

Biosynthesis of Glucaric Acid by Recombinant Strain of Escherichia coli Expressing Two Different Urinate Dehydrogenases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- (1)
- We determined the assay method to determine the content of glucosinolates in the fermentation broth and conducted research on the stability of the assay method.
- (2)
- The conditions of the carbon source in the fermentation medium, pH of the culture initiation, inoculum amount and induction intensity were optimized to produce glucosinolates with a high yield. The recombinant bacteria were also supplemented and fermented in a 30 L fermenter according to the optimized conditions in shake flasks.
- (3)
- The permeability of the cell membrane of the high-yielding recombinant bacteria was changed by physical and chemical methods. The physical method used cell freezing and thawing and ultrasonication to change the permeability of the cell membrane and optimize conditions such as the buffer system, buffer system pH and so on. The chemical method used a Cetyltrimethylamine bromide (CTAB) surfactant and toluene organic reagent to change the cell permeability and optimize the concentration of the permeabilizer CTAB and toluene.
2. Materials and Methods
2.1. Plasmids, Strains and the Culture Medium
2.2. Reagents and Chemicals
2.3. Construction of the Expression Vector pETDuet-2 × Udh
2.4. Expression of the Udh Genes in E. coli BL21
2.5. Purification of Recombinant Urinate Dehydrogenase Proteins
2.6. Optimization of Recombinant Protein Expression Conditions
2.7. Biosynthesis of Glucaric Acid from Glucuronic Acid with Recombinant BL21 Harboring the Expression Vector pETDuet-2 × Udh
2.8. MS and HPLC Analysis of the Reaction Product
2.9. Preliminary Fermentation of Recombinant BL21 for Glucaric Acid Biosynthesis in a 30 L Tank
3. Results
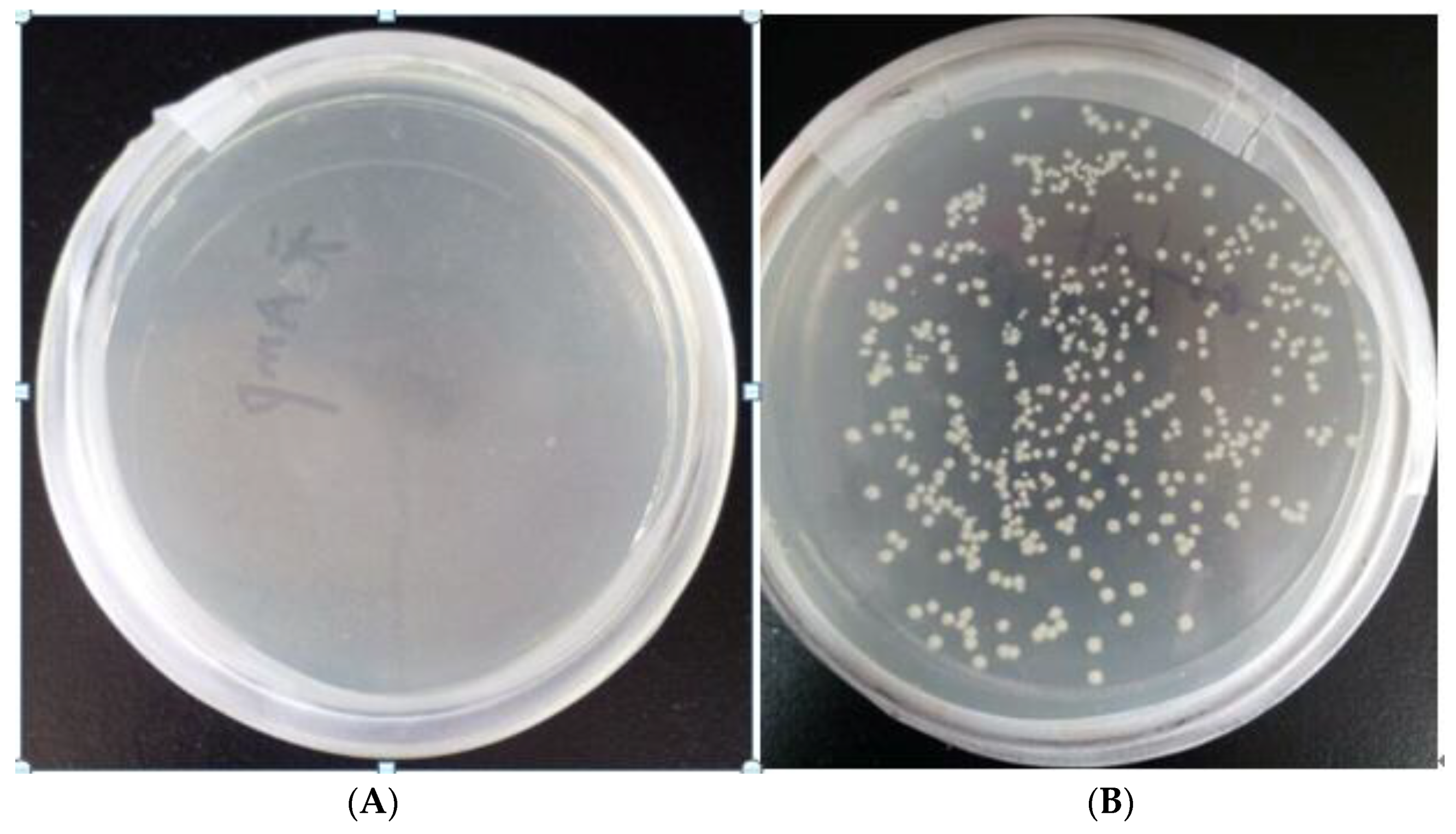
3.1. Screening Results of Recombinant Bacteria
3.2. Growth Curve of Recombinant Escherichia coli
3.3. Comparative Analysis of the Conversion Ability of Different Large Intestine Hosts to Produce Glucose Dicarboxylic Acid
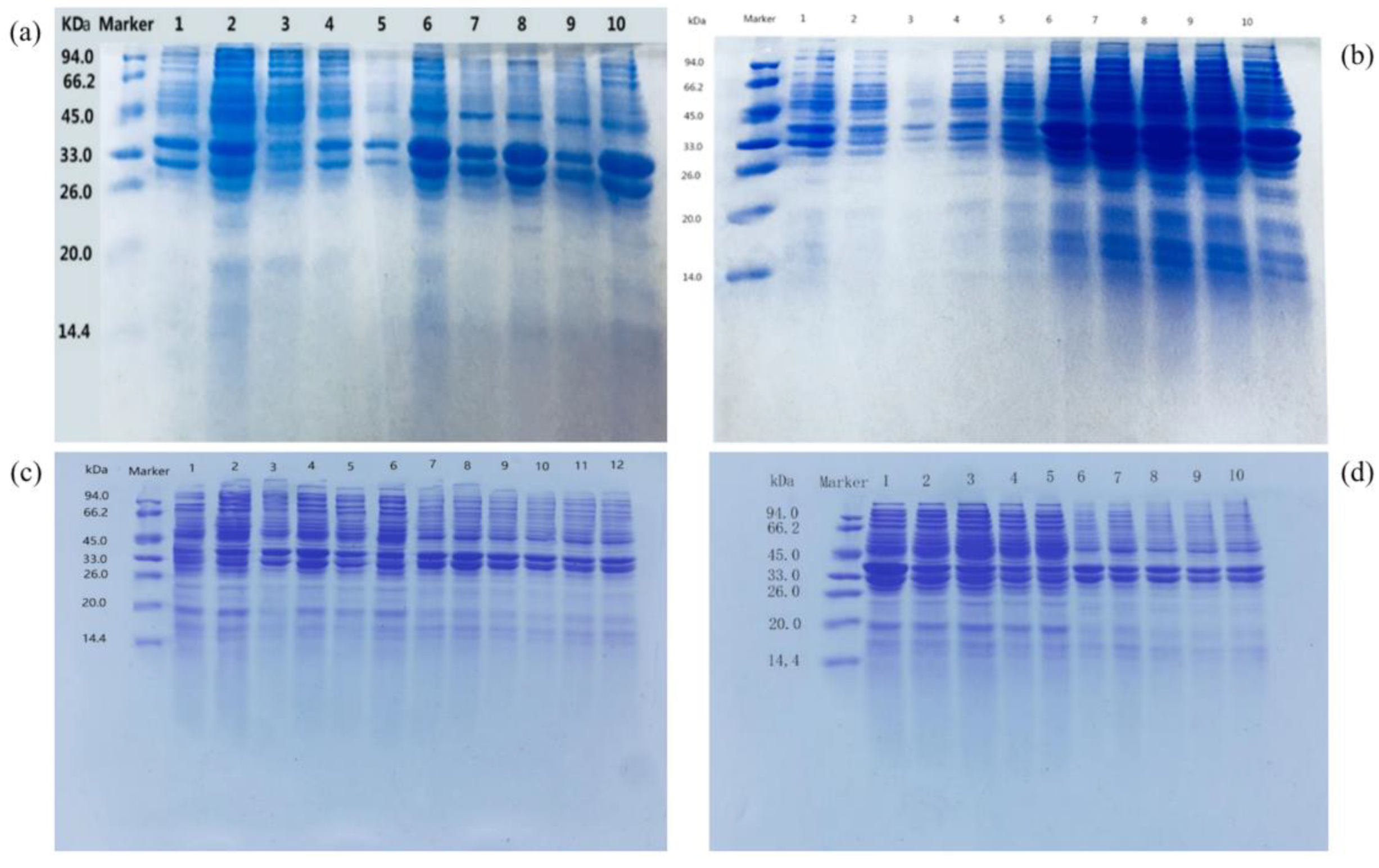
3.4. Analysis of the Expression of Recombinant Protein
3.5. Purification and Induction Conditions of Udh Expression Construct
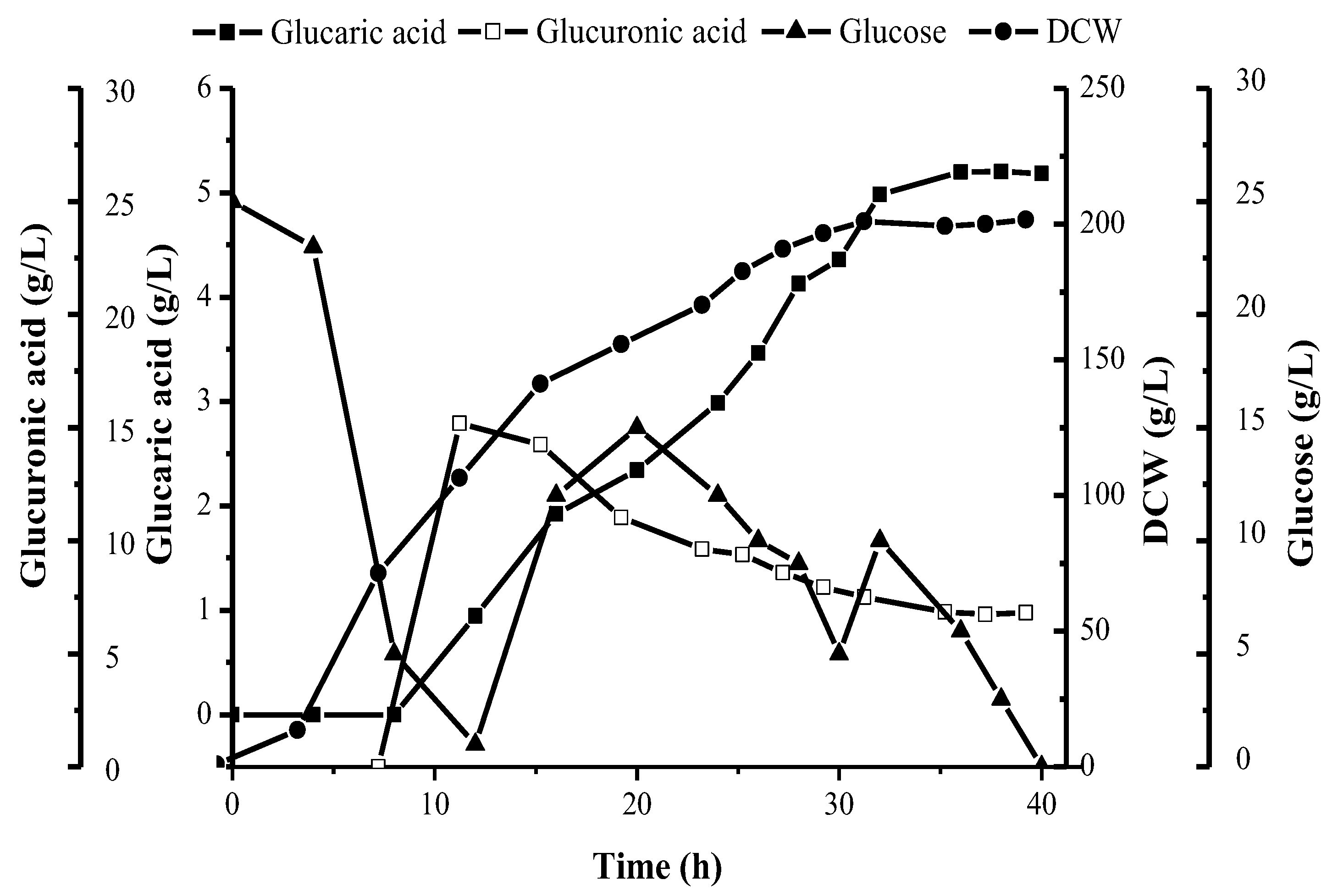
3.6. Production Analysis of Glucaric Acid
3.7. Production of Glucaric Acid with 30 L Tank Fermentation Strategy
4. Conclusions
- (1)
- Replacement of safe strains. In this experiment, glucuronide dehydrogenase was constructed and expressed in E. coli, but E. coli is not regarded as a safe strain by GRAS, which has limited its application in the production of food-grade chemicals. Therefore, safe microbial strains can be considered for glucuronic acid production.
- (2)
- Multifactorial analysis of the method for cell membrane permeabilization. Scholars should combine the permeabilization methods to improve the production of glucosinolates to increase the permeability of cell membranes as much as possible without breaking the integrity of the cells in the conversation so as to further improve the production of glucosinolates.
- (3)
- Addition of nicotinamide to the culture medium. The production of glucosinolate requires NAD+ cofactors, and nicotinamide is a precursor for synthesizing NAD+, which is the most direct way to replenish NAD+. Compared to other precursors, nicotinamide bypasses the nicotinamide phosphoribosyltransferase (NAMPT) restriction enzyme and is able to replenish NAD+ rapidly, and the addition of nicotinamide to the fermentation broth has the potential to increase the content of glucosinolates.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, T.; Denton, T.; Zhang, J. Synthesis and characterization of higher molecular weight stereorandom poly (d-glucaramides) from 1:1 alkylenediammonium d-glucaric acid. In Proceedings of the 234th ACS National Meeting, Boston, MA, USA, 19–23 August 2007. [Google Scholar]
- Liu, Y.; Gong, X.; Wang, C. Production of glucaric acid from myoinositol in engineered Pichia pastoris. Enzym. Microb. Technol. 2016, 34, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wang, J.; Zhao, Y. Metabolic engineering of Saccharomyces cerevisiae for efficient production of glucaric acid at high titer. Appl. Biochem. Biotechnol. 2018, 17, 67. [Google Scholar]
- Bauer, F.; Coenen, L.; Hansen, T. Technological innovation systems for biorefineries: A review of the literature. Biofuels Bioprod. Biorefining 2017, 11, 534–548. [Google Scholar] [CrossRef]
- Dwivedi, C.; Heck, W.J.; Downie, A.A. Effect of calcium glucarate on beta-glucuronidase activity and glucarate content of certain vegetables and fruits. Biochem. Med. Metab. Biol. 1990, 43, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.J. A large-scale purification of beta-glucuronidase from human liver by immunoaffinity chromatography. Biotechnol. Appl. Biochem. 2011, 14, 296–305. [Google Scholar]
- Moon, T.S.; Yoon, S.H.; Ching, M.J.T.M. Enzymatic assay of d-glucuronate using uronate dehydrogenase. Anal. Biochem. 2009, 392, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Gupta, K.P. Induction of apoptosis by calcium D-glucarate in 7,12-dimethyl benz [a] anthracene-exposed mouse skin. J. Environ. Pathol. Toxicol. Oncol. 2007, 26, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Su, H.H.; Guo, Z.W.; Wu, X.L. Efficient bioconversion of sucrose to high-value-added glucaric acid by in vitro metabolic engineering. ChemSusChem 2019, 12, 2278–2285. [Google Scholar] [CrossRef] [PubMed]
- Tae Seok, M.; Sang-Hwal, Y.; Lanza, A.M. Production of glucaric acid from a synthetic pathway in recombinant Escherichia coli. Appl. Environ. Microbiol. 2009, 75, 589. [Google Scholar]
- Shiue, E.; Prather, K.L.J. Improving d-glucaric acid production from myo-inositol in E. coli by increasing MIOX stability and myo-inositol transport. Metab. Eng. 2014, 22, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Yang, J.E.; Ha, J.Y. Bio-based production of monomers and polymers by metabolically engineered microorganisms. Curr. Opin. Biotechnol. 2015, 36, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Nielsen, J. Biobased organic acids production by metabolically engineered microorganisms. Curr. Opin. Biotechnol. 2016, 37, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.N.; Yan, H.J.; Guo, Q. Biosynthesis of d-glucaric acid from sucrose with routed carbon distribution in metabolically engineered Escherichia coli. Metab. Eng. 2018, 47, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Hicks, M.A.; Manchester, S.P. Porting the synthetic D-glucaric acid pathway from Escherichia coli to saccharomyces cerevisiae. Biotechnol. J. 2016, 11, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.; You, R.; Hu, M. Production of d-glucuronic acid from myo-inositol using Escherichia coli whole-cell biocatalyst overexpressing a novel myo-inositol oxygenase from thermothelomyces thermophile. Enzym. Microb. Technol. 2019, 127, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xue, Y.; Cao, Z. Characterization of a uronate dehydrogenase from thermobispora bispora for production of glucaric acid from hemicellulose substrate. World J. Microbiol. Biotechnol. 2018, 34, 102. [Google Scholar] [CrossRef] [PubMed]
- Janarthini, R.; Wang, X.; Chen, L. A tobacco-derived thymosin beta4 concatemer promotes cell proliferation and wound healing in mice. Biomed Res. Int. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Rubiyana, Y.; Santoso, A.; Batubara, I. Comparison of immobilized metal affinity chromatography Ni-NTA and Co-TALON for the purification of recombinant human erythropoietin. Makara J. Sci. 2015, 19, 137. [Google Scholar] [CrossRef]
- Yang, Y.; Mitri, K.; Zhang, C. Promiscuity of host cell proteins in the purification of histidine tagged recombinant xylanase a by IMAC procedures: A case study with a Ni2+-tacn-based IMAC system. Protein Expr. Purif. 2019, 162, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.; Villeneuve, D.C.; Chu, I. HPLC determination of d-glucaric acid in human urine. J. Anal. Toxicol. 1993, 17, 146–150. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Niu, L.; Ye, C.; Wang, Y.; Liu, Y.; Wang, F.; Sun, N. Biosynthesis of Glucaric Acid by Recombinant Strain of Escherichia coli Expressing Two Different Urinate Dehydrogenases. Fermentation 2023, 9, 764. https://doi.org/10.3390/fermentation9080764
Yang X, Niu L, Ye C, Wang Y, Liu Y, Wang F, Sun N. Biosynthesis of Glucaric Acid by Recombinant Strain of Escherichia coli Expressing Two Different Urinate Dehydrogenases. Fermentation. 2023; 9(8):764. https://doi.org/10.3390/fermentation9080764
Chicago/Turabian StyleYang, Xinchao, Linlin Niu, Chunjiang Ye, Yuanxiu Wang, Yuehui Liu, Fang Wang, and Naxin Sun. 2023. "Biosynthesis of Glucaric Acid by Recombinant Strain of Escherichia coli Expressing Two Different Urinate Dehydrogenases" Fermentation 9, no. 8: 764. https://doi.org/10.3390/fermentation9080764
APA StyleYang, X., Niu, L., Ye, C., Wang, Y., Liu, Y., Wang, F., & Sun, N. (2023). Biosynthesis of Glucaric Acid by Recombinant Strain of Escherichia coli Expressing Two Different Urinate Dehydrogenases. Fermentation, 9(8), 764. https://doi.org/10.3390/fermentation9080764