADPH regeneration system. Front. Bioeng. Biotechnol. 2021, 9, 647176. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaans, S.K.; Weusthuis, R.A.; van der Oost, J.; Kengen, S.W. NADPH-generating systems in bacteria and archaea. Front. Microbiol. 2015, 6, 742. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.M.; Qin, Z.; Li, F.L.; Zeng, B.B.; Zheng, G.W.; Xu, J.H. Development of an engineered ketoreductase with simultaneously improved thermostability and activity for making a bulky atorvastatin precursor. ACS Catal. 2019, 9, 147–153. [Google Scholar] [CrossRef]
- Rao, J.; Zhang, R.; Xu, G.; Li, L.; Xu, Y. Efficient production of (S)-1-phenyl-1,2-ethanediol using xylan as co-substrate by a coupled multi-enzyme Escherichia coli system. Microb. Cell Fact. 2020, 19, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Wang, Y.; Xu, J.; Chen, J.; Wang, L.; Qi, B. Enantioselective biosynthesis of L-phenyllactic acid by whole cells of recombinant Escherichia coli. Molecules 2017, 22, 1966. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.D.; Budgen, N.; Bungard, S.J.; Danson, M.J.; Hough, D.W. Purification and characterization of glucose dehydrogenase from the thermoacidophilic archaebacterium Thermoplasma acidophilum. Biochem. J. 1989, 261, 973–977. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.T.; Du, Y.Q.; Liu, D.F.; Li, Z.L.; Chen, X.J.; Zhao, Y.H. Cloning and expression in E. coli of an organic solvent-tolerant and alkali-resistant glucose 1-dehydrogenase from Lysinibacillus sphaericus G10. Bioresour. Technol. 2011, 102, 1528–1536. [Google Scholar] [CrossRef]
- Park, H.J.; Jung, J.; Choi, H.; Uhm, K.N.; Kim, H.K. Enantioselective bioconversion using Escherichia coli cells expressing Saccharomyces cerevisiae reductase and Bacillus subtilis glucose dehydrogenase. J. Microbiol. Biotechnol. 2010, 20, 1300–1306. [Google Scholar] [CrossRef] [Green Version]
- Borriss, R.; Chen, X.H.; Rueckert, C.; Blom, J.; Becker, A.; Baumgarth, B.; Fan, B.; Pukall, R.; Schumann, P.; Sproer, C. Relationship of Bacillus amyloliquefaciens clades associated with strains DSM 7T and FZB42T: A proposal for Bacillus amyloliquefaciens subsp. amyloliquefaciens subsp. nov. and Bacillus amyloliquefaciens subsp. plantarum subsp. nov. based on complete genome sequence comparisons. Int. J. Syst. Evol. Microbiol. 2011, 61, 1786–1801. [Google Scholar]
- Postma, P.W.; Lengeler, J.W.; Jacobson, G.R. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol. Rev. 1993, 57, 543–594. [Google Scholar] [CrossRef] [PubMed]
- Snoep, J.L.; Arfman, N.; Yomano, L.P.; Fliege, R.K.; Conway, T.; Ingram, L.O. Reconstruction of glucose uptake and phosphorylation in a glucose-negative mutant of Escherichia coli by using Zymomonas mobilis genes encoding the glucose facilitator protein and glucokinase. J. Bacteriol. 1994, 176, 2133–2135. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.H.; Kim, M.D.; Jin, Y.S.; Seo, J.H. Engineering of NADPH regenerators in Escherichia coli for enhanced biotransformation. Appl. Microbiol. Biotechnol. 2013, 97, 2761–2772. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Montalvo, V.; Martinez, A.; Hernandez-Chavez, G.; Bolivar, F.; Valle, F.; Gosset, G. Expression of galP and glk in a Escherichia coli PTS mutant restores glucose transport and increases glycolytic flux to fermentation products. Biotechnol. Bioeng. 2003, 83, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Tanaka, Y.; Hiraga, K.; Inui, M.; Yukawa, H. Characterization of shikimate dehydrogenase homologues of Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2013, 97, 8139–8149. [Google Scholar] [CrossRef]
- Muir, R.M.; Ibanez, A.M.; Uratsu, S.L.; Ingham, E.S.; Leslie, C.A.; McGranahan, G.H.; Batra, N.; Goyal, S.; Joseph, J.; Jemmis, E.D.; et al. Mechanism of gallic acid biosynthesis in bacteria (Escherichia coli) and walnut (Juglans regia). Plant Mol. Biol. 2011, 75, 555–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bontpart, T.; Marlin, T.; Vialet, S.; Guiraud, J.L.; Pinasseau, L.; Meudec, E.; Sommerer, N.; Cheynier, V.; Terrier, N. Two shikimate dehydrogenases, VvSDH3 and VvSDH4, are involved in gallic acid biosynthesis in grapevine. J. Exp. Bot. 2016, 67, 3537–3550. [Google Scholar] [CrossRef] [Green Version]
- Oelschlaeger, P. β-Lactamases: Sequence, structure, function, and inhibition. Biomolecules 2021, 11, 986. [Google Scholar] [CrossRef]
- Li, K.; Mikola, M.R.; Draths, K.M.; Worden, R.M.; Frost, J.W. Fed-batch fermentor synthesis of 3-dehydroshikimic acid using recombinant Escherichia coli. Biotechnol. Bioeng. 1999, 64, 61–73. [Google Scholar] [CrossRef]
- Yi, J.; Li, K.; Draths, K.M.; Frost, J.W. Modulation of phosphoenolpyruvate synthase expression increases shikimate pathway product yields in E. coli. Biotechnol. Prog. 2002, 18, 1141–1148. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | DHS Titer (g/L, with Cells) | DHS Broth Volume (L) | SA Titer (g/L, with Cells) | SA Broth Volume (L) | Yield (mol/mol, %) | SA Titer (g/L, Supernatant) | Productivity (g/L/h) |
|---|---|---|---|---|---|---|---|
| 1 | 89.8 | 2.24 | 78.8 | 2.56 | 99.0 | 83.0 | 41.5 |
| 2 | 87.5 | 2.90 | 76.1 | 3.33 | 98.7 | 80.0 | 40.0 |
| 3 | 88.3 | 3.09 | 76.5 | 3.52 | 97.5 | 81.8 | 40.9 |
| Mean ± SD | 88.6 ± 1.2 | 77.1 ± 1.5 | 98.4 ± 0.8 | 81.6 ± 1.5 | 40.8 ± 0.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wu, F.; Zhou, D.; Song, G.; Chen, W.; Zhang, C.; Wang, Q. Cofactor Self-Sufficient Whole-Cell Biocatalysts for the Relay-Race Synthesis of Shikimic Acid. Fermentation 2022, 8, 229. https://doi.org/10.3390/fermentation8050229
Wang X, Wu F, Zhou D, Song G, Chen W, Zhang C, Wang Q. Cofactor Self-Sufficient Whole-Cell Biocatalysts for the Relay-Race Synthesis of Shikimic Acid. Fermentation. 2022; 8(5):229. https://doi.org/10.3390/fermentation8050229
Chicago/Turabian StyleWang, Xiaoshuang, Fengli Wu, Dan Zhou, Guotian Song, Wujiu Chen, Cuiying Zhang, and Qinhong Wang. 2022. "Cofactor Self-Sufficient Whole-Cell Biocatalysts for the Relay-Race Synthesis of Shikimic Acid" Fermentation 8, no. 5: 229. https://doi.org/10.3390/fermentation8050229
APA StyleWang, X., Wu, F., Zhou, D., Song, G., Chen, W., Zhang, C., & Wang, Q. (2022). Cofactor Self-Sufficient Whole-Cell Biocatalysts for the Relay-Race Synthesis of Shikimic Acid. Fermentation, 8(5), 229. https://doi.org/10.3390/fermentation8050229