Early Stages of Biomineral Formation—A Solid-State NMR Investigation of the Mandibles of Minipigs

 ,
,
Abstract
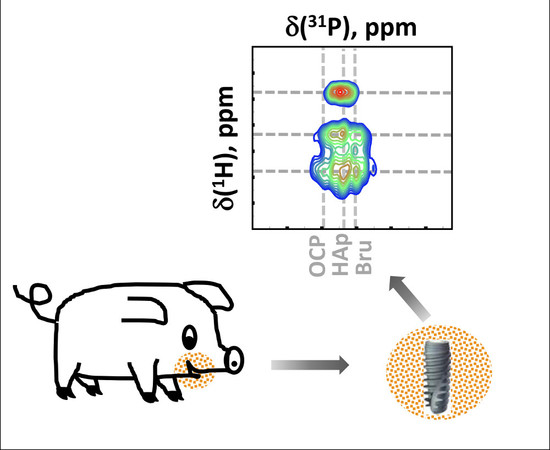
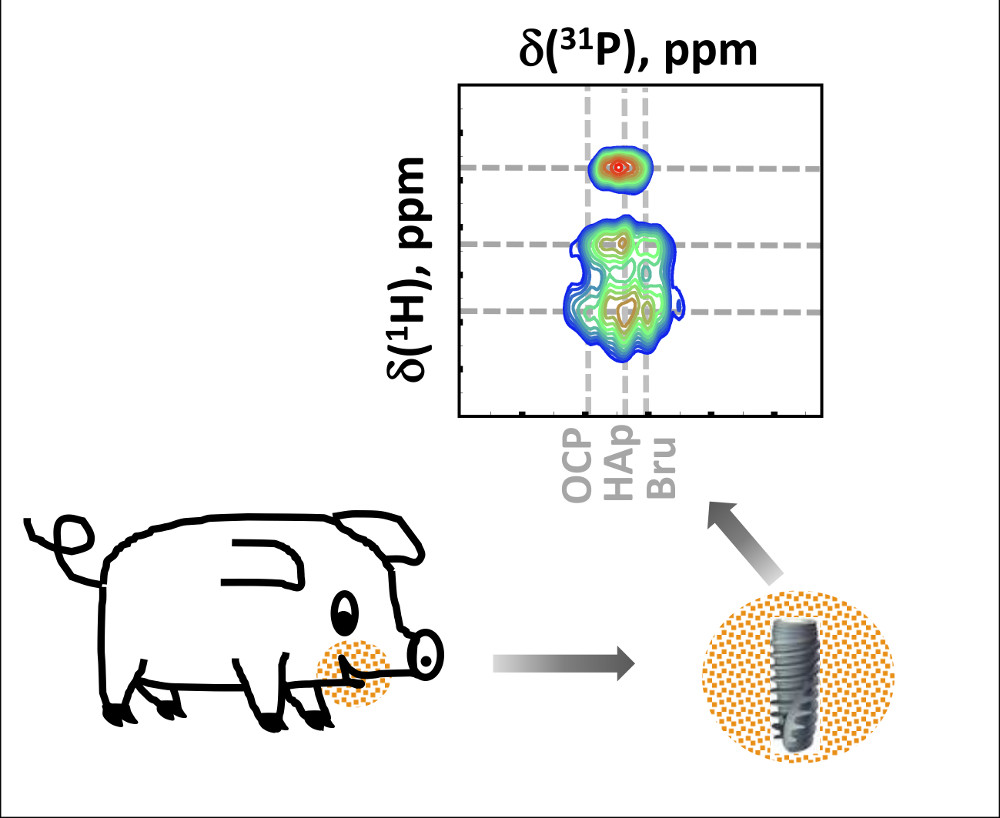
:
1. Introduction
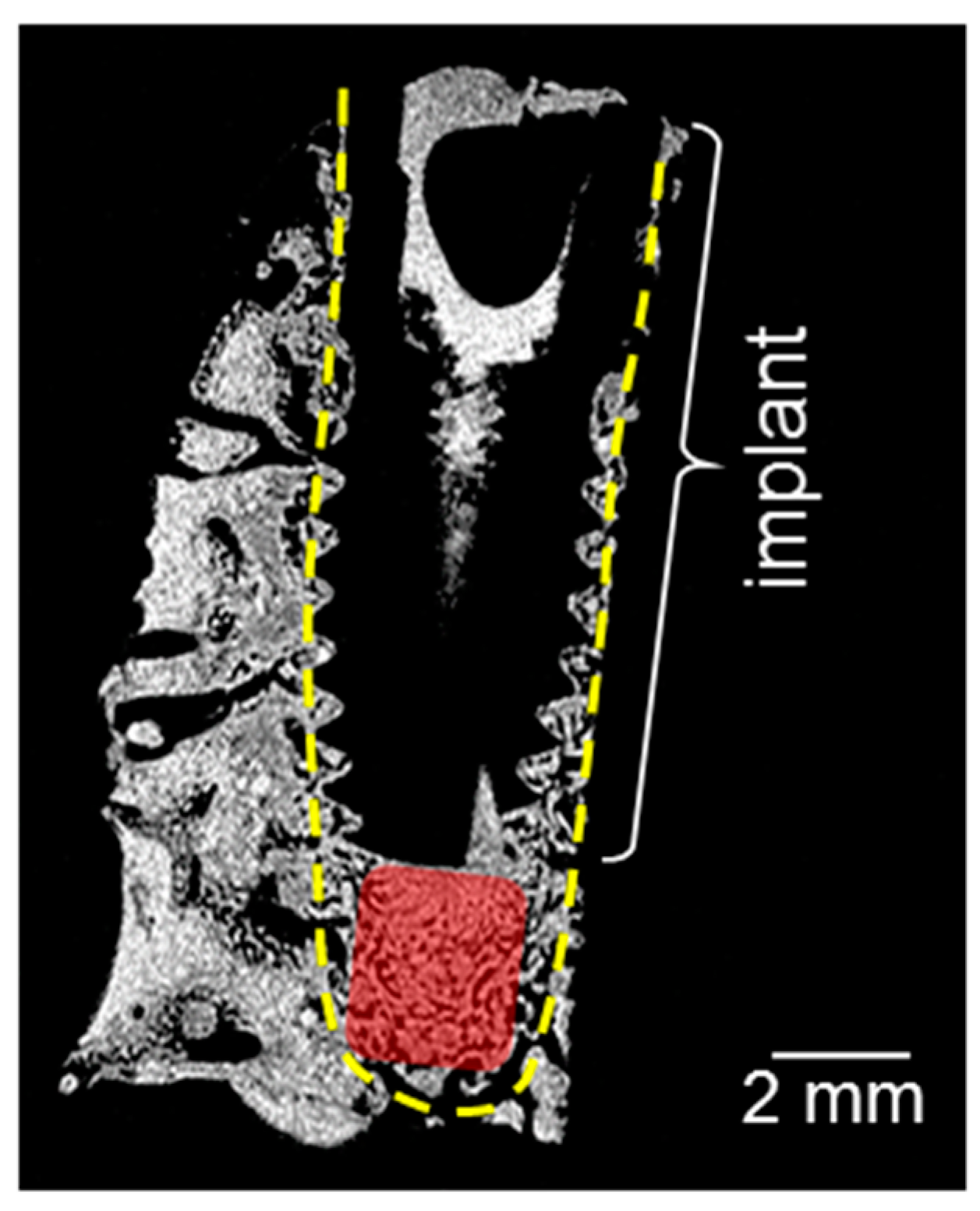
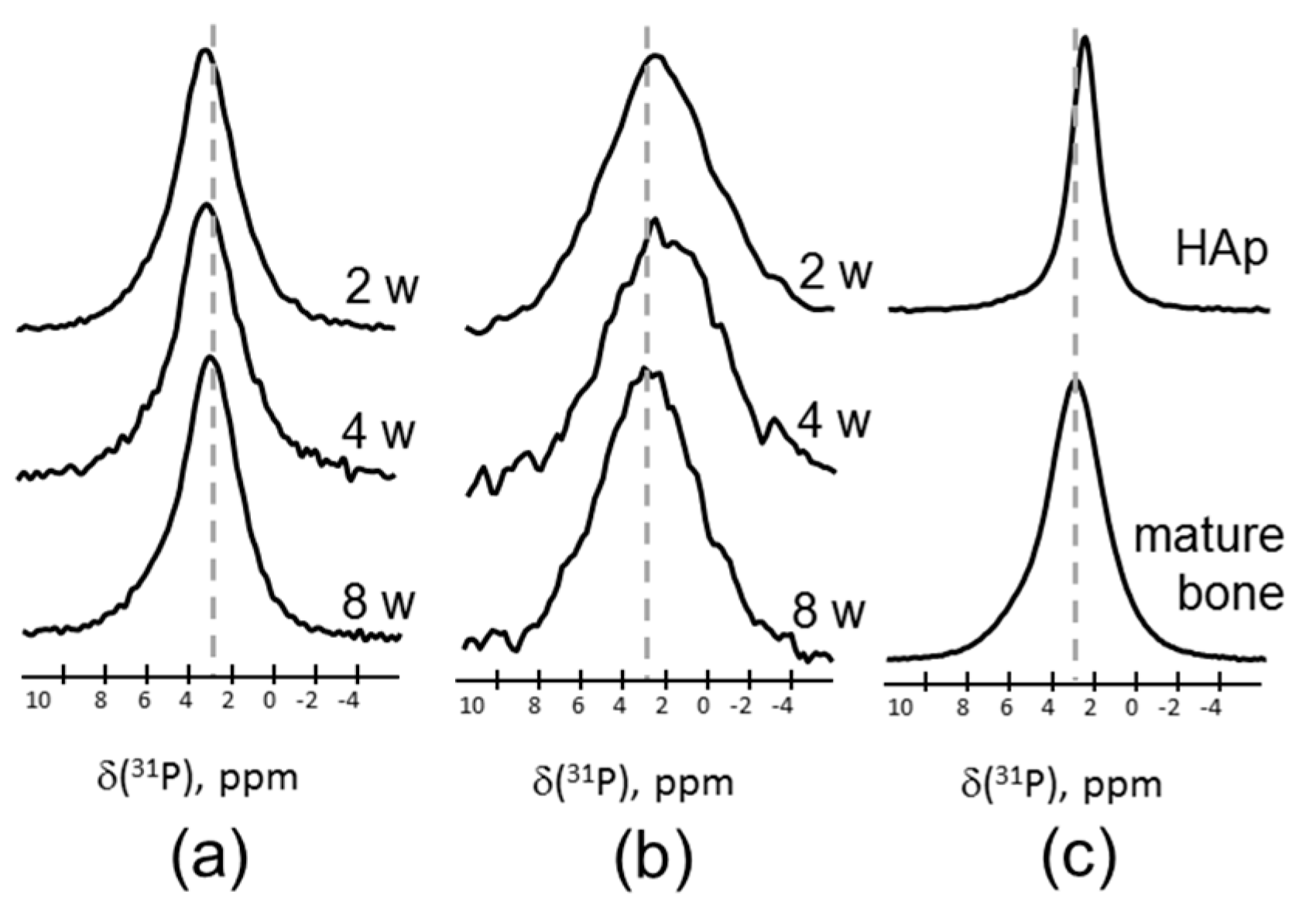
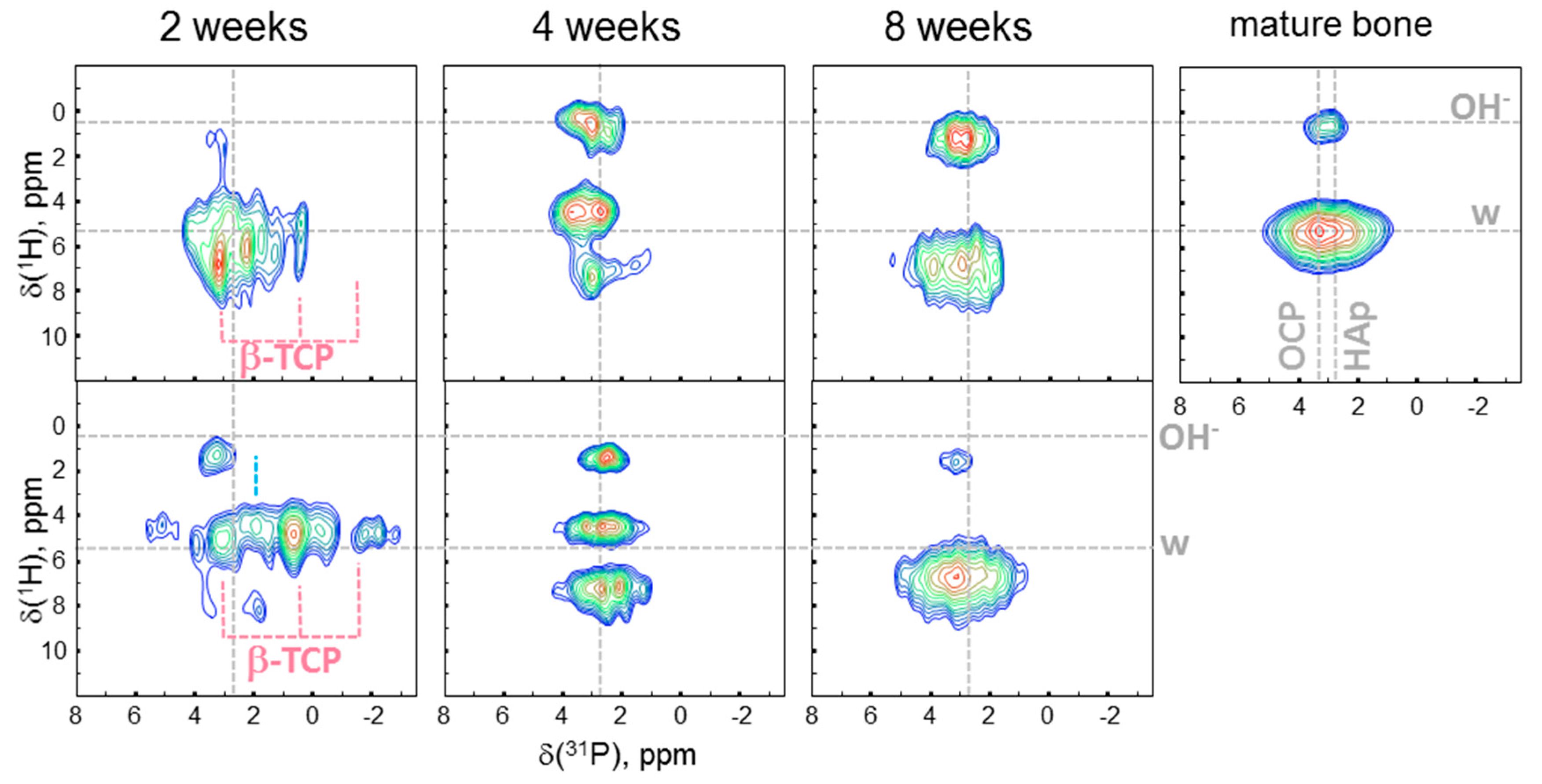
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. NMR Imaging Measurements
4.3. Solid State NMR
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, L.; Nancollas, G.H. Pathways to biomineralization and biodemineralization of calcium phosphates: The thermodynamic and kinetic controls. Dalton Trans. 2009, 2665–2672. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D.; Völkel, A.; Cölfen, H. Stable prenucleation calcium carbonate clusters. Science 2008, 322, 1819–1822. [Google Scholar] [CrossRef] [PubMed]
- Pouget, E.M.; Bomans, P.H.H.; Goos, J.A.C.M.; Frederik, P.M.; de With, G.; Sommerdijk, N.A.J.M. The initial stages of template-controlled CaCO3 formation revealed by cryo-TEM. Science 2009, 323, 1455–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nudelman, F.; Pieterse, K.; George, A.; Bomans, P.H.H.; Friedrich, H.; Brylka, L.J.; Hilbers, P.A.J.; de With, G.; Sommerdijk, N.A.J.M. The role of collagen in bone apatite formation in the presence of hydroxyapatite nucleation inhibitors. Nat. Mater. 2010, 9, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Bomans, P.H.H.; Müller, F.A.; Will, J.; Frederik, P.M.; de With, G.; Sommerdijk, N.A.J.M. The role of prenucleation clusters in surface-induced calcium phosphate crystallization. Nat. Mater. 2010, 9, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Mahamid, J.; Sharir, A.; Addadi, L.; Weiner, S. Amorphous calcium phosphate is a major component of the forming fin bones of zebrafish: Indications for an amorphous precursor phase. Proc. Natl. Acad. Sci. USA 2008, 105, 12748–12753. [Google Scholar] [CrossRef] [PubMed]
- Beniash, E.; Metzler, R.A.; Lam, R.S.; Gilbert, P.U. Transient amorphous calcium phosphate in forming enamel. J. Struct. Biol. 2009, 166, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Boivin, G.; Meunier, P.J. The degree of mineralization of bone tissue measured by computerized quantitative contact microradiography. Calcif. Tissue Int. 2002, 70, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Bala, Y.; Farlay, D.; Delmas, P.D.; Meunier, P.J.; Boivin, G. Time sequence of secondary mineralization and microhardness in cortical and cancellous bone from ewes. Bone 2010, 46, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Posner, A.S.; Betts, F. Synthetic amorphous calcium phosphate and its relation to bone mineral structure. Acc. Chem. Res. 2002, 8, 273–281. [Google Scholar] [CrossRef]
- Crane, N.J.; Popescu, V.; Morris, M.D.; Steenhuis, P.; Ignelzi, M.A. Raman spectroscopic evidence for octacalcium phosphate and other transient mineral species deposited during intramembranous mineralization. Bone 2006, 39, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-H.; Tsai, Y.-L.; Tsai, T.W.T.; Chao, J.C.H.; Lin, C.-P.; Huang, S.-H.; Mou, C.-Y.; Chan, J.C.C. Characterization of the Phosphate Units in Rat Dentin by Solid-State NMR Spectroscopy. Chem. Mater. 2007, 19, 6088–6094. [Google Scholar] [CrossRef]
- Huang, S.-J.; Tsai, Y.-L.; Lee, Y.-L.; Lin, C.-P.; Chan, J.C.C. Structural Model of Rat Dentin Revisited. Chem. Mater. 2009, 21, 2583–2585. [Google Scholar] [CrossRef]
- Duer, M.J.; Friscić, T.; Murray, R.C.; Reid, D.G.; Wise, E.R. The mineral phase of calcified cartilage: Its molecular structure and interface with the organic matrix. Biophys. J. 2009, 96, 3372–3378. [Google Scholar] [CrossRef] [PubMed]
- Cho, G.; Wu, Y.; Ackerman, J.L. Detection of hydroxyl ions in bone mineral by solid-state NMR spectroscopy. Science 2003, 300, 1123–1127. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejski, W. Solid-state NMR studies of bone. Top. Curr. Chem. 2005, 246, 235–270. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.E.; Awonusi, A.; Morris, M.D.; Kohn, D.H.; Tecklenburg, M.M.J.; Beck, L.W. Three structural roles for water in bone observed by solid-state NMR. Biophys. J. 2006, 90, 3722–3731. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, C.; Groom, N.S.; Bowe, E.A.; Horner, A.; Davies, M.E.; Murray, R.C.; Duer, M.J. Investigation of the Nature of the Protein−Mineral Interface in Bone by Solid-State NMR. Chem. Mater. 2005, 17, 3059–3061. [Google Scholar] [CrossRef]
- Tseng, Y.-H.; Mou, C.-Y.; Chan, J.C.C. Solid-state NMR study of the transformation of octacalcium phosphate to hydroxyapatite: A mechanistic model for central dark line formation. J. Am. Chem. Soc. 2006, 128, 6909–6918. [Google Scholar] [CrossRef] [PubMed]
- Nassif, N.; Martineau, F.; Syzgantseva, O.; Gobeaux, F.; Willinger, M.; Coradin, T.; Cassaignon, S.; Azaïs, T.; Giraud-Guille, M.M. In Vivo Inspired Conditions to Synthesize Biomimetic Hydroxyapatite. Chem. Mater. 2010, 22, 3653–3663. [Google Scholar] [CrossRef]
- Ndao, M.; Ash, J.T.; Stayton, P.S.; Drobny, G.P. The Role of Basic Amino Acids in the Molecular Recognition of Hydroxyapatite by Statherin using Solid State NMR. Surf. Sci. 2010, 604, L39–L42. [Google Scholar] [CrossRef] [PubMed]
- Vyalikh, A.; Simon, P.; Kollmann, T.; Kniep, R.; Scheler, U. Local Environment in Biomimetic Hydroxyapatite−Gelatin Nanocomposites As Probed by NMR Spectroscopy. J. Phys. Chem. C 2011, 115, 1513–1519. [Google Scholar] [CrossRef]
- Vyalikh, A.; Simon, P.; Rosseeva, E.; Buder, J.; Kniep, R.; Scheler, U. Intergrowth and interfacial structure of biomimetic fluorapatite-gelatin nanocomposite: A solid-state NMR study. J. Phys. Chem. B 2014, 118, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Korn, P.; Elschner, C.; Schulz, M.C.; Range, U.; Mai, R.; Scheler, U. MRI and dental implantology: Two which do not exclude each other. Biomaterials 2015, 53, 634–645. [Google Scholar] [CrossRef] [PubMed]
- Aue, W.P.; Roufosse, A.H.; Glimcher, M.J.; Griffin, R.G. Solid-state phosphorus-31 nuclear magnetic resonance studies of synthetic solid phases of calcium phosphate: Potential models of bone mineral. Biochemistry 2002, 23, 6110–6114. [Google Scholar] [CrossRef]
- Combes, C.; Rey, C. Amorphous calcium phosphates: Synthesis, properties and uses in biomaterials. Acta Biomater. 2010, 6, 3362–3378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yesinowski, J.P.; Eckert, H. Hydrogen environments in calcium phosphates: Proton MAS NMR at high spinning speeds. J. Am. Chem. Soc. 1987, 109, 6274–6282. [Google Scholar] [CrossRef]
- Vyalikh, A.; Simon, P.; Rosseeva, E.; Buder, J.; Scheler, U.; Kniep, R. An NMR Study of Biomimetic Fluorapatite—Gelatine Mesocrystals. Sci. Rep. 2015, 5, 15797. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Wind, R.A.; Bronnimann, C.E. 1H CRAMPS and 1H-31P HetCor Experiments on Bone, Bone Mineral, and Model Calcium Phosphate Phases. J. Magn. Reson. Ser. B 1994, 105, 183–187. [Google Scholar] [CrossRef]
- Rothwell, W.P.; Waugh, J.S.; Yesinowski, J.P. High-resolution variable-temperature phosphorus-31 NMR of solid calcium phosphates. J. Am. Chem. Soc. 1980, 102, 2637–2643. [Google Scholar] [CrossRef]
- Bak, M.; Thomsen, J.K.; Jakobsen, H.J.; Petersen, S.E.; Petersen, T.E.; Nielsen, N.C. Solid-state 13C and 31P NMR analysis of urinary stones. J. Urol. 2000, 164, 856–863. [Google Scholar] [CrossRef]
- Kaflak-Hachulska, A.; Samoson, A.; Kolodziejski, W. 1H MAS and 1H→31P CP/MAS NMR study of human bone mineral. Calcif. Tissue Int. 2003, 73, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Legrand, A.P.; Sfihi, H.; Lequeux, N.; Lemaître, J. 31P Solid-State NMR study of the chemical setting process of a dual-paste injectable brushite cements. J. Biomed. Mater. Res. B 2009, 91, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.; Duer, M.J.; Ashbrook, S.E.; Griffin, J.M. Applications of NMR crystallography to problems in biomineralization: Refinement of the crystal structure and 31P solid-state NMR spectral assignment of octacalcium phosphate. J. Am. Chem. Soc. 2012, 134, 12508–12515. [Google Scholar] [CrossRef] [PubMed]
- Obadia, L.; Deniard, P.; Alonso, B.; Rouillon, T.; Jobic, S.; Guicheux, J.; Julien, M.; Massiot, D.; Bujoli, B.; Bouler, J.-M. Effect of Sodium Doping in β-Tricalcium Phosphate on Its Structure and Properties. Chem. Mater. 2006, 18, 1425–1433. [Google Scholar] [CrossRef]
- Wu, Y.; Ackerman, J.L.; Strawich, E.S.; Rey, C.; Kim, H.-M.; Glimcher, M.J. Phosphate ions in bone: Identification of a calcium-organic phosphate complex by 31P solid-state NMR spectroscopy at early stages of mineralization. Calcif. Tissue Int. 2003, 72, 610–626. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.P.; Kirkpatrick, R.J.; Howell, D.; Risbud, S.H. 31P Magic-angle spinning nuclear magnetic resonance spectroscopy of calcium phosphate glasses. J. Chem. Soc. Faraday Trans. 1993, 89, 3297. [Google Scholar] [CrossRef]
- Neuman, W.F.; Bareham, B.J. Evidence for the presence of secondary calcium phosphate in bone and its stabilization by acid production. Calcif. Tissue Res. 1975, 18, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Kodaka, T.; Ohohara, Y.; Debari, K. Scanning electron microscopy and energy-dispersive X-ray microanalysis studies of early dental calculus on resin plates exposed to human oral cavities. Scanning Microsc. 1992, 6, 475–486. [Google Scholar] [PubMed]
- LeGeros, R.Z.; Orly, I.; LeGeros, J.P.; Gomez, C.; Kazimiroff, J.; Tarpley, T.; Kerebel, B. Scanning electron microscopy and electron probe microanalyses of the crystalline components of human and animal dental calculi. Scanning Microsc. 1988, 2, 345–356. [Google Scholar] [PubMed]
- Jin, Y.; Yip, H.-K. Supragingival Calculus: Formation and Control. Crit. Rev. Oral Biol. Med. 2002, 13, 426–441. [Google Scholar] [CrossRef] [PubMed]
- Loong, C.-K.; Rey, C.; Kuhn, L.T.; Combes, C.; Wu, Y.; Chen, S.-H.; Glimcher, M.J. Evidence of hydroxyl-ion deficiency in bone apatites: An inelastic neutron-scattering study. Bone 2000, 26, 599–602. [Google Scholar] [CrossRef]
- Vyalikh, A.; Mai, R.; Scheler, U. OH− deficiency in dental enamel, crown and root dentine as studied by 1H CRAMPS. Bio-Med. Mater. Eng. 2013, 23, 507–512. [Google Scholar] [CrossRef]
- Vyalikh, A.; Emmler, T.; Grünberg, B.; Xu, Y.; Shenderovich, I.; Findenegg, G.H.; Limbach, H.-H.; Buntkowsky, G. Hydrogen Bonding of Water Confined in Controlled-Pore Glass 10–75 Studied by 1H-Solid State NMR. Z. Phys. Chem. 2007, 221, 155–168. [Google Scholar] [CrossRef]
- Stadlinger, B.; Hintze, V.; Bierbaum, S.; Möller, S.; Schulz, M.C.; Mai, R.; Kuhlisch, E.; Heinemann, S.; Scharnweber, D.; Schnabelrauch, M.; et al. Biological functionalization of dental implants with collagen and glycosaminoglycans-A comparative study. J. Biomed. Mater. Res. B 2012, 100, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling one- and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Chemical Shift | References | |
|---|---|---|---|
| 31P, ppm | 1H, ±0.05 ppm | ||
| HAp, hydroxyapatite | 2.8 ± 0.2 | 0.2 | [25,30,31] |
| 5.5 | |||
| Bru, brushite (dicalcium phosphate, dehydrate) | 1.7 ± 0.3 | 4.1 | [25,30,31,32,33] |
| 6.4 | |||
| 10.4 | |||
| Monetite (dicalcium phosphate, anhydrous) | 0.0 ± 0.4 | [25,30,31,33] | |
| −1.5 ± 0.4 | |||
| ACP, amorphous calcium phosphate | 3.0 | 5.5 | [25,26] |
| OCP, octacalcium phosphate | −0.2 ± 0.4 | 0.18 | [30,31,34] |
| 1.9 | 5.5 | ||
| 3.3 ± 0.3 | 13.6 | ||
| 3.7 | |||
| β-TCP, tricalcium phosphate | −1.6 | [31,35] | |
| 0.2 | |||
| 2.9 | |||
| Sample | Mineral Components | ||||
|---|---|---|---|---|---|
| ACP | OCP | Bru | β-TCP | HAp | |
| 2 w | x | x | x | ||
| x | x | x | x | ||
| 4 w | x | x | x | x | |
| x | x | x | x | ||
| 8 w | x | x | x | ||
| x | x | x | x | ||
| Mature bone (reference) | x | x | x | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vyalikh, A.; Elschner, C.; Schulz, M.C.; Mai, R.; Scheler, U. Early Stages of Biomineral Formation—A Solid-State NMR Investigation of the Mandibles of Minipigs. Magnetochemistry 2017, 3, 39. https://doi.org/10.3390/magnetochemistry3040039
Vyalikh A, Elschner C, Schulz MC, Mai R, Scheler U. Early Stages of Biomineral Formation—A Solid-State NMR Investigation of the Mandibles of Minipigs. Magnetochemistry. 2017; 3(4):39. https://doi.org/10.3390/magnetochemistry3040039
Chicago/Turabian StyleVyalikh, Anastasia, Cindy Elschner, Matthias C. Schulz, Ronald Mai, and Ulrich Scheler. 2017. "Early Stages of Biomineral Formation—A Solid-State NMR Investigation of the Mandibles of Minipigs" Magnetochemistry 3, no. 4: 39. https://doi.org/10.3390/magnetochemistry3040039
APA StyleVyalikh, A., Elschner, C., Schulz, M. C., Mai, R., & Scheler, U. (2017). Early Stages of Biomineral Formation—A Solid-State NMR Investigation of the Mandibles of Minipigs. Magnetochemistry, 3(4), 39. https://doi.org/10.3390/magnetochemistry3040039