1. Introduction
The characterization of rare cells has become a major topic in applied research since addressing personalized medicinal approaches is seen as the future to oppose complex pathologies. For example, the enrichment, detection and isolation of circulating tumor cells (CTCs) reveals crucial information on prognostic [
1], diagnostic [
2] and even therapeutic aspects [
3] that can support treatment decision-making in oncology. It is known that CTCs detach from the primary epithelial tumor tissue and intravasate into the cardiovascular system to foster secondary tumor formation as disease-driving precursor at another site [
4].
For malignant neoplasia of the breast tissue in particular, liquid biopsy of biomarkers such as cell-free circulating tumor DNA (ctDNA), exosomes and CTCs has attracted immense interest in the scientific community. Based on the current incidence, one out of eight women will be diagnosed with breast cancer throughout her life. Moreover, one in six of these women will be younger than 50 years at the time of the diagnosis. Although breast cancer is by far the most common solid tumor type in females, it has a relatively high 10-year survival rate of 83% when diagnosed at early stage, ideally prior to metastasis, and treated appropriately [
5]. Comparable to most cancers, the systemic spread of secondary tumors (metastasis) is the major cause of death due to multi-organ failure or opportunistic infections in metastatic breast cancer.
Since the hormone receptor status and surface antigen distribution among different subtypes of breast cancer occupy a critical role for disease invasiveness, tumor burden and therapy decision, the characterization of CTC biomarker features at the point-of-care (PoC) has become a subject of intense research. An association of CTCs with poor prognosis due to metastasis could already be shown in breast cancer as well as in various other cancers. More detailed, the 3-year survival of patients with triple negative breast cancer (TNBC) is significantly higher at prior-surgery CTC counts below 5 cells in 7.5 mL whole blood [
6]. TNBC is highly invasive due to cancer cells that lost expression of estrogen, progesterone and human epidermal growth factor receptors ESR, PGR and HER-2 [
7] and is hence especially difficult to tackle without hormone therapy with a strong tendency to therapy-resistant metastization [
8].
Besides size exclusion, magnetic cell separation has become a key technique for the isolation of CTC subpopulations from biological suspensions like blood and leucapheresis products. However, improvement in selectivity, yield, and sensitivity of the separation protocols with cost reduction and standardization is still needed to fully implement magnetic cell separation in breast cancer diagnostics or therapeutic decision-making. Magnetic cell separation of CTCs from complex body fluids like whole blood often results in high background signal of white blood cell contamination (1:1000) hampering downstream molecular analysis. Interdisciplinary research to combine immunomagnetic separation, microfluidic cell sorting and system engineering in biological validation can tackle the challenge to lower the signal-to-noise ratio. We developed an automated benchtop platform (CTCelect) for the single cell isolation of CTCs using immunomagnetic enrichment, chip-based microfluidic cell sorting and single cell dispensing in droplets on a 96-well plate to overcome these current limitations. The device was characterized in-depth and optimized in our previous works [
9,
10]. The platform consists of an immunomagnetic enrichment module and a microfluidic fluorescence-activated cell sorting subunit with a disposable cartridge (see
Supplementary Figure S1). Fluid control and sample transfer is managed by a pipetting robot and syringe pumps. Epithelial cell adhesion molecule (EpCAM)-positive selection of tumor cells from a 7.5 mL sample with subsequent depletion of leukocytes takes place in the enrichment module of the device. The enriched and stained sample is then transferred to the cell sorting subunit into the microfluidic chip for detection and single cell dispensing in microliter droplets.
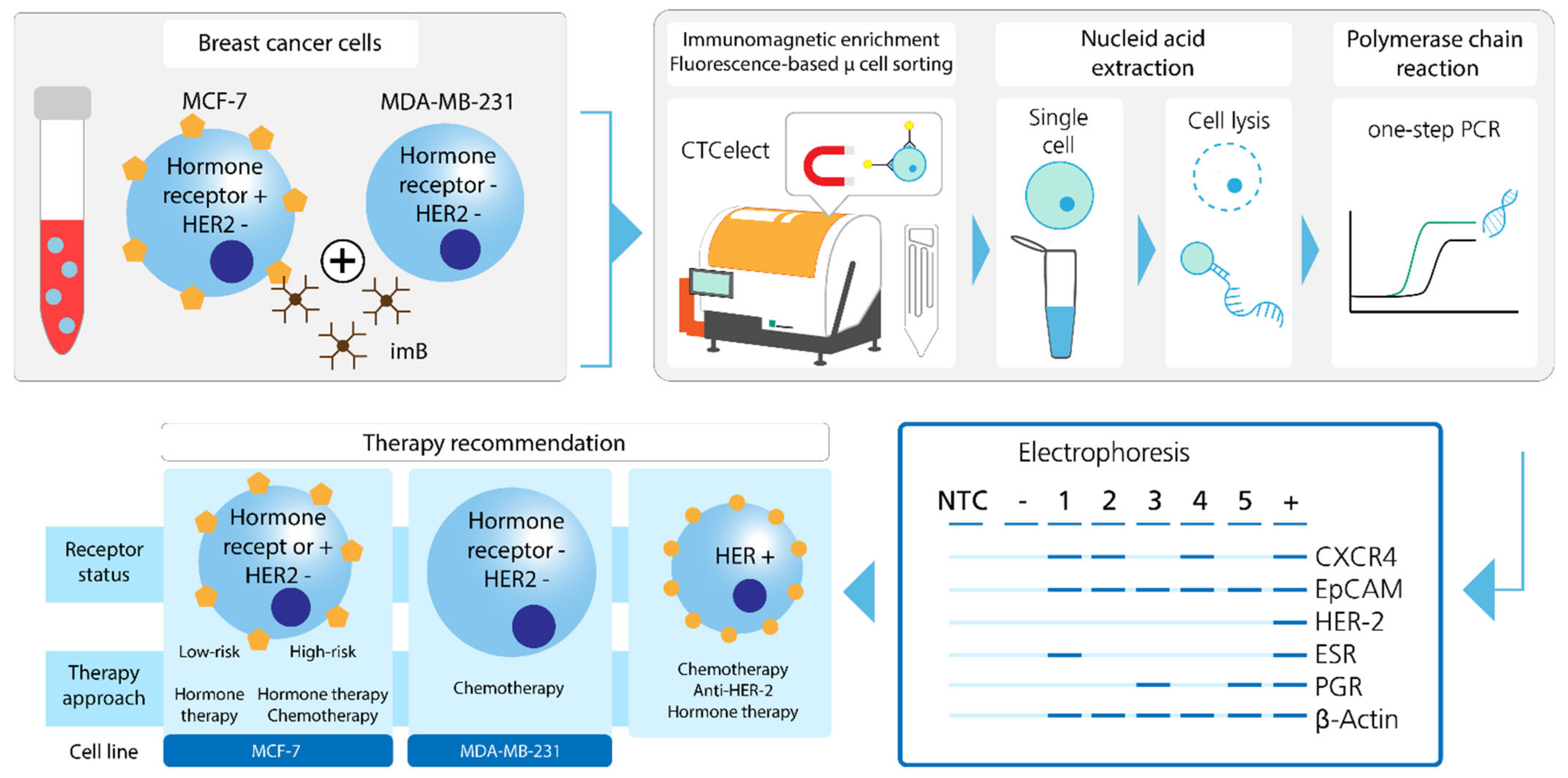
In this study, we investigated the flow properties of magnetic bead-bound cells in the microfluidic demonstrator and highlight the bottlenecks of immunomagnetic microfluidic cell separation respectively. Additionally, we showcase a proof-of-concept for a simplified detection and distinction of hormone-related tumor markers in single hormone receptor-positive and triple-negative breast cancer cells developing a protocol for combining automated cell enrichment, detection and dispensing with one-step PCR analyses (
Figure 1). These results contribute to sharpen the overall picture of an individual cancer pathology and to pave the way towards additional therapy suggestions from liquid biopsy.
3. Discussion
CTCs are systemic tumor components and hence are of high clinical value, not only to access minimal-invasive tumor components but also to correlate the cell count with disease staging and monitor treatment efficacy as personalized measure. An additional benefit of isolating CTCs from liquid samples is that the molecular profile is obtained from relevant single tumor cells that include all information on RNA and DNA level and is not highly diluted in the blood-like cell-free tumor DNA. Thus, the isolation of single tumor cells can be of great advantage for personalized therapies. However, current isolation methods are error-prone, laborious and cost intensive precluding CTC analysis for larger patient cohorts. Immunomagnetic separation is widely used to pre-enrich target CTCs from body fluids. We can use this pre-enrichment step in combination with an automated microfluidic detection and dispensing process to further purify the cells of interest from contaminating blood cells. Thus, the immunomagnetic enrichment step requires accuracy, specificity and adaptability to the needs of the microfluidic detection and dispensing step. By doing that, we are able to combine both processes in one automated benchtop device. In previous publications, the advantages and disadvantages of our process were qualitatively compared to existing methods [
9,
10].
We knew that certain obstacles arise by introducing relatively dense magnetic particles in microfluidic systems. The aim of this study was to adapt the immunomagnetic enrichment and handling of the pre-enriched cell-bead suspension. Even though 1 µm magnetic beads have a relatively low sedimentation rate which makes them useable for automated isolations, they still gravitate after a certain period of time, especially in low-density aqueous solutions. This is this reason why we formulated a polysaccharide buffer that prevented visible bead sedimentation for at least 30 min (
Figure 2A) enabling improved sample flow and consequently the overall recovery rate of the isolated cells. Effects of fluid inertia and viscosity on the settling of particles in a viscous buffer are described in detail in the literature [
12,
13]. Glycerol as an agent to prevent the sinking of beads was only successful using a 100% concentrated solution in our investigations. Studies report an already significant cytotoxic influence on mammalian cell volume due to osmotic pressure and decreased viability in 3.5% concentrated glycerol [
14], excluding this solution as potential transfer buffer for microfluidic CTC isolation.
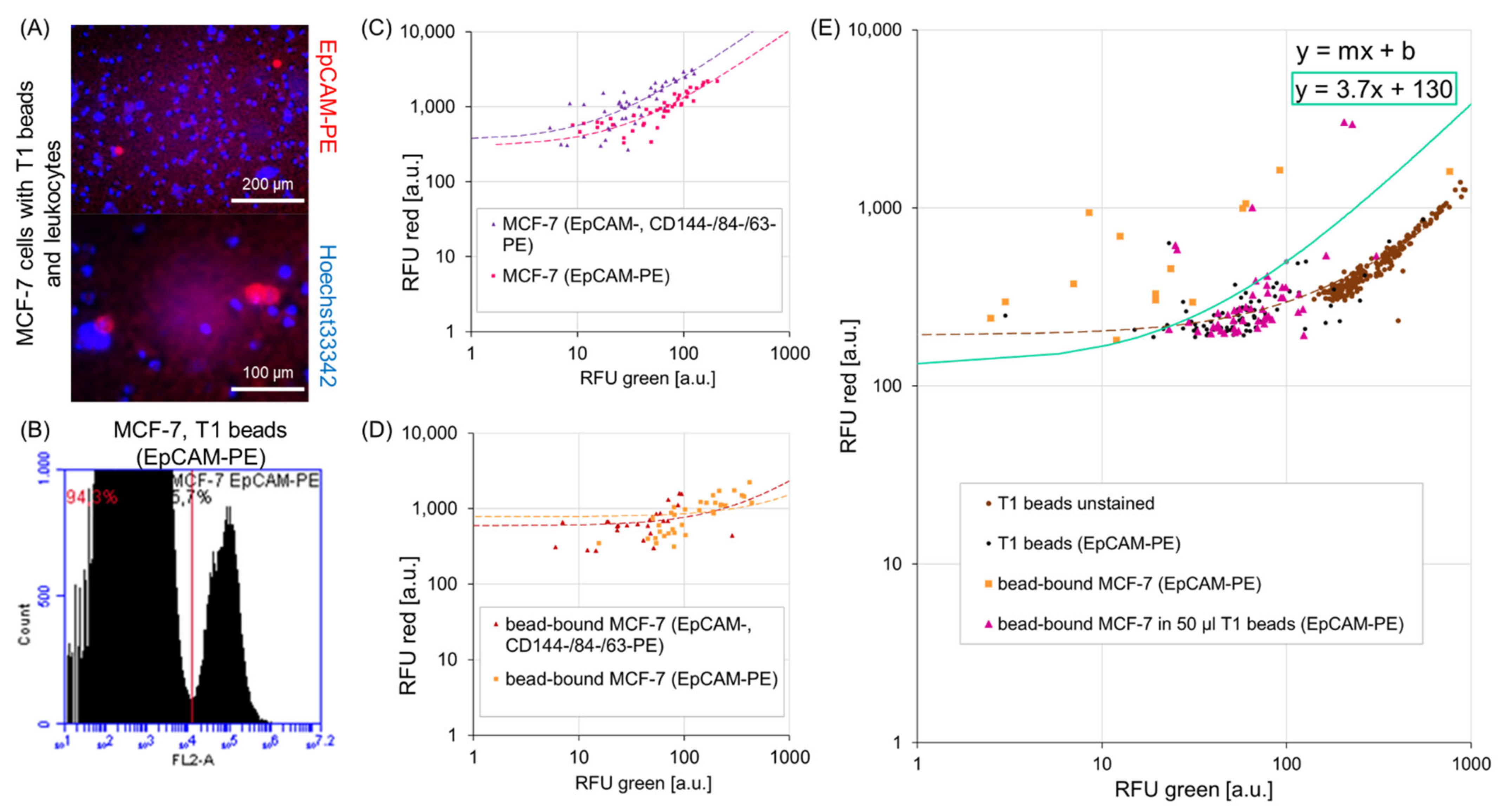
In addition, when working with high proportions of excess magnetic beads, a few effects hamper fluorescence-based cell detection. For example, Andree et al. showed that in flow cytometry an unstained population overlaps with the fluorescence signal of a target cell population due to uneven probability distributions. A ratio of 1:1000 between a wanted (stained) and an unwanted (unstained) population already led to an overlap of 48.9% in fluorescence intensity [
15]. On one hand, they block epitopes for antibody staining and quench the fluorescence on the cell surface (see
Supplementary Figure S2). On the other hand, beads exhibit a certain autofluorescence at 488 nm excitation as shown on the microscopic image in
Figure 4A and described, i.e., by Roth et al. [
16]. If not mixed properly or due to suboptimal buffers, beads may form agglomerates and falsify the detection process. We observed these influences in our experiments and adjusted the parameters accordingly to provide fluorescence-based single cell dispensing. These effects are more severe using antibody staining which generally emits lower fluorescence than synthetic dyes and relies on EpCAM expression only. In the literature, it was stated that a cocktail of fluorescent-labelled antibodies could improve the detection of cells [
17]. With our detection set-up, we could not confirm a clear advantage of using a mixture of PE-conjugated antibodies to enhance fluorescence intensity of bead bound cells. For this purpose we developed a gating function that was implemented in the CTCelect software. We managed a distinction between the autofluorescent beads population and the fluorescent cell population to selectively trigger cell dispensing (
Figure 4E).
Finally, molecular analysis of isolated tumor cells was optimized and accelerated by means of one-step qPCR to detect hormone-related breast cancer-associated targets. We found that under the same conditions, the efficacy of the one-step PCR method is not as high as that of the two-step method. Nevertheless, it was possible to successfully display the hormone receptor status of hormone receptor positive, Her-2
− MCF-7 cells (as defined by [
18,
19]) on a single cell level with one-step PCR after complete CTCelect enrichment, detection and dispensing (
Figure 5C,E). Additionally, a comparable concept was developed for triple negative MDA-MB-231 cells which will be further investigated in the future (
Figure 5D). Eventually, it should be the user’s choice to perform one- or two-step PCR depending on time constraints, transcriptomic frequency and target number. Multiplexing in single cell PCR is undoubtedly limited by low sample input, lysis buffer dilution and RNA degradation. The 4-fold multiplexing Adnatest was not applicable using 1:4 diluted unpurified RNA from <20 cells (
Figure 5C,E). In comparison with column-based nucleic acid extraction, chemical lysis also risks potential sample loss by inefficient pipetting. However, by keeping lysis buffer volumes low and therefore RNA concentrations high, an assay of up to 3 cancer- or hormone-associated targets from one cell was realized (
Figure 5E).
From a clinical point of view, the hormone receptor status is a key checkpoint in breast cancer diagnostics as hospital guidelines suggest hormone and/or antibody therapy depending on a positive receptor status, replacing or accompanied by radio-chemotherapy. Without axillary lymph node infiltration, stage I breast cancer has high curative chances by mastectomic resection and prophylactic hormone therapy only. In the opposite viewpoint, metastatic breast cancer is characterized by lymph node, bone, lung, brain or liver invasion and the respective therapy contains harsh radiation and cytotoxic chemical agents with severe side-effects for the patient. Not least because of that, personalized and, above all, correct therapy administration has become a major topic in basic and clinical research. Alix-Panabières and Pantel have impressively reviewed that CTC characterization can provide answers to these in-depth questions [
20]. The isolation of ctDNA for Next Generation Sequencing is exploited and extensively evolved to monitor cancer dynamics [
21]. Improved single tumor cell isolation as shown in the present study might be useful to overcome limitations in the clinical use of these methodologies due to standardization in automation and lowered costs. In theory, it is also possible to use the residual blood sample for ctDNA analysis after the pre-enrichment of CTCs with the workflow of the CTCelect benchtop system or separate the plasma from the cell phase beforehand. This would enable a parallel isolation of ctDNA and CTCs to provide encompassing diagnostics from the same sample in a clinical setting or for central laboratories. Although magnetic cell separation is a straight-forward, combinable and automatable technique, commercial bead systems mostly use batch analysis (PCR) for tumor markers. The herein presented combination-of-combinations principle could reduce background signal for detailed examination of molecular tumor features.
To conclude, our improved protocol to directly isolate and amplify single cell RNA allows for a swift single CTC analysis resulting from immunomagnetic enrichment in combination with microfluidic dispensing, highlighting the potential for future therapeutic decision-making.
4. Materials and Methods
If not otherwise specified, reagents and supplements were purchased at Thermo Fisher Scientific, Darmstadt, DE.
4.1. Cell Lines and Blood Samples
The MCF-7 cell line was purchased from AdnaGen (Langenhagen, Germany) and cultured in RPMI1640 medium with L-Glutamine (Capricorn Scientific, Ebsdorfergrund, Germany) supplemented with 10% fetal calf serum (FCS; Merck, Darmstadt, Germany). MDA-MB-231 cells were kindly provided by Dr. Pierpaolo Moscariello (Max-Planck-Institute for Polymer Research, Mainz, Germany) and cultured in Gibco™ DMEM (low glucose, pyruvate) medium supplemented with 10% FCS. Cell lines were split at subconfluence and incubated at 37 °C in a humidified atmosphere in the presence of 5% CO2.
Whole blood bags from healthy donors were purchased from the local Blood Transfusion Center (University Medical Center Mainz, Germany) in 500 mL CompoFlex® blood bags (Fresenius Kabi, Bad Homburg, Germany) with CPD-1 anticoagulant and stored at room temperature for a maximum of three days. Informed consent was obtained from the donors as usual in clinical blood donation guidelines.
4.2. Immobilization of Biotinylated Antibodies on Streptavidin Coupled Magnetic Microbeads
Dynabeads™ MyOne™ Streptavidin T1 superparamagnetic beads were tumor-specifically coated with biotinylated monoclonal mouse anti-human EpCAM (CD326; 20 µg/mL) antibody 1B7. EpCAM antibody was immobilized on the bead surface with an extended incubation time of 1 h. After immobilization, a saturated biotin/PBS solution was added for 30 min with gentle rotation of the tube to block free streptavidin binding sites and prevent clumping. After an additional washing step, anti-EpCAM magnetic beads (herein abbreviated as “beads”) were stored in PBS/0.1% BSA at 4 °C for several weeks.
4.3. Bead Sedimentation Test
Different solutions of PBS, 0.125, 0.05 and 0.01% polysaccharide transfer buffer (TB, subject to confidentiality), 23, 50 and 100% glycerol and 23% glycerol/5% mannitol were produced. An amount of 5 × 109/mL 1 µm beads were added to 1 mL test solution in a reaction tube, respectively. The visual sedimentation was observed over the course of 30 min.
4.4. Spike-in Experiments and Automated CTCelect Single Cell Isolation of Tumor Cells
Spike-in experiments and automated CTCelect assays were performed as described in [
9]. Cell recovery rates were determined using fluorescence microscopy. CFSE staining was conducted as directed in the CellTrace
TM CFSE Cell Proliferation kit manual.
4.5. Immunostaining of Pre-Enriched Samples
Immunomagnetically enriched samples were stained with monoclonal antibody EpCAM (1B7)-, CD144-, CD84-, CD63-PE eBioscience™ antibody (1:30) to label epithelial cells and with nuclear dye Hoechst33342 (1:1000) for 10 min at room temperature. Washing steps were performed in a magnetic separator. Stained samples were processed in the microfluidic chip of the CTCelect device or measured using the Accuri C6 flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
4.6. Nucleic Acid Extraction and Single Cell RT-qPCR of Isolated Tumor Cells
To confirm cancer-relevant mRNAs in cell bulks and in single cell isolates, two-step and one-step real time quantitative PCR (RT-qPCR) was performed. β-actin (RPLP0) served as housekeeping RNA control. RNA was extracted from dispensed droplets immediately after CTCelect isolation using 2× or 10× guanidine salt-free lysis buffer (LB; 1:1 to the droplet volume/1:9 in pooled samples) or transferred in purification columns using either the RNeasy Micro kit or AllPrep DNA/mRNA Nano kit (both QIAGEN, Hilden, DE) by following the manuals of the manufacturer. Elution was performed at the lowest volume possible to maintain highest RNA concentrations. For two-step protocols, total RNA was reverse transcribed into cDNA with the SensiFAST™ cDNA Synthesis kit (Meridian Bioscience, Luckenwalde, Germany) and qPCR was conducted using 5 µL cDNA and QuantiFast SYBR Green RT-PCR kit (QIAGEN, Hilden, DEGermanyin a 96 well cycler (BioRad CFX96 Touch Real-Time PCR Detection System, Feldkirchen, Germany). One-step qPCR was performed by means of the SensiFAST™ Probe No-ROX One-Step kit (Meridian Bioscience, Luckenwalde, Germany) whereas reverse transcription was extended to 20 min and extension time was set to 10 s. BreastCancer Detect Adnatest (QIAGEN, Hilden, Germany) primers were added as described in the manual. Threshold cycle values (C
T) were obtained. Primers are available in the
Supplementary Materials (see
Table S1) and were designed with Primer-BLAST (National Institutes of Health, Bethesda, MD, USA;
https://www.ncbi.nlm.nih.gov/tools/primer-blast/ accessed on 29 July 2022). Primer sequences of the Adnatest are non-disclosed by QIAGEN.
4.7. Gel Electrophoresis and Bioanalyzer
To analyze PCR product sizes, gel electrophoresis and automated electrophoresis using the Bioanalyzer (Agilent Technologies) was performed. For gel electrophoresis, we used a 2% agarose gel in tris-acetate-EDTA buffer. DNA was diluted 1:1 in DEPC H2O, added 1:5 6× Orange G loading dye per sample and run for 45 min at 100 V. Ladders were used at 5 µL (100 bp DNA Ladder, puC19 Ladder, both Carl Roth, Karlsruhe, Germany). Gels were displayed in the Fujifilm LAS-3000 Luminescent Image Analyzer. Automated electrophoresis in the Bioanalyzer was performed using the Agilent DNA 1000 kit as indicated in the manual.
4.8. Statistical Analyses
Each experiment was repeated as indicated in the text, figures or legends. CTCelect data were collected from the output files of the software and further analyzed using Microsoft Excel 2016. Data are depicted as means with SD, and statistical analysis was done using GraphPad PRISM 8.2.0 for Windows (GraphPad Software, San Diego, CA, USA,
www.graphpad.com). The
p values of the statistical tests given in the respective figures were reported as not significant (ns) when P
ns > 0.05 and as significant when *
p ≤ 0.05.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




