


Dehydrogenation of Alkali Metal Aluminum Hydrides MAlH4 (M = Li, Na, K, and Cs): Insight from First-Principles Calculations

Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
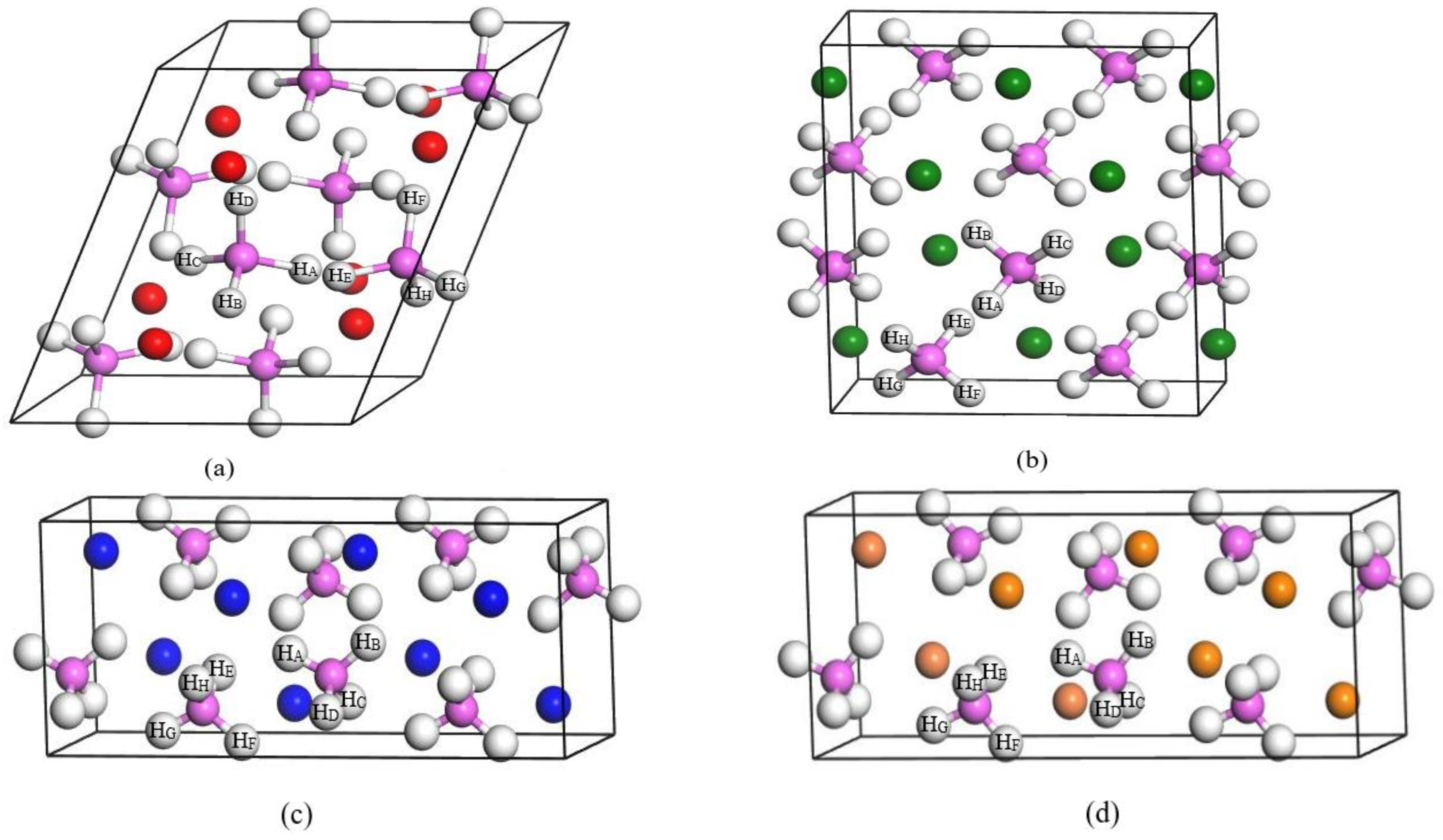
3.1. Thermal Stability
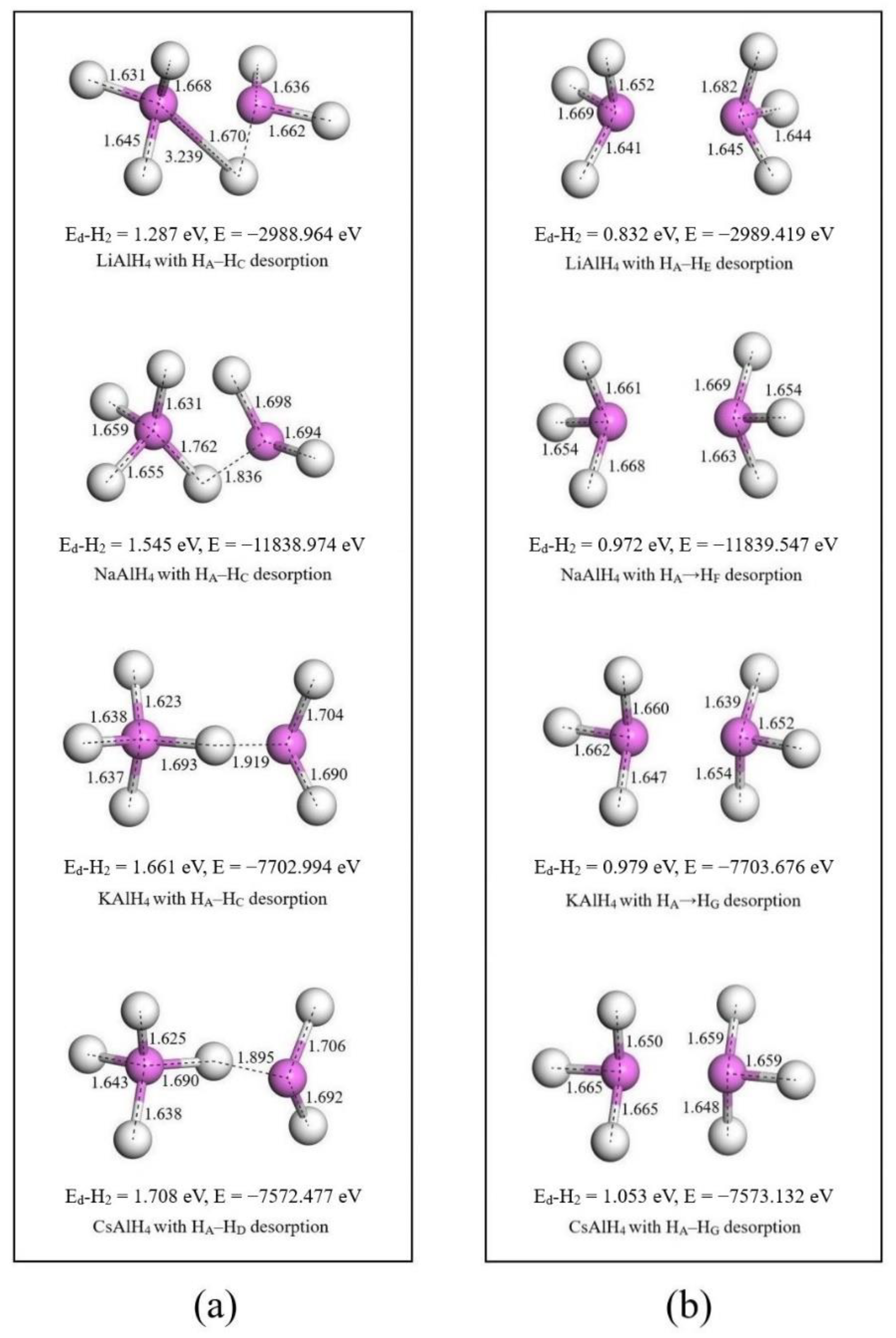
3.2. Hydrogen Dissociation Energy
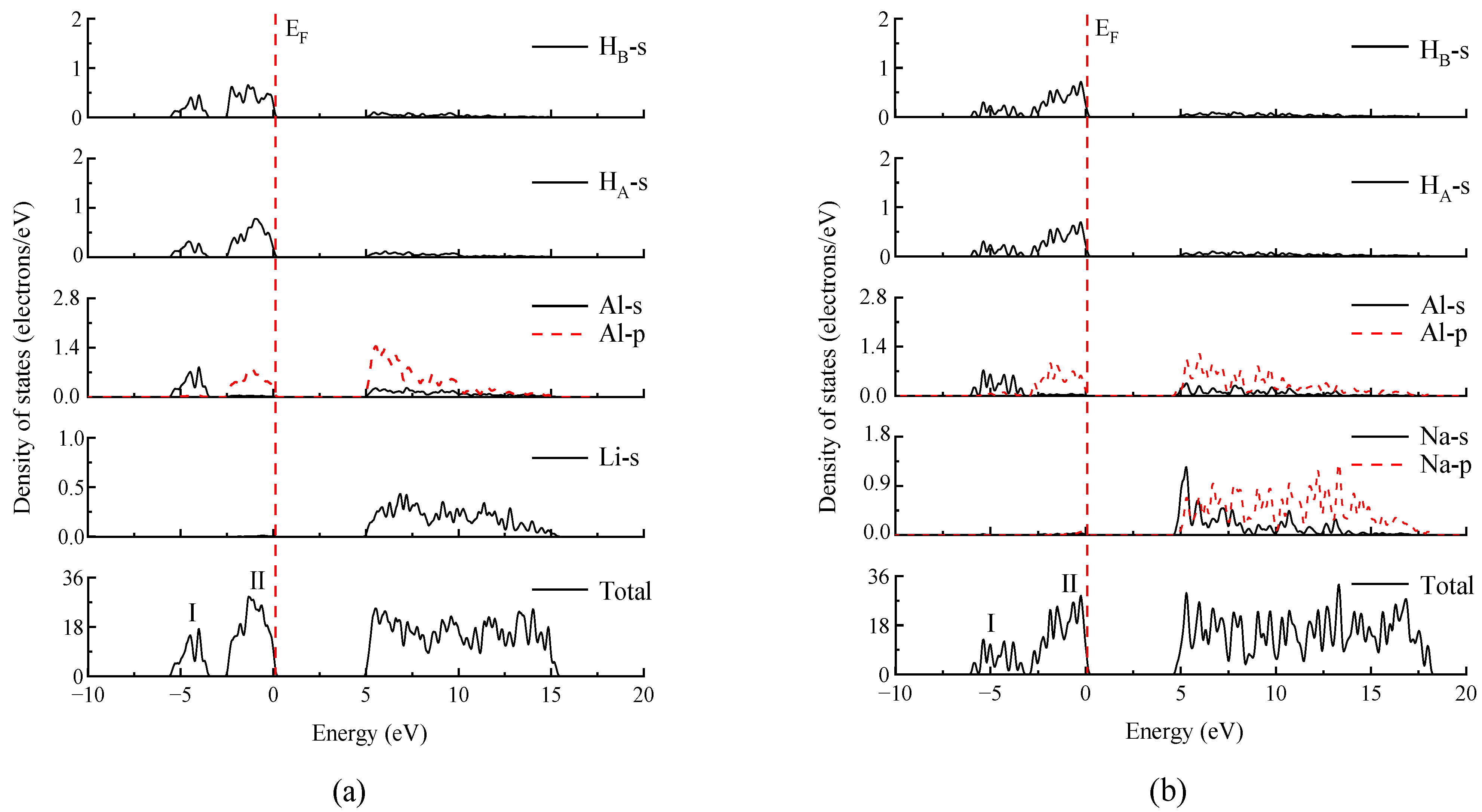
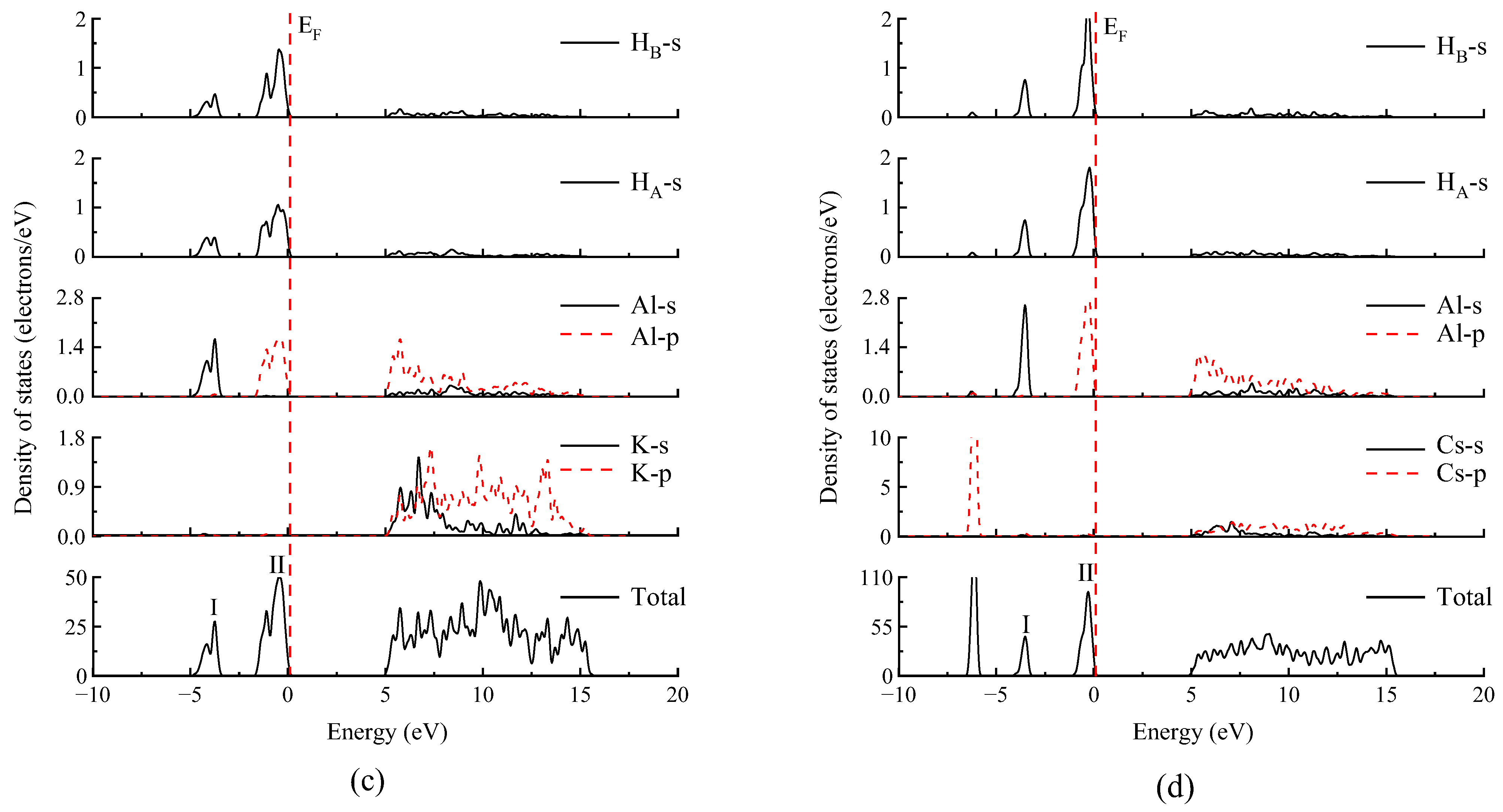
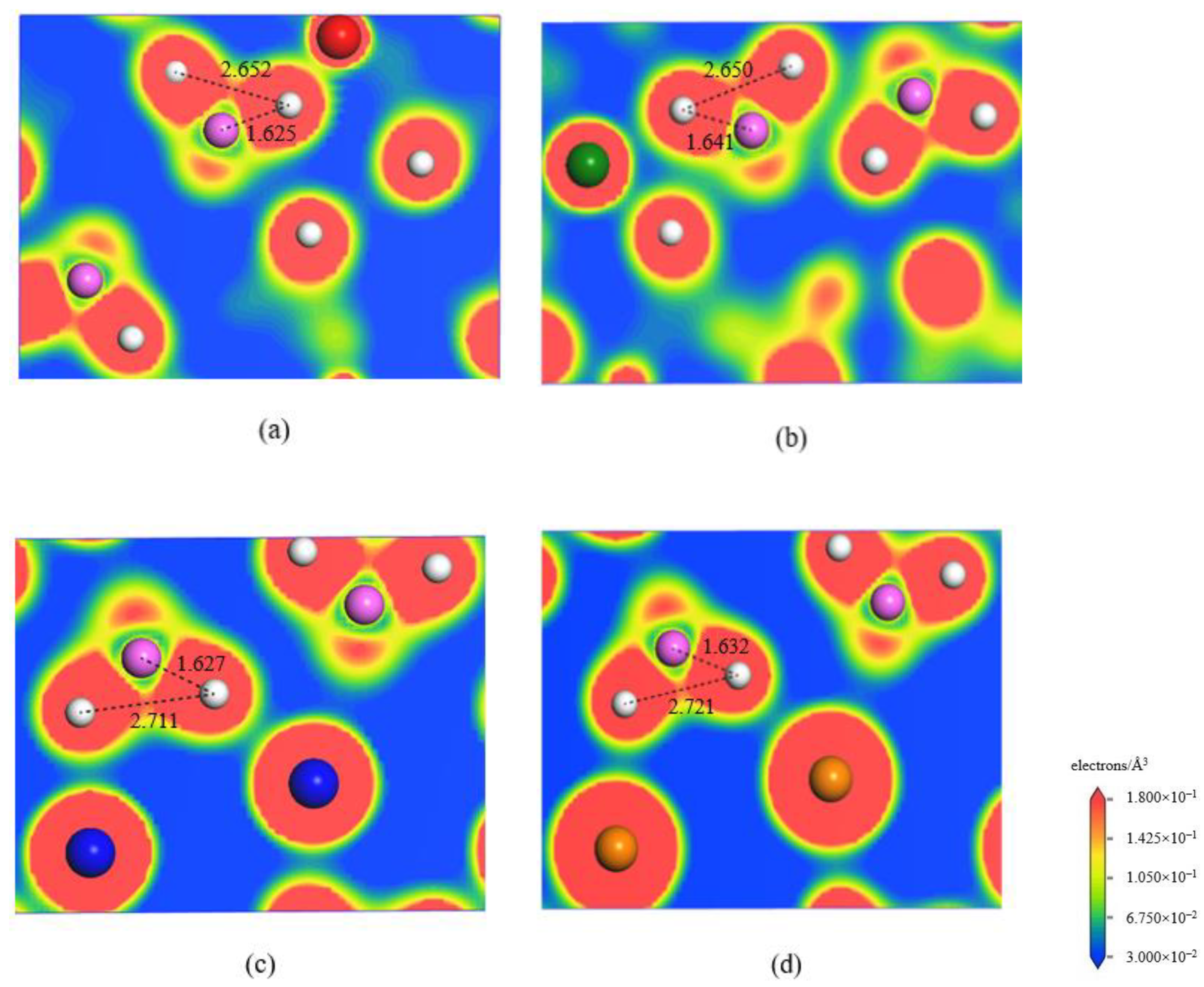
3.3. Electronic Structures
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Lu, Y.S.; Ouyang, L.Z.; Wang, H. Thermodynamic tuning of Mg-based hydrogen storage alloys: A review. Materials 2013, 6, 4654–4674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusman, N.A.A.; Dahari, M. A review on the current progress of metal hydrides material for solid-state hydrogen storage applications. Int. J. Hydrogen Energy 2016, 41, 12108–12126. [Google Scholar] [CrossRef]
- Li, L.; Huang, Y.K.; An, C.H.; Wang, Y.J. Lightweight hydrides nanocomposites for hydrogen storage: Challenges, progress and prospects. Sci. China Mater. 2019, 62, 1597–1625. [Google Scholar] [CrossRef] [Green Version]
- Ali, N.A.; Ismail, M. Modification of NaAlH4 properties using catalysts for solid-state hydrogen storage: A review. Int. J. Hydrogen Energy 2021, 46, 766–782. [Google Scholar] [CrossRef]
- Li, H.W.; Orimo, S.; Nakamori, Y.; Miwa, K.; Ohba, N.; Towata, S.; Züttel, A. Materials designing of metal borohydrides: Viewpoints from thermodynamical stabilities. J. Alloys Compd. 2007, 446–447, 315–318. [Google Scholar] [CrossRef] [Green Version]
- Ren, Z.H.; Zhang, X.; Huang, Z.G.; Hu, J.J.; Li, Y.Z.; Zheng, S.Y.; Gao, M.X.; Pan, H.G.; Liu, Y.F. Controllable synthesis of 2D TiH2 nanoflakes with superior catalytic activity for low-temperature hydrogen cycling of NaAlH4. Chem. Eng. J. 2022, 427, 131546. [Google Scholar] [CrossRef]
- Guo, Y.H.; Yu, X.B.; Gao, L.; Xia, G.L.; Guo, Z.P.; Liu, H.K. Significantly improved dehydrogenation of LiBH4 destabilized by TiF3. Energy Environ. Sci. 2010, 3, 465–470. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhang, Y.; Gao, M.X.; Pan, H.G.; Liu, Y.F. Preparation and catalytic effect of porous Co3O4 on the hydrogen storage properties of a Li-B-N-H system. Prog. Nat. Sci.-Mater. 2017, 27, 132–138. [Google Scholar] [CrossRef]
- Wei, S.; Liu, J.X.; Xia, Y.P.; Zhang, H.Z.; Cheng, R.G.; Sun, L.X.; Xu, F.; Huang, P.R.; Rosei, F.; Pimerzin, A.A.; et al. Remarkable catalysis of spinel ferrite XFe2O4 (X = Ni, Co, Mn, Cu, Zn) nanoparticles on the dehydrogenation properties of LiAlH4: An experimental and theoretical study. J. Mater. Sci. Technol. 2022, 111, 189–203. [Google Scholar] [CrossRef]
- Nakamori, Y.; Miwa, K.; Ninomiya, A.; Li, H.W.; Ohba, N.; Towata, S.; Züttel, A.; Orimo, S. Correlation between thermodynamical stabilities of metal borohydrides and cation electronegativites: First-principles calculations and experiments. Phys. Rev. B 2006, 74, 045126. [Google Scholar] [CrossRef] [Green Version]
- Nakamori, Y.; Li, H.W.; Kikuchi, K.; Aoki, M.; Miwa, K.; Towata, S.; Orimo, S. Thermodynamical stabilities of metal-borohydrides. J. Alloys Compd. 2007, 446–447, 296–300. [Google Scholar] [CrossRef]
- Zhang, B.J.; Liu, B.H. Hydrogen desorption from LiBH4 destabilized by chlorides of transition metal Fe, Co, and Ni. Int. J. Hydrogen Energy 2010, 35, 7288–7294. [Google Scholar] [CrossRef]
- Mo, X.H.; Jiang, W.Q. Dehydrogenation properties of LiBH4 modified by Mg from first-principles calculations. J. Alloys Compd. 2018, 735, 668–676. [Google Scholar] [CrossRef]
- Cai, W.T.; Yang, Y.Z.; Tao, P.J.; Ouyang, L.Z.; Wang, H. Correlation between structural stability of LiBH4 and cation electronegativity in metal borides: An experimental insight for catalyst design. Dalton Trans. 2018, 47, 4987–4993. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Z.; Zhang, Y.; Liu, D.M.; Wang, C.Y.; Li, Y.T.; Si, T.Z.; Zhang, Q.G. Enhanced low-temperature hydrogen storage in nanoporous Ni-based alloy supported LiBH4. Front. Chem. 2020, 8, 283. [Google Scholar] [CrossRef] [PubMed]
- Nakamori, Y.; Orimo, S.I. Destabilization of Li-based complex hydrides. J. Alloys Compd. 2004, 370, 271–275. [Google Scholar] [CrossRef]
- Bai, Y.; Pei, Z.W.; Wu, F.; Wu, C. Role of metal electronegativity in the dehydrogenation thermodynamics and kinetics of composite metal borohydride-LiNH2 hydrogen storage materials. ACS Appl. Mater. Interfaces 2018, 10, 9514–9521. [Google Scholar] [CrossRef] [PubMed]
- Weidenthaler, C. Crystal structure evolution of complex metal aluminum hydrides upon hydrogen release. J. Energy Chem. 2020, 42, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Mamatha, M.; Weidenthaler, C.; Pommerin, A.; Felderhoff, M.; Schüth, F. Comparative studies of the decomposition of alanates followed by in situ XRD and DSC methods. J. Alloys Compd. 2006, 416, 303–314. [Google Scholar] [CrossRef]
- Finholt, A.E.; Barbaras, G.D.; Barbaras, G.K.; Urry, G.; Wartik, T.; Schlesinger, H.I. The preparation of sodium and calcium aluminum hydrides. J. Inorg. Nucl. Chem. 1955, 1, 317–325. [Google Scholar] [CrossRef]
- Nyahuma, F.M.; Zhang, L.T.; Song, M.C.; Lu, X.; Xiao, B.B.; Zheng, J.G.; Wu, F.Y. Significantly improved hydrogen storage behaviors in MgH2 with Nb nanocatalyst. Int. J. Min. Met. Mater. 2022, 29, 1788–1797. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, Z.N.; Gao, X.; Wang, Y.Q.; Wang, F.; Zheng, S.S.; Guan, S.N.; Yan, H.L.; Yang, X.; Jia, W.H. Effect of novel La-based alloy modification on hydrogen storage performance of magnesium hydride: First-principles calculation and experimental investigation. J. Power Sources 2022, 551, 232187. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.D.; Probert, M.J.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condes. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Hauback, B.C.; Brinks, H.W.; Fjellvåg, H. Accurate structure of LiAlD4 studied by combined powder neutron and X-ray diffraction. J. Alloys Compd. 2002, 346, 184–189. [Google Scholar] [CrossRef]
- Ozolins, V.; Majzoub, E.H.; Udovic, T.J. Electronic structure and Rietveld refinement parameters of Ti-doped sodium alanates. J. Alloys Compd. 2004, 375, 1–10. [Google Scholar] [CrossRef]
- Hauback, B.C.; Brinks, H.W.; Heyn, R.H.; Blom, R.; Fjellvåg, H. The crystal structure of KAlD4. J. Alloys Compd. 2005, 394, 35–38. [Google Scholar] [CrossRef]
- Bernert, T.; Krech, D.; Kockelmann, W.; Felderhoff, M.; Frankcombe, T.J.; Weidenthaler, C. Crystal structure relation between tetragonal and orthorhombic CsAlD4: DFT and time-of-flight neutron powder diffraction studies. Eur. J. Inorg. Chem. 2015, 2015, 5545–5550. [Google Scholar] [CrossRef]
- Sirsch, P.; Che, F.N.; Titah, J.T.; McGrady, G.S. Hydride-hydride bonding interactions in the hydrogen stoage materials AlH3, MgH2, and NaAlH4. Chem. Eur. J. 2012, 18, 9476–9480. [Google Scholar] [CrossRef]
- Sorte, E.G.; Emery, S.B.; Majzoub, E.H.; Ellis-Caleo, T.; Ma, Z.L.; Hammann, B.A.; Hayes, S.E.; Bowman, R.C.; Conradi, M.S. NMR Study of Anion Dynamics in Solid KAlH4. J. Phys. Chem. C 2014, 118, 5725–5732. [Google Scholar] [CrossRef]
- Ravindran, P.; Vajeeston, P.; Vidya, R.; Fjellvåg, H.; Kjekshus, A. Modeling of hydrogen storage materials by density-functional calculations. J. Power Sources 2006, 159, 88–99. [Google Scholar] [CrossRef]
- Setten, M.J.V. Electronic structure and formation enthalpy of hydroaluminates and hydroborates. Encycl. Mater. Sci. Technol. 2008, 043152, 1–6. [Google Scholar]
- Fukai, Y. The Metal-Hydrogen System; Volume 21 of Springer Series in Material Science; Springer: Berlin/Heidelberg, Germany, 1993. [Google Scholar]
- Shi, B.; Song, Y. Influence of transition metals Fe, Ni, and Nb on dehydrogenation characteristics of Mg(BH4)2: Electronic structure mechanisms. Int. J. Hydrogen Energy 2013, 38, 6417–6424. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, L.Q.; Zhou, Y.C.; Peng, P. Dehydrogenation thermodynamics of magnesium hydride doped with transition metals: Experimental and theoretical studies. Comput. Mater. Sci. 2015, 98, 211–219. [Google Scholar] [CrossRef]
- Kumar, A.; Muthukumar, P.; Sharma, P.; Kumar, E.A. Absorption based solid state hydrogen storage system: A review. Sustain. Energy Technol. Assess. 2022, 52, 102204. [Google Scholar] [CrossRef]
- Zhao, L.; Xu, F.; Zhang, C.C.; Wang, Z.Y.; Ju, H.Y.; Gao, X.; Zhang, X.X.; Sun, L.X.; Liu, Z.W. Enhanced hydrogen storage of alanates: Recent progress and future perspectives. Prog. Nat. Sci. 2021, 31, 165–179. [Google Scholar] [CrossRef]
- Mo, X.H.; Liang, J.Q.; Wang, W.H.; Jiang, W.Q. First-principles study on the dehydrogenation of Li4BN3H10 modified by Co. Int. J. Hydrogen Energy 2021, 46, 11815–11823. [Google Scholar] [CrossRef]
- Jiang, W.Q.; Chen, Y.J.; Mo, X.H.; Li, X.L. First-principles investigation of LaMg2Ni and its hydrides. Sci. Rep. 2020, 10, 12167. [Google Scholar] [CrossRef]
- Jiang, W.Q.; Qin, C.S.; Zhu, R.R.; Guo, J. Annealing effect on hydrogen storage property of Co-free La1.8Ti0.2MgNi8.7Al0.3 alloy. J. Alloys Compd. 2013, 565, 37–43. [Google Scholar] [CrossRef]
- Wang, H.Y.; Zhang, N.; Wang, C.; Jiang, Q.C. First-principles study of the generalized stacking fault energy in Mg-3Al-3Sn alloy. Scr. Mater. 2011, 65, 723–726. [Google Scholar] [CrossRef]
- Løvvik, O.M.; Opalka, S.M.; Brinks, H.W.; Hauback, B.C. Crystal structure and thermodynamic stability of the lithium alanates LiAlH4 and Li3AlH6. Phys. Rev. B 2004, 69, 134117. [Google Scholar] [CrossRef]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 80th ed.; CRC Press: Boca Raton, FL, USA, 2000. [Google Scholar]
- Wang, X.; Yu, X.H.; Zhong, Y.; Rong, J.; Li, X.Y.; Feng, J.; Zhan, Z.L. Stability and mechanical peroperties of high-La content La-Ni phases by first-principles study. Int. J. Mod. Phys. B 2018, 32, 1850242. [Google Scholar] [CrossRef]
- Miwa, K.; Ohba, N.; Towata, S.; Nakamori, Y.; Orimo, S. First-principles study on lithium borohydride LiBH4. Phys. Rev. B 2004, 69, 245120. [Google Scholar] [CrossRef]
- Miwa, K.; Ohba, N.; Towata, S.; Nakamori, Y.; Orimo, S. First-principles study on copper-substituted lithium borohydride, (Li1−xCux)BH4. J. Alloys Compd. 2005, 404–406, 140–143. [Google Scholar] [CrossRef]
- Dai, J.H.; Song, Y.; Yang, R. Intrinsic mechanisms on enhancement of hydrogen desorption from MgH2 by (001) surface doping. Int. J. Hydrogen Energy 2011, 36, 12939–12949. [Google Scholar] [CrossRef]
- Mo, X.H.; Long, L.X.; Tan, W.B.; Huang, Y.P.; Lu, J.W.; Zhao, Y.W.; Jiang, W.Q. First-principles investigation of dehydrogenation of Cu-doped LiBH4. Solid State Commun. 2021, 326, 114184. [Google Scholar] [CrossRef]
- Mo, X.H.; Jiang, W.Q.; Cao, S.L. First-principles study on the dehydrogenation characteristics of LiBH4 modified by Ti. Results Phys. 2017, 7, 3236–3242. [Google Scholar] [CrossRef]
- Shi, Q.; Voss, J.; Jacobsen, H.S.; Lefmann, K.; Zamponi, M.; Vegge, T. Point defect dynamics in sodium aluminum hydrides–a combined quasielastic neutron scattering and density functional theory study. J. Alloys Compd. 2007, 446–447, 469–473. [Google Scholar] [CrossRef]
- Araújo, C.M.; Li, S.; Ahuja, R.; Jena, P. Vacancy-mediated hydrogen desorption in NaAlH4. Phys. Rev. B 2005, 72, 165101. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Tezuka, A.; Ogawa, H.; Ikeshoji, T. First-principles study of hydrogen vacancies in sodium alanate with Ti substitution. J. Phys. Condes. Matter 2010, 22, 205503. [Google Scholar] [CrossRef]
- Araújo, C.M.; Ahuja, R.; Guillén, J.M.O.; Jena, P. Role of titanium in hydrogen desorption in crystalline sodium alanate. Appl. Phys. Lett. 2005, 86, 251913. [Google Scholar] [CrossRef]
- Zhang, X.; Ren, Z.H.; Zhang, X.L.; Gao, M.X.; Pan, H.G.; Liu, Y.F. Triggering highly stable catalytic activity of metallic titanium for hydrogen storage in NaAlH4 by preparing ultrafine nanoparticles. J. Mater. Chem. A 2019, 7, 4651–4659. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Liu, J.X.; Wei, S.; Xia, Y.P.; Cheng, R.G.; Sun, L.X.; Xu, F.; Huang, P.R.; Bu, Y.T.; Cheng, J.; et al. Improved hydrogen storage properties and mechanisms of LiAlH4 doped with Ni/C nanoparticles anchored on large-size Ti3C2Tx. J. Alloys Compd. 2023, 931, 167353. [Google Scholar] [CrossRef]
- Jiang, W.Q.; Cao, S.L. Effect of Al on the dehydrogenation of LiBH4 from first-principles calculations. Int. J. Hydrogen Energy 2017, 42, 6181–6188. [Google Scholar]
- Segall, M.D.; Shah, R.; Pickard, C.J.; Payne, M.C. Population analysis of plane-wave electronic structure calculations of bulk materials. Phys. Rev. B 1996, 54, 16317–16320. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.P.; Wang, X.M.; Ge, F.F.; Zhou, M.J.; Wu, W.D.; Lu, T.C. Density function study of H2 adsorption on LiB (010) surface. Phys. B 2010, 405, 1792–1795. [Google Scholar]
- Zhu, C.Y.; Liu, Y.H.; Duan, D.F.; Cui, T. Structural transitions of NaAlH4 under high pressure by first-principles calculations. Phys. B 2011, 406, 1612–1614. [Google Scholar] [CrossRef]
- Berseth, P.A.; Harter, A.G.; Zidan, R.; Blomqvist, A.; Araújo, C.M.; Scheicher, R.H.; Ahuja, R.; Jena, P. Carbon nanomaterials as catalysts for hydrogen uptake and release in NaAlH4. Nano Lett. 2009, 9, 1501–1505. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | SP | R (Ǻ) | V (Ǻ3) | ΔH (kJ/mol) | Ecoh (kJ/mol) | C (e) | χP | ||
|---|---|---|---|---|---|---|---|---|---|
| a | b | c | |||||||
| LiAlH4 | P21/C | 4.908 | 8.031 | 7.953 | 289.762 | −117.156 | −1684.32 | 0.21 | 0.98 |
| NaAlH4 | I41/A | 4.986 | 4.986 | 11.178 | 277.905 | −120.468 | −1684.704 | 0.28 | 0.93 |
| KAlH4 | Pnma | 8.896 | 5.810 | 7.399 | 382.448 | −157.2 | −1719.552 | 0.35 | 0.82 |
| CsAlH4 | Pnma | 10.018 | 6.163 | 8.077 | 498.704 | −171.9 | −1740.768 | 0.42 | 0.79 |
| Hydrogen Dissociation Energy (eV) | LiAlH4 | NaAlH4 | KAlH4 | CsAlH4 | |
|---|---|---|---|---|---|
| Atomic hydrogen desorption Ed-H | HA | 1.815 | 1.719 | 1.702 | 1.720 |
| HB | −0.080 | −0.129 | 0.080 | 0.142 | |
| HC | −0.526 | −0.077 | −0.038 | −0.006 | |
| HD | −0.019 | −0.077 | −0.037 | 0.000 | |
| HE | −0.862 | −0.736 | −0.721 | −0.658 | |
| HF | −0.971 | −0.747 | −0.722 | −0.663 | |
| HG | −0.788 | −0.637 | −0.723 | −0.664 | |
| HH | −0.786 | −0.634 | −0.723 | −0.665 | |
| Molecular hydrogen desorption Ed-H2 | HA→HB (HA–HB) | 1.735 (1.733) | 1.590 (1.592) | 1.782 (1.773) | 1.862 (1.866) |
| HA→HC (HA–HC) | 1.289 (1.287) | 1.642 (1.545) | 1.664 (1.661) | 1.714 (1.726) | |
| HA→HD (HA–HD) | 1.796 (1.704) | 1.642 (1.660) | 1.665 (1.664) | 1.720 (1.708) | |
| HA→HE (HA–HE) | 0.953 (0.832) | 0.983 (0.997) | 0.981 (0.984) | 1.062 (1.057) | |
| HA→HF (HA–HF) | 0.844 (0.844) | 0.972 (0.975) | 0.980 (0.982) | 1.057 (1.055) | |
| HA→HG (HA–HG) | 1.027 (0.856) | 1.082 (1.085) | 0.979 (0.986) | 1.056 (1.053) | |
| HA→HH (HA–HH) | 1.029 (1.029) | 1.085 (1.081) | 0.979 (0.993) | 1.055 (1.065) | |
| Minimum value | 0.832 | 0.972 | 0.979 | 1.053 | |
| Average value | 1.211 | 1.281 | 1.291 | 1.361 | |
| Compounds | Al-H | H-H | ||||
|---|---|---|---|---|---|---|
| BO | BL (Å) | BOS (Å−1) | BO | BL (Å) | BOS (Å−1) | |
| LiAlH4 | 0.778 | 1.634 | 0.476 | −0.036 | 2.751 | −0.013 |
| NaAlH4 | 0.855 | 1.642 | 0.521 | −0.039 | 2.755 | −0.014 |
| KAlH4 | 0.906 | 1.634 | 0.555 | −0.055 | 2.668 | −0.021 |
| CsAlH4 | 0.893 | 1.637 | 0.545 | −0.058 | 2.673 | −0.022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, R.; Mo, X.; Huang, Y.; Hu, C.; Zuo, X.; Ma, Y.; Wei, Q.; Jiang, W. Dehydrogenation of Alkali Metal Aluminum Hydrides MAlH4 (M = Li, Na, K, and Cs): Insight from First-Principles Calculations. Batteries 2023, 9, 179. https://doi.org/10.3390/batteries9030179
Zhou R, Mo X, Huang Y, Hu C, Zuo X, Ma Y, Wei Q, Jiang W. Dehydrogenation of Alkali Metal Aluminum Hydrides MAlH4 (M = Li, Na, K, and Cs): Insight from First-Principles Calculations. Batteries. 2023; 9(3):179. https://doi.org/10.3390/batteries9030179
Chicago/Turabian StyleZhou, Rui, Xiaohua Mo, Yong Huang, Chunyan Hu, Xiaoli Zuo, Yu Ma, Qi Wei, and Weiqing Jiang. 2023. "Dehydrogenation of Alkali Metal Aluminum Hydrides MAlH4 (M = Li, Na, K, and Cs): Insight from First-Principles Calculations" Batteries 9, no. 3: 179. https://doi.org/10.3390/batteries9030179
APA StyleZhou, R., Mo, X., Huang, Y., Hu, C., Zuo, X., Ma, Y., Wei, Q., & Jiang, W. (2023). Dehydrogenation of Alkali Metal Aluminum Hydrides MAlH4 (M = Li, Na, K, and Cs): Insight from First-Principles Calculations. Batteries, 9(3), 179. https://doi.org/10.3390/batteries9030179