
Metal Substitution versus Oxygen-Storage Modifier to Regulate the Oxygen Redox Reactions in Sodium-Deficient Three-Layered Oxides



Abstract
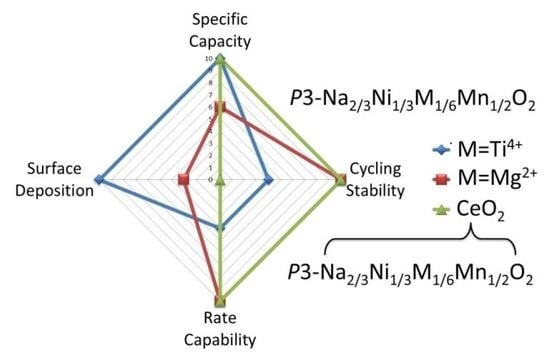
:
1. Introduction
2. Results and Discussions
2.1. Structure of Metal-Substituted and CeO2-Treated Oxides
2.2. Electrochemical Behavior of Metal-Substituted and CeO2-Treated Oxides
2.2.1. Sodium-Ion Cells
2.2.2. Lithium-Ion Cells
2.3. Surface Deposition after Na+ and Li+ Intercalation
2.3.1. Sodium Electrolyte
2.3.2. Lithium Electrolyte
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gür, T.M. Review of electrical energy storage technologies, materials and systems: Challenges and prospects for large-scale grid storage. Energy Environ. Sci. 2018, 11, 2696–2767. [Google Scholar] [CrossRef]
- Whittingham, M.S. Special editorial perspective: Beyond Li-ion battery chemistry. Chem. Rev. 2020, 120, 6328–6330. [Google Scholar] [CrossRef] [PubMed]
- Abraham, K.M. How comparable are sodium-ion batteries to lithium-ion counterparts? ACS Energy Lett. 2020, 5, 3544–3547. [Google Scholar] [CrossRef]
- Rahman, M.M.; Lin, F. Oxygen redox chemistry in rechargeable Li-ion and Na-ion batteries. Matter 2021, 4, 490–527. [Google Scholar] [CrossRef]
- Lee, G.; Lau, V.W.; Yang, W.; Kang, Y. Utilizing oxygen redox in layered cathode materials from multiscale perspective. Adv. Energy Mater. 2021, 11, 2003227. [Google Scholar] [CrossRef]
- Delmas, C. Sodium and sodium-ion batteries: 50 years of research. Adv. Energy Mater. 2018, 8, 1703137. [Google Scholar] [CrossRef]
- Koga, H.; Croguennec, L.; Ménétrier, M.; Mannessiez, P.; Weill, F.; Delmas, C. Different oxygen redox participation for bulk and surface: A possible global explanation for the cycling mechanism of Li1.20Mn0.54Co0.13Ni0.13O2. J. Power Sources 2013, 236, 250–258. [Google Scholar] [CrossRef]
- He, W.; Guo, W.; Wu, H.; Lin, L.; Liu, Q.; Han, X.; Xie, Q.; Liu, P.; Zheng, H.; Wang, L.; et al. Challenges and recent advances in high capacity Li-rich cathode materials for high energy density lithium-ion batteries. Adv. Mater. 2021, 33, 2005937. [Google Scholar] [CrossRef]
- De La Llave, E.; Talaie, E.; Levi, E.; Nayak, P.K.; Dixit, M.; Rao, P.T.; Hartmann, P.; Chesneau, F.; Major, D.T.; Greenstein, M.; et al. Improving Energy Density and Structural Stability of Manganese Oxide Cathodes for Na-Ion Batteries by Structural Lithium Substitution. Chem. Mater. 2016, 28, 9064–9076. [Google Scholar] [CrossRef]
- Ma, C.; Alvarado, J.; Xu, J.; Clément, R.J.; Kodur, M.; Tong, W.; Grey, C.P.; Meng, Y.S. Exploring oxygen activity in the high energy P2-type Na0.78Ni0.23Mn0.69O2 cathode material for Na-ion batteries. J. Am. Chem. Soc. 2017, 139, 4835–4845. [Google Scholar] [CrossRef] [Green Version]
- Maitra, U.; House, R.A.; Somerville, J.W.; Tapia-Ruiz, N.; Lozano, J.G.; Guerrini, N.; Hao, R.; Luo, K.; Jin, L.; Pérez-Osorio, M.A.; et al. Oxygen redox chemistry without excess alkali-metal ions in Na2/3[Mg0.28Mn0.72]O2. Nat. Chem. 2018, 10, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Sathiya, M.; Mendoza-Sánchez, B.; Iadecola, A.; Vergnet, J.; Dedryvère, R.; Saubanère, M.; Abakumov, A.M.; Rozier, P.; Tarascon, J.M. Anionic redox activity in a newly Zn-doped sodium layered oxide P2-Na2/3Mn1−y Zn y O2 (0 < y < 0.23). Adv. Energy Mater. 2018, 8, 1802379. [Google Scholar]
- Wang, Y.; Kim, S.; Lu, J.; Feng, G.; Li, X. A study of Cu doping effects in P2-Na0.75Mn0.6Fe0.2(CuxNi0.2-x)O2 layered cathodes for sodium-ion batteries. Batter. Supercaps 2020, 3, 376–387. [Google Scholar] [CrossRef]
- Zhao, C.; Yao, Z.; Wang, J.; Lu, Y.; Bai, X.; Aspuru-Guzik, A.; Chen, L.; Hu, Y.-S. Ti Substitution facilitating oxygen oxidation in Na2/3Mg1/3Ti1/6Mn1/2O2 cathode. Chem. 2019, 5, 2913–2925. [Google Scholar] [CrossRef]
- Mortemard de Boisse, B.; Nishimura, S.-I.; Watanabe, E.; Lander, L.; Tsuchimoto, A.; Kikkawa, J.; Kobayashi, E.; Asakura, D.; Okubo, M.; Yamada, A. Highly reversible oxygen-redox chemistry at 4.1 V in Na4/7–x[□1/7Mn6/7]O2 (□: Mn vacancy). Adv. Energy Mater. 2018, 8, 1800409. [Google Scholar] [CrossRef]
- Kubota, K.; Komaba, S. Review-practical issues and future perspective for Na-ion batteries. J. Electrochem. Soc. 2015, 162, A2538–A2550. [Google Scholar] [CrossRef]
- Xu, J.; Sun, M.; Qiao, R.; Renfrew, S.; Ma, L.; Wu, T.; Hwang, S.; Nordlund, D.; Su, D.; Amine, K.; et al. Elucidating anionic oxygen activity in lithium-rich layered oxides. Nat. Commun. 2018, 9, 947. [Google Scholar] [CrossRef]
- Lee, J.; Papp, J.K.; Clément, R.J.; Sallis, S.; Kwon, D.-H.; Shi, T.; Yang, W.; McCloskey, B.; Ceder, G. Mitigating oxygen loss to improve the cycling performance of high capacity cation-disordered cathode materials. Nat. Commun. 2017, 8, 981. [Google Scholar] [CrossRef]
- Zhou, K.; Zheng, S.; Ren, F.; Wu, J.; Liu, H.; Luo, M.; Liu, X.; Xiang, Y.; Zhang, C.; Yang, W.; et al. Fluorination effect for stabilizing cationic and anionic redox activities in cation-disordered cathode materials. Energy Storage Mater. 2020, 32, 234–243. [Google Scholar] [CrossRef]
- Li, Z.; Kong, W.; Yu, Y.; Zhang, J.; Wong, D.; Xu, Z.; Chen, Z.; Schulz, C.; Bartkowiak, M.; Liu, X. Tuning bulk O2 and nonbonding oxygen state for reversible anionic redox chemistry in P2-layered cathodes. Angew. Chem. Int. Ed. Engl. 2022, 134, e202115552. [Google Scholar]
- Kaur, G.; Gates, B.D. Review—Surface coatings for cathodes in lithium ion batteries: From crystal structures to electro-chemical performance. J. Electrochem. Soc. 2022, 169, 043504. [Google Scholar] [CrossRef]
- Nisar, U.; Muralidharan, N.; Essehli, R.; Amin, R.; Belharouak, I. Valuation of surface coatings in high-energy density lithium-ion battery cathode materials. Energy Storage Mater. 2021, 38, 309–328. [Google Scholar] [CrossRef]
- Shi, C.; Wang, L.; Chen, X.; Wang, S.; Wang, J.; Jin, H. Challenges of layer-structured cathodes for sodium-ion bat-teries. Nanoscale Horiz. 2022, 7, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-Y.; Myung, S.-T.; Choi, J.U.; Yoon, C.S.; Yashiro, H.; Sun, Y.-K. Resolving the degradation pathways of the O3-type layered oxide cathode surface through the nano-scale aluminum oxide coating for high-energy density sodium-ion batteries. J. Mater. Chem. A 2017, 5, 23671–23680. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, K.; Li, X.; Yu, R.; Zhang, X.; Huang, Y.; Chen, G.; Jamil, S.; Cao, S.; Xie, X.; et al. Improved cycle and air stability of P3-Na0.65Mn0.75Ni0.25O2 electrode for sodium-ion batteries coated with metal phosphates. Chem. Eng. J. 2019, 372, 1066–1076. [Google Scholar] [CrossRef]
- Yu, Y.; Kong, W.; Li, Q.; Ning, D.; Schuck, G.; Schumacher, G.; Su, C.; Liu, X. Understanding the multiple effect of TiO2 coating on NaMn0.33Fe0.33Ni0.33O2 cathode material for Na-ion batteries. ACS Appl. Energy Mater. 2020, 3, 933–942. [Google Scholar] [CrossRef] [Green Version]
- House, R.A.; Maitra, U.; Jin, L.; Lozano, J.G.; Somerville, J.W.; Rees, N.H.; Naylor, A.J.; Duda, L.C.; Massel, F.; Chadwick, A.V.; et al. What triggers oxygen loss in oxygen redox cathode materials? Chem. Mater. 2019, 31, 3293–3300. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Pei, Y.; Chen, H.; Song, Y.; Zhen, L.; Xu, C.-Y.; Xiao, P.; Henkelman, G. Highly reversible oxygen redox in layered compounds enabled by surface polyanions. Nat. Commun. 2020, 11, 3411. [Google Scholar] [CrossRef]
- Hwang, J.-Y.; Yu, T.-Y.; Sun, Y.-K. Simultaneous MgO coating and Mg doping of NaNi0.5Mn0.5O2 cathode: Facile and customizable approach to high-voltage sodium-ion batteries. J. Mater. Chem. A 2018, 6, 16854–16862. [Google Scholar] [CrossRef]
- Shin, Y.; Kan, W.H.; Aykol, M.; Papp, J.K.; McCloskey, B.D.; Chen, G.; Persson, K.A. Alleviating oxygen evolution from Li-excess oxide materials through theory-guided surface protection. Nat. Commun. 2018, 9, 4597. [Google Scholar] [CrossRef] [Green Version]
- Kalapsazova, M.L.; Kostov, K.L.; Kukeva, R.R.; Zhecheva, E.N.; Stoyanova, R.K. Oxygen-storage materials to stabilize the oxygen redox activity of three-layered sodium transition metal oxides. J. Phys. Chem. Lett. 2021, 12, 7804–7811. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Hu, E.; Liu, J.; Zhang, Y.; Yang, X.-Q.; Nanda, J.; Huq, A.; Page, K. A novel P3-type Na2/3Mg1/3Mn2/3O2 as high capacity sodium-ion cathode using reversible oxygen redox. J. Mater. Chem. A 2019, 7, 1491–1498. [Google Scholar] [CrossRef]
- Kalapsazova, M.; Markov, P.; Kostov, K.; Zhecheva, E.; Nihtianova, D.; Stoyanova, R. Controlling at elevated temperature the sodium intercalation capacity and rate capability of P3-Na2/3Ni1/2Mn1/2O2 through the selective substitution of nickel with magnesium. Batter. Supercaps 2020, 3, 1329–1340. [Google Scholar] [CrossRef]
- Lee, J.; Koo, S.; Lee, J.; Kim, D. Rational design of Ti-based oxygen redox layered oxides for advanced sodium-ion batteries. J. Mater. Chem. A 2021, 9, 11762–11770. [Google Scholar] [CrossRef]
- Yang, J.; Maughan, A.E.; Teeter, G.; de Villers, B.J.T.; Bak, S.; Han, S. Structural stabilization of P2-type sodium iron manganese oxides by electrochemically inactive Mg substitution: Insights of redox behavior and voltage decay. ChemSusChem 2020, 13, 5972–5982. [Google Scholar] [CrossRef] [PubMed]
- Stansby, J.H.; Sharma, N.; Goonetilleke, D. Probing the charged state of layered positive electrodes in sodium-ion batteries: Reaction pathways, stability and opportunities. J. Mater. Chem. A 2020, 8, 24833–24867. [Google Scholar] [CrossRef]
- Zhu, Y.-F.; Xiao, Y.; Dou, S.-X.; Chou, S.-L. Dynamic structural evolution and controllable redox potential for abnormal high-voltage sodium layered oxide cathodes. Cell Rep. Phys. Sci. 2021, 2, 100631. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, W.; Wang, W.; Wang, S.; Li, B. Comprehensive review of P2-type Na2/3Ni1/3Mn2/3O2, a potential cathode for practical application of Na-ion batteries. ACS Appl. Mater. Interfaces 2019, 11, 22051–22066. [Google Scholar] [CrossRef]
- Yosida, T.; Aoki, H.; Takeuchi, H.; Arakawa, M.; Horai, K. EPR, 19F-ENDOR and 55Mn-ENDOR of Mn2+ impurity center in MgF2 single crystal. J. Phys. Soc. Jpn. 1991, 60, 625–635. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, L.; Li, L.; Xie, M.; Wu, F.; Chen, R. Electrolytes and electrolyte/electrode interfaces in sodium-ion batteries: From scientific research to practical application. Adv. Mater. 2019, 31, e1808393. [Google Scholar] [CrossRef]
- Kumar, A.; Hong, J.; Yun, Y.; Bhardwaja, A.; Song, S.-J. The role of surface lattice defects of CeO2−δ nanoparticles as a scav-enging redox catalyst in polymer electrolyte membrane fuel cells. J. Mater. Chem. A 2020, 8, 26023–26034. [Google Scholar] [CrossRef]
- Leggesse, E.G.; Lin, R.T.; Teng, T.-F.; Chen, C.-L.; Jiang, J.-C. Oxidative decomposition of propylene carbonate in lithium ion batteries: A DFT study. J. Phys. Chem. A 2013, 117, 7959–7969. [Google Scholar] [CrossRef] [PubMed]
- Kalapsazova, M.; Kostov, K.; Zhecheva, E.; Stoyanova, R. Hybrid Li/Na ion batteries: Temperature-induced reactivity of three-layered oxide (P3-Na2/3Ni1/3Mg1/6Mn1/2O2) toward lithium ionic liquid electrolytes. Front. Chem. 2020, 8, 600140. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
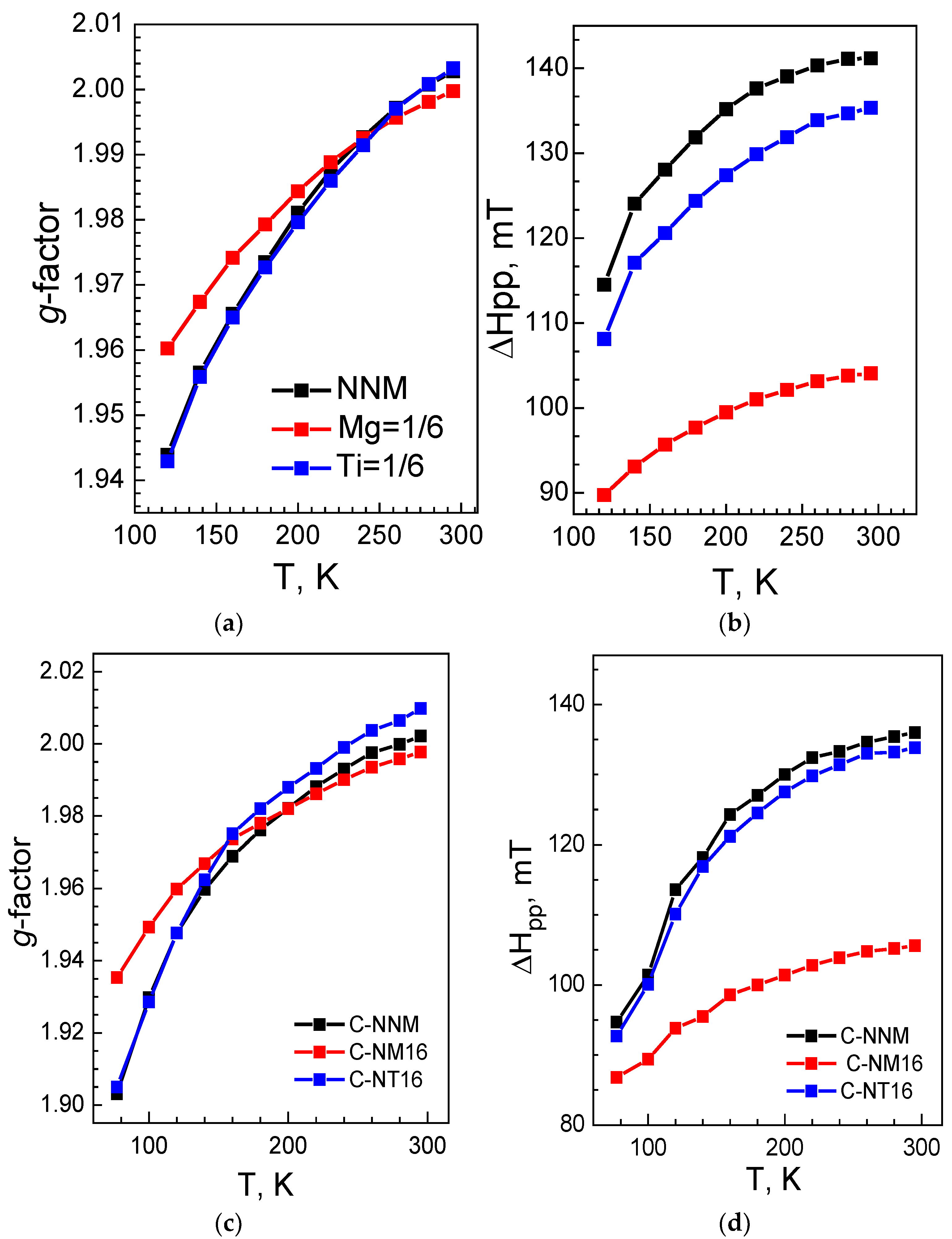{kind=link}
{kind=link}
{kind=link}
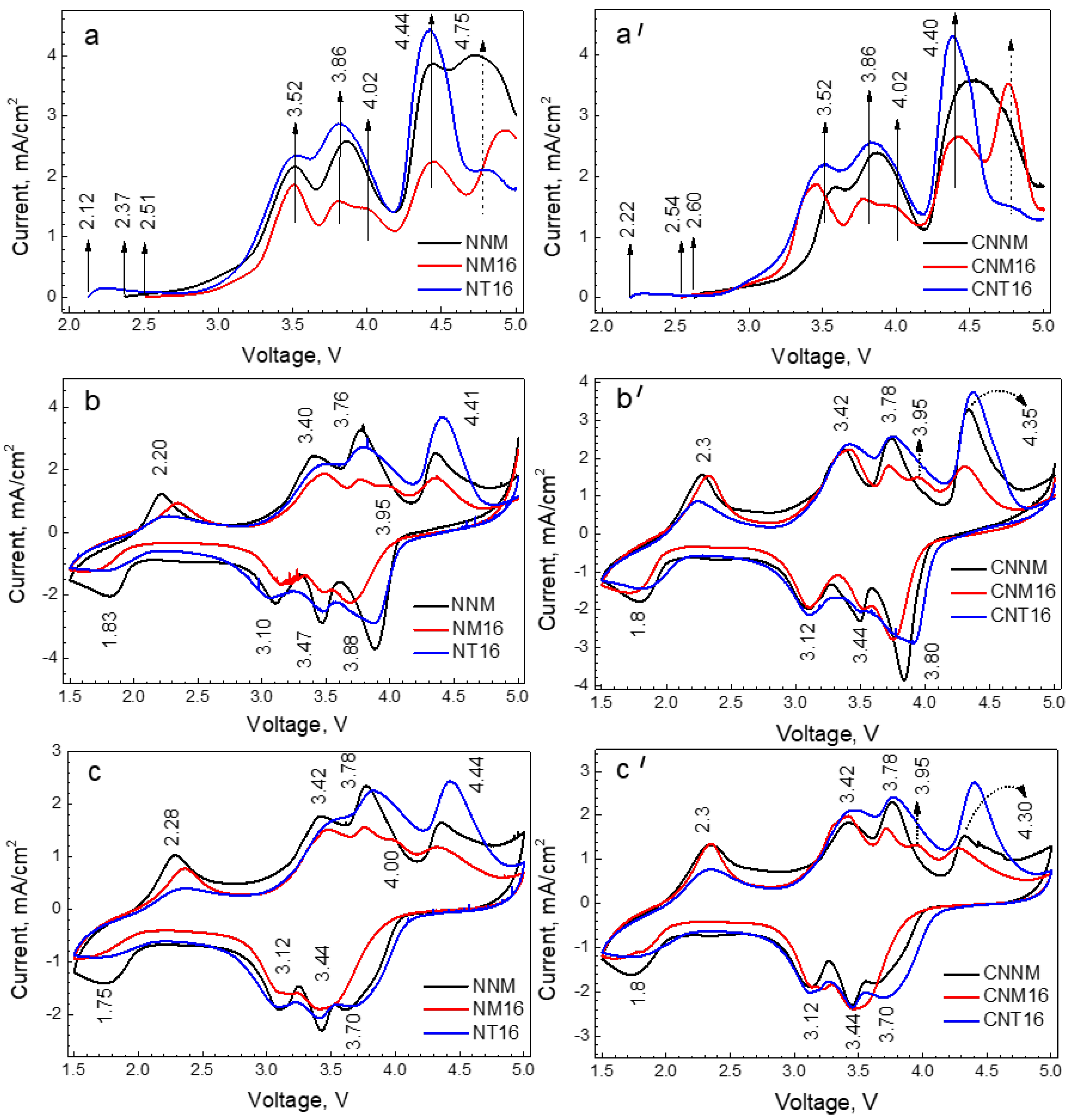{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
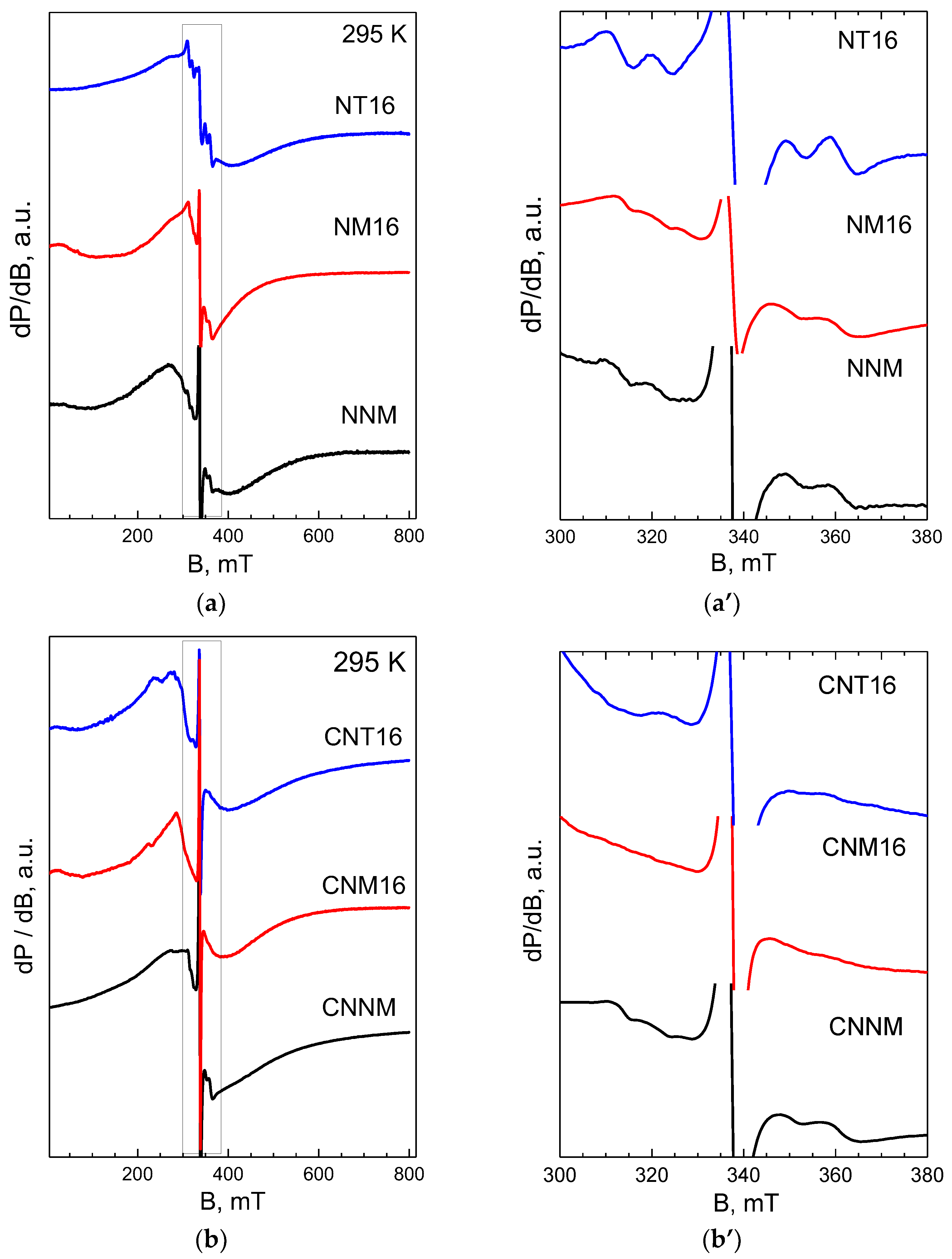{kind=link}
{kind=link}
| Samples | a ± 0.0001, Å | с ± 0.002, Å | V ± 0.02, Å3 |
|---|---|---|---|
| NNM | 2.8867 | 16.7692 | 121.01 |
| NM16 | 2.8889 | 16.7789 | 121.27 |
| NT16 | 2.8992 | 16.8237 | 122.46 |
| CNNM | 2.8872 | 16.7724 | 121.09 |
| CNM16 | 2.8867 | 16.7732 | 121.29 |
| CNT16 | 2.8889 | 16.8236 | 122.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalapsazova, M.; Kukeva, R.; Zhecheva, E.; Stoyanova, R. Metal Substitution versus Oxygen-Storage Modifier to Regulate the Oxygen Redox Reactions in Sodium-Deficient Three-Layered Oxides. Batteries 2022, 8, 56. https://doi.org/10.3390/batteries8060056
Kalapsazova M, Kukeva R, Zhecheva E, Stoyanova R. Metal Substitution versus Oxygen-Storage Modifier to Regulate the Oxygen Redox Reactions in Sodium-Deficient Three-Layered Oxides. Batteries. 2022; 8(6):56. https://doi.org/10.3390/batteries8060056
Chicago/Turabian StyleKalapsazova, Mariya, Rositsa Kukeva, Ekaterina Zhecheva, and Radostina Stoyanova. 2022. "Metal Substitution versus Oxygen-Storage Modifier to Regulate the Oxygen Redox Reactions in Sodium-Deficient Three-Layered Oxides" Batteries 8, no. 6: 56. https://doi.org/10.3390/batteries8060056
APA StyleKalapsazova, M., Kukeva, R., Zhecheva, E., & Stoyanova, R. (2022). Metal Substitution versus Oxygen-Storage Modifier to Regulate the Oxygen Redox Reactions in Sodium-Deficient Three-Layered Oxides. Batteries, 8(6), 56. https://doi.org/10.3390/batteries8060056