The Magnetic Band-Structures of Ordered PtxFe1−x, PtxCo1−x, and PtxNi1−x (x = 0.25, 0.50, and 0.75)


Abstract
:1. Introduction
2. Results
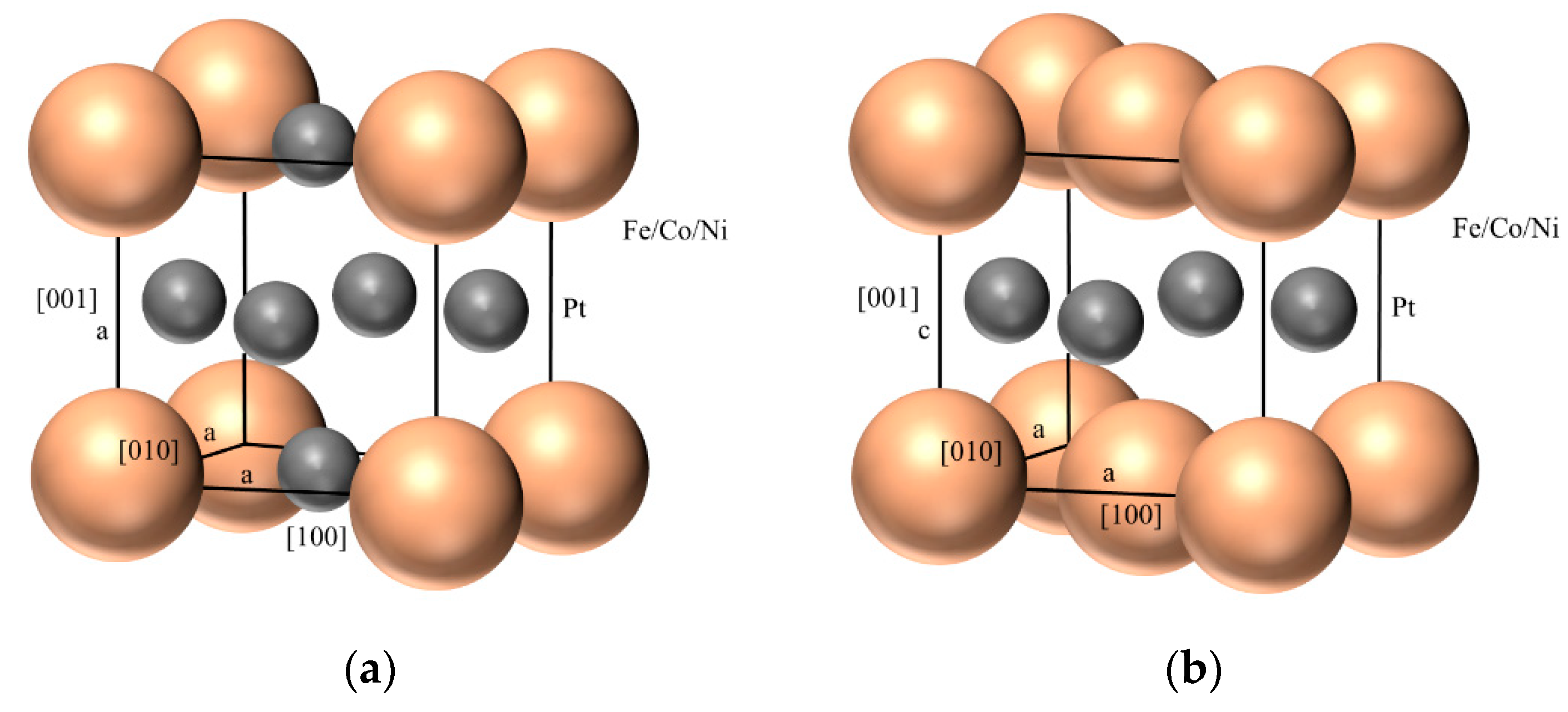
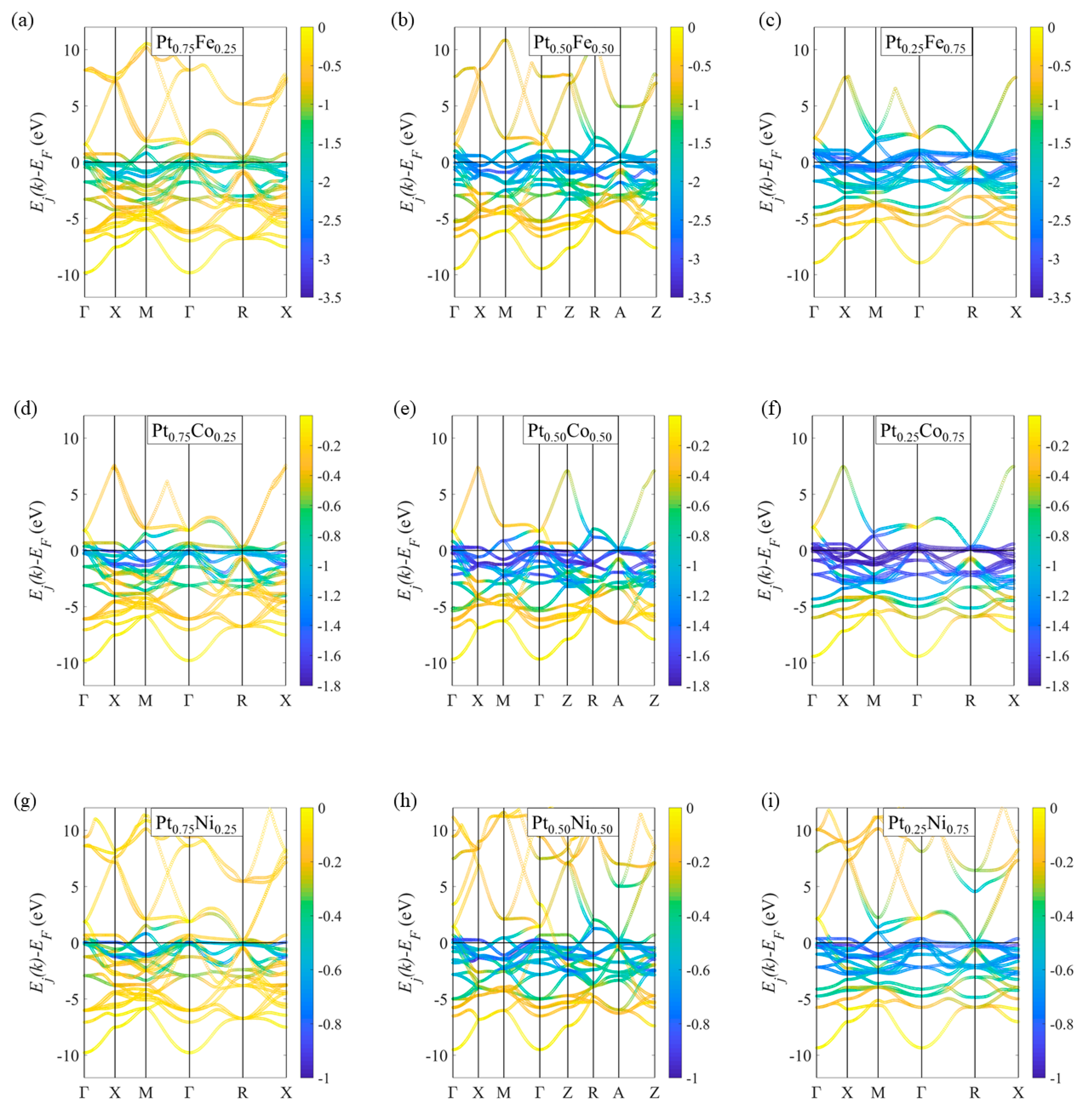
2.1. Spin-Resolved Electron Band Structures for the Ordered Ptxm1−X Alloys
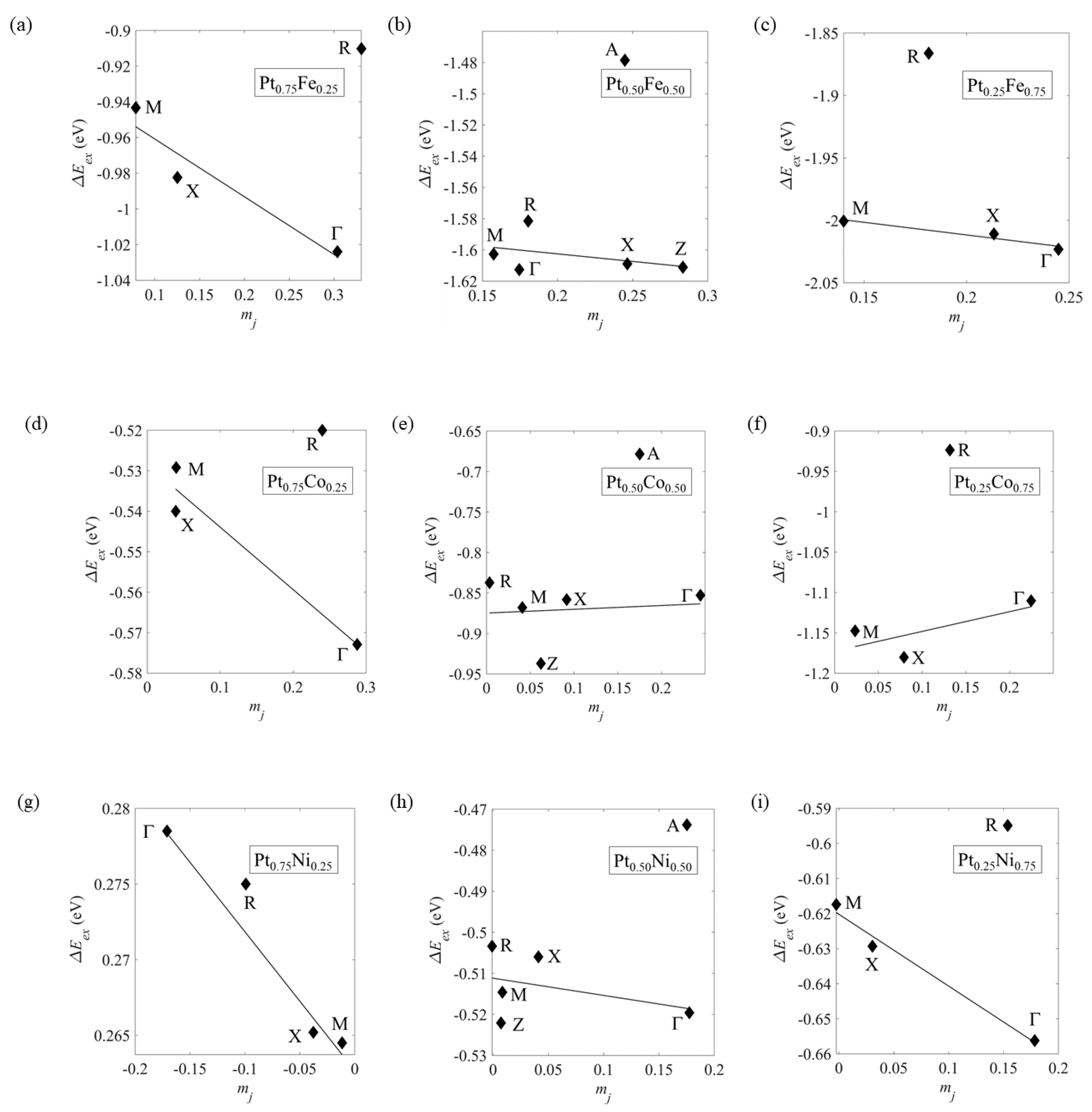
2.2. Exchange Splitting ΔEex for the Ordered Ptxm1−X Alloys
3. Discussion
4. Materials and Methods
5. Conclusions
Funding
Conflicts of Interest
References
- Singamaneni, S.; Bliznyuk, V.N.; Binek, C.; Tsymbal, E.Y. Magnetic nanoparticles: Recent advances in synthesis, self-assembly and applications. J. Mater. Chem. 2011, 21, 16819–16845. [Google Scholar] [CrossRef] [Green Version]
- Ferrando, R.; Jellinek, J.; Johnston, R.L. Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles. Chem. Rev. 2008, 108, 845–910. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Q.; Xu, M.; Dhawan, U.; Liu, W.-C.; Wu, K.-T.; Liu, X.-R.; Lin, C.; Zhao, G.; Wu, Y.-C.; Chung, R.-J. Iron–gold alloy nanoparticles serve as a cornerstone in hyperthermia-mediated controlled drug release for cancer therapy. Int. J. Nanomed. 2018, 13, 5499–5509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jong, W.H. Drug delivery and nanoparticles: Applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, K.M.; Pakhomov, A.B.; Bao, Y.; Blomqvist, P.; Chun, Y.; Gonzales, M.; Griffin, K.; Ji, X.; Roberts, B.K. Nanomagnetism and spin electronics: Materials, microstructure and novel properties. J. Mater. Sci. 2006, 41, 793–815. [Google Scholar] [CrossRef]
- Koh, I.; Blois, J. Magnetic Nanoparticle Sensors. Sensors 2009, 9, 8130–8145. [Google Scholar] [CrossRef]
- Rocha-Santos, T.A. Sensors and biosensors based on magnetic nanoparticles. TrAC Trends Anal. Chem. 2014, 62, 28–36. [Google Scholar] [CrossRef]
- Neamtu, M.; Nadejde, C.; Hodoroaba, V.-D.; Schneider, R.J.; Verestiuc, L.; Panne, U. Functionalized magnetic nanoparticles: Synthesis, characterization, catalytic application and assessment of toxicity. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rossi, L.M.; Costa, N.J.S.; Silva, F.P.; Wojcieszak, R. Magnetic nanomaterials in catalysis: Advanced catalysts for magnetic separation and beyond. Green Chem. 2014, 16, 2906–2933. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; Da Silva, A.G.M.; Camargo, P.H.C. Nanocatalysis by noble metal nanoparticles: Controlled synthesis for the optimization and understanding of activities. J. Mater. Chem. A 2019, 7, 5857–5874. [Google Scholar] [CrossRef] [Green Version]
- Massalski, T.B.; Okamoto, H.; Subramanian, P.R.; Kacprzak, L. (Eds.) Binary Alloy Phase Diagrams, 2nd ed.; ASM International: Materials Park, OH, USA, 1990; p. 2844. [Google Scholar] [CrossRef]
- Cadeville, M.; Morán-López, J. Magnetism and spatial order in transition metal alloys: Experimental and theoretical aspects. Phys. Rep. 1987, 153, 331–399. [Google Scholar] [CrossRef]
- Cadeville, M.; Dahmani, C.; Kern, F. Magnetism and spatial order in Ni-Pt and Co-Pt alloys. J. Magn. Magn. Mater. 1986, 54–57, 1055–1056. [Google Scholar] [CrossRef]
- Sadhukhan, B.; Nayak, A.; Mookerjee, A. Effect of disorder on the optical response of NiPt and Ni3Pt alloys. Comput. Mater. Sci. 2017, 140, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Andreazza, P.; Pierron-Bohnes, V.; Tournus, F.; Andreazza-Vignolle, C.; Dupuis, V. Structure and order in cobalt/platinum-type nanoalloys: From thin films to supported clusters. Surf. Sci. Rep. 2015, 70, 188–258. [Google Scholar] [CrossRef]
- Alam, A.; Kraczek, B.; Johnson, D.D. Structural, magnetic, and defect properties of Co-Pt-type magnetic-storage alloys: Density-functional theory study of thermal processing effects. Phys. Rev. B 2010, 82, 024435. [Google Scholar] [CrossRef]
- Wen, Y.-H.; Zhang, L.-H.; Wang, J.-B.; Huang, R. Atomic-scale insights into thermal stability of Pt3Co nanoparticles: A comparison between disordered alloy and ordered intermetallics. J. Alloys Compd. 2019, 776, 629–635. [Google Scholar] [CrossRef]
- Shuttleworth, I.G. Magnetism in the strained ordered phases of PtxFe1−x and PtxCo1−x (x = 0.25, 0.5, and 0.75). J. Phys. Chem. Solids 2018, 114, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Makushko, P.V.; Verbytska, M.Y.; Shamis, M.N.; Verbytska, T.I.; Beddies, G.; Safonova, N.Y.; Albrecht, M.; Makogon, I.M. Effect of initial stress/strain state on the L10 phase formation of FePt in FePt/Au/FePt trilayers. Appl. Nanosci. 2019, 10, 2775–2780. [Google Scholar] [CrossRef]
- Shuttleworth, I.G. The effects of strain on the ordered phases of NixPt1−x (x = 0.25, 0.5, and 0.75). Chem. Phys. Lett. 2017, 689, 41–47. [Google Scholar] [CrossRef]
- Zhang, S.; Johnson, D.D.; Shelton, W.A.; Xu, Y. Hydrogen Adsorption on Ordered and Disordered Pt-Ni Alloys. Top. Catal. 2020, 63, 714–727. [Google Scholar] [CrossRef]
- Zhao, Z.; Fisher, A.; Shen, Y.; Cheng, D. Magnetic Properties of Pt-Based Nanoalloys: A Critical Review. J. Clust. Sci. 2016, 27, 817–843. [Google Scholar] [CrossRef]
- Karoui, S.; Amara, H.; Legrand, B.; Ducastelle, F. Magnetism: The driving force of order in CoPt, a first-principles study. J. Phys. Condens. Matter 2013, 25, 056005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunger, A.; Wei, S.-H.; Ferreira, L.G.; Bernard, J.E. Special quasirandom structures. Phys. Rev. Lett. 1990, 65, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Soven, P. Coherent-Potential Model of Substitutional Disordered Alloys. Phys. Rev. 1967, 156, 809–813. [Google Scholar] [CrossRef]
- Rowlands, D.A.; Ernst, A.; Györffy, B.L.; Staunton, J.B. Density functional theory for disordered alloys with short-range order: Systematic inclusion of charge-correlation effects. Phys. Rev. B 2006, 73, 165122. [Google Scholar] [CrossRef] [Green Version]
- Staunton, J.B.; Marmodoro, A.; Ernst, A. Using density functional theory to describe slowly varying fluctuations at finite temperatures: Local magnetic moments in Gd and the ‘not so local’ moments of Ni. J. Phys. Condens. Matter 2014, 26, 274210. [Google Scholar] [CrossRef] [Green Version]
- Guevara, U.; López, R.; Blanco, J.; Núñez, J.; Hernández, U.J.G. Theoretical study of electronic properties and spin density in Pt-Co alloys. Mater. Res. Express 2019, 6, 096514. [Google Scholar] [CrossRef]
- Alsaad, A.; Ahmad, A.; Qattous, H.A. Structural, electronic and magnetic properties of MxPt1−X, (M = Co, Ni and V) binary alloys. Heliyon 2019, 5, e02433. [Google Scholar] [CrossRef]
- Shuttleworth, I.G. The quasiparticle band structures of ordered NixPt1−x. Heliyon 2018, 4, e01000. [Google Scholar] [CrossRef]
- Pisanty, A.; Amador-Bedolla, C.; Ruiz, Y.; De La Vega, M. Band structures of Ni3Pt and NiPt3. Eur. Phys. J. B 1990, 80, 237–239. [Google Scholar] [CrossRef]
- Yonezawa, F.; Morigaki, K. Coherent Potential Approximation. Prog. Theor. Phys. Suppl. 1973, 53, 1–76. [Google Scholar] [CrossRef] [Green Version]
- Sternik, M.; Couet, S.; Łażewski, J.; Jochym, P.; Parlinski, K.; Vantomme, A.; Temst, K.; Piekarz, P. Dynamical properties of ordered Fe–Pt alloys. J. Alloys Compd. 2015, 651, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Pearson, W.B. A Handbook of Lattice Spacings and Structures of Metals and Alloys; Pergamon Press Ltd.: Oxford, UK, 1964. [Google Scholar]
- Leroux, C.; Cadeville, M.C.; Pierron-Bohnes, V.; Inden, G.; Hinz, F. Comparative investigation of structural and transport properties of L10NiPt and CoPt phases; the role of magnetism. J. Phys. F Met. Phys. 1988, 18, 2033–2051. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corso, A.D. Pseudopotentials periodic table: From H to Pu. Comput. Mater. Sci. 2014, 95, 337–350. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Alloy | Phase | a | c | ||
|---|---|---|---|---|---|
| Theory | Exptl | Theory | Exptl | ||
| Pt0.75Fe0.25 | L12 | 3.911 | 3.866 [33] | - | - |
| Pt0.50Fe0.50 | L10 | 3.894 | 3.852 [33] | 3.705 | 3.713 [33] |
| Pt0.25Fe0.75 | L12 | 3.740 | 3.750 [33] | - | - |
| Pt0.75Co0.25 | L12 | 3.890 | 3.831 [34] | - | - |
| Pt0.50Co0.50 | L10 | 3.817 | 3.810 [35] 3.812 [34] | 3.727 | 3.710 [35] 3.708 [34] |
| Pt0.25Co0.75 | L12 | 3.666 | 3.66 [34] | - | - |
| Pt0.75Ni0.25 | L12 | 3.878 | 3.837 [33] | - | - |
| Pt0.50Ni0.50 | L10 | 3.895 | 3.840 [35] | 3.540 | 3.610 [35] |
| Pt0.25Ni0.75 | L12 | 3.657 | 3.646 [33] | - | - |
| Alloy | Phase | μPt | μM |
|---|---|---|---|
| Pt0.75Fe0.25 | L12 | 0.378 (0.388) | 3.309 (3.386) |
| Pt0.50Fe0.50 | L10 | 0.408 (0.400) | 3.007 (3.051) |
| Pt0.25Fe0.75 | L12 | 0.401 (0.381) | 2.762 (2.677) |
| Pt0.75Co0.25 | L12 | 0.337 (0.324) | 2.041 (2.135) |
| Pt0.50Co0.50 | L10 | 0.423 (0.411) | 1.965 (1.908) |
| Pt0.25Co0.75 | L12 | 0.406 (0.386) | 1.848 (1.838) |
| Pt0.75Ni0.25 | L12 | 0.168 (0.161) | 0.749 (0.712) |
| Pt0.50Ni0.50 | L10 | 0.376 (0.387) | 0.792 (0.762) |
| Pt0.25Ni0.75 | L12 | 0.360 (0.364) | 0.752 (0.764) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shuttleworth, I. The Magnetic Band-Structures of Ordered PtxFe1−x, PtxCo1−x, and PtxNi1−x (x = 0.25, 0.50, and 0.75). Magnetochemistry 2020, 6, 61. https://doi.org/10.3390/magnetochemistry6040061
Shuttleworth I. The Magnetic Band-Structures of Ordered PtxFe1−x, PtxCo1−x, and PtxNi1−x (x = 0.25, 0.50, and 0.75). Magnetochemistry. 2020; 6(4):61. https://doi.org/10.3390/magnetochemistry6040061
Chicago/Turabian StyleShuttleworth, Ian. 2020. "The Magnetic Band-Structures of Ordered PtxFe1−x, PtxCo1−x, and PtxNi1−x (x = 0.25, 0.50, and 0.75)" Magnetochemistry 6, no. 4: 61. https://doi.org/10.3390/magnetochemistry6040061
APA StyleShuttleworth, I. (2020). The Magnetic Band-Structures of Ordered PtxFe1−x, PtxCo1−x, and PtxNi1−x (x = 0.25, 0.50, and 0.75). Magnetochemistry, 6(4), 61. https://doi.org/10.3390/magnetochemistry6040061