
The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
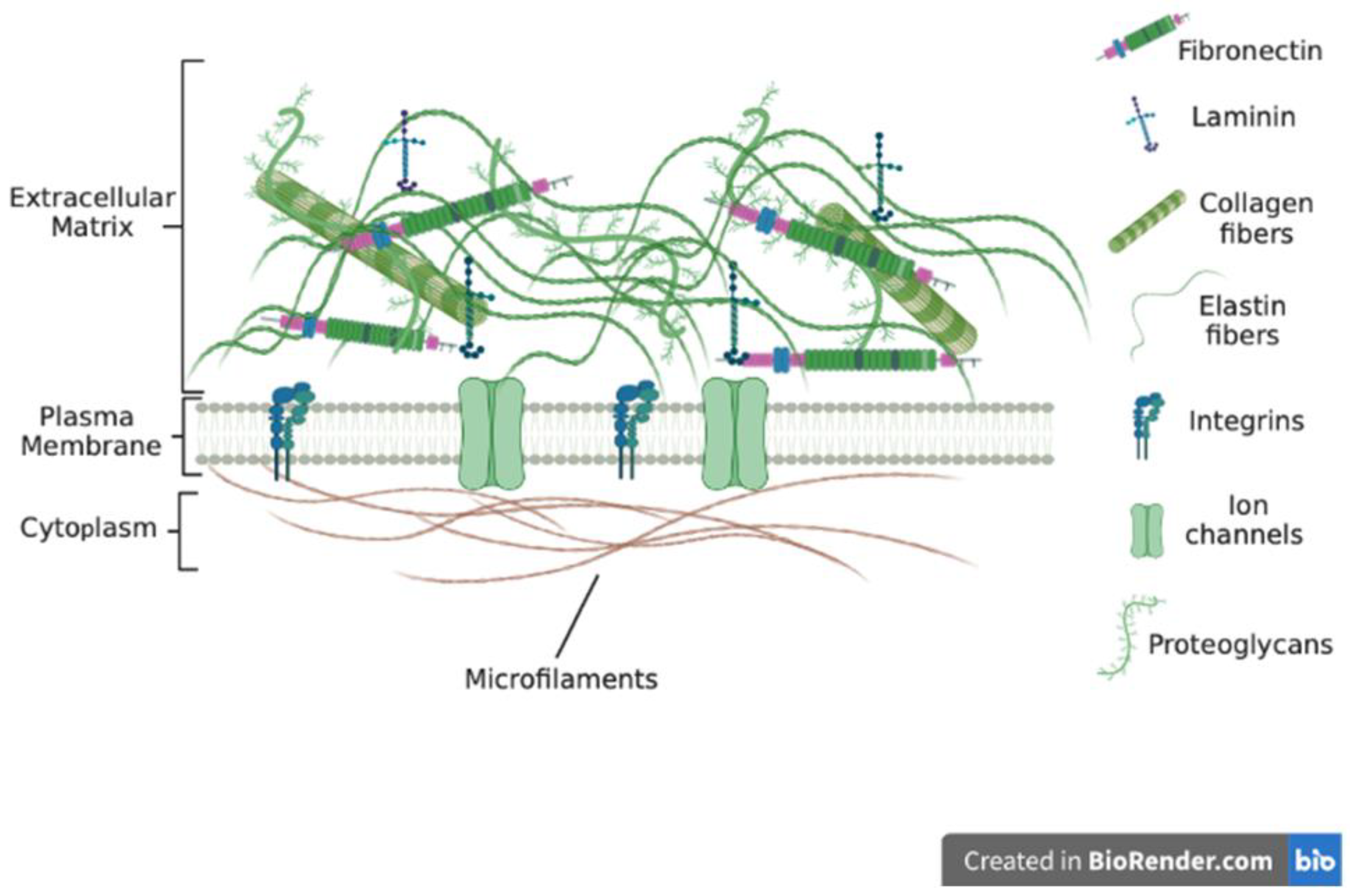
2. The Extracellular Matrix Macromolecules
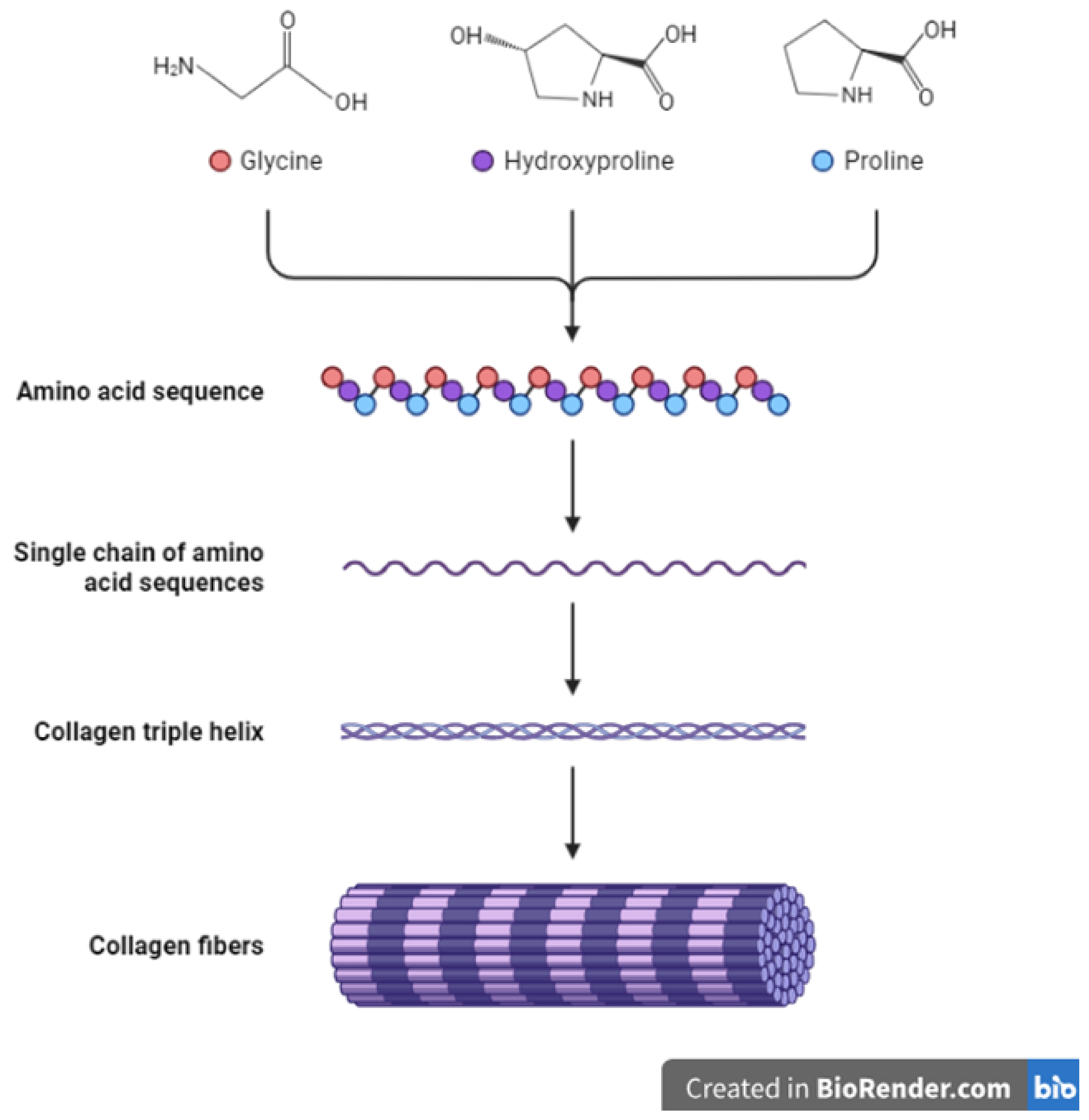
3. Collagens
4. Other Extracellular Matrix Macromolecules
4.1. Laminin
4.2. Fibronectin
4.3. Periostin
4.4. Hyaluronic Acid
5. Extracellular Matrix Function
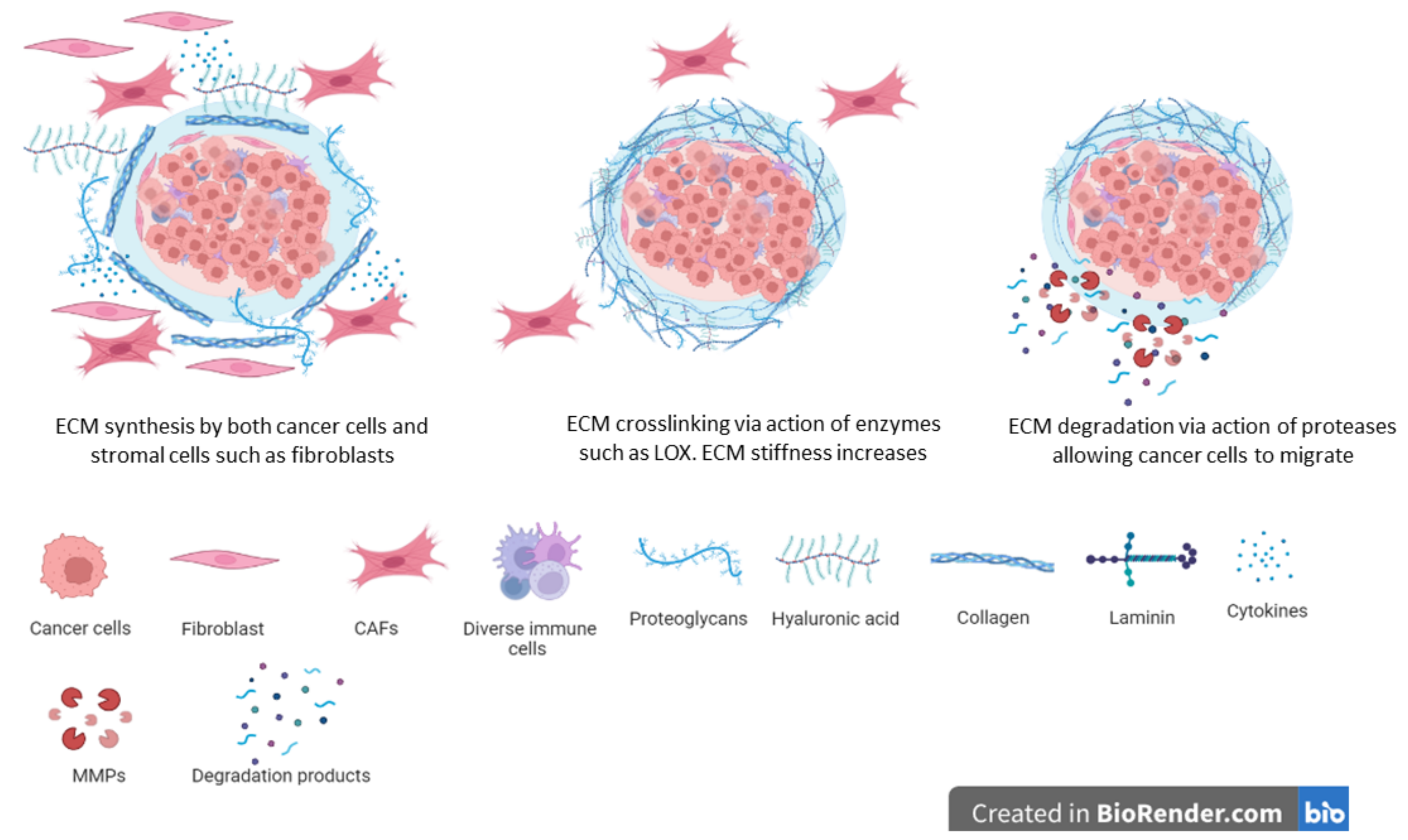
6. Extracellular Matrix Modifications
7. Proteolytic Degradation of the Extracellular Matrix
8. Fibroblasts and Extracellular Matrix Remodeling
9. Extracellular Matrix Signaling
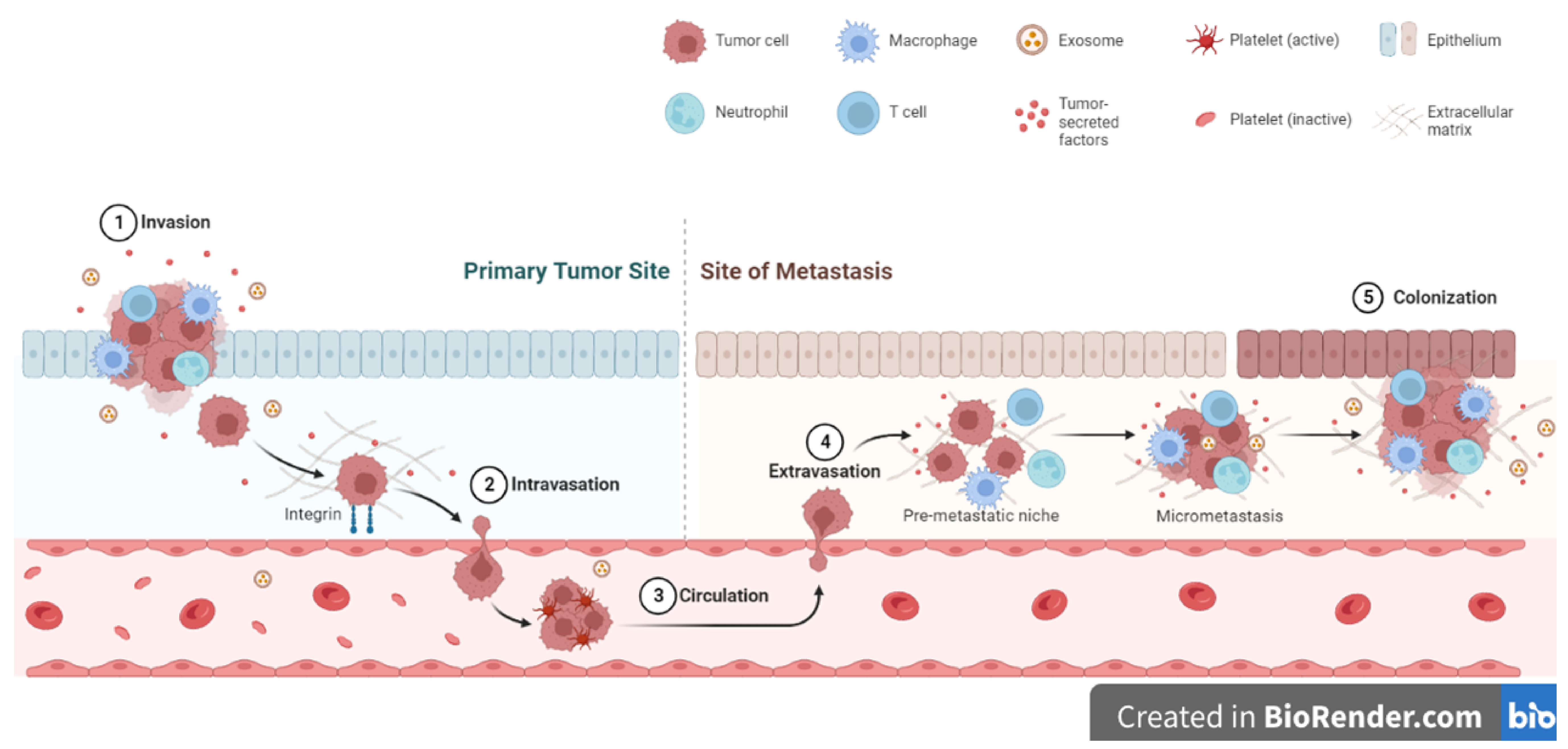
10. Extracellular Matrix and Cell Invasion and Metastasis
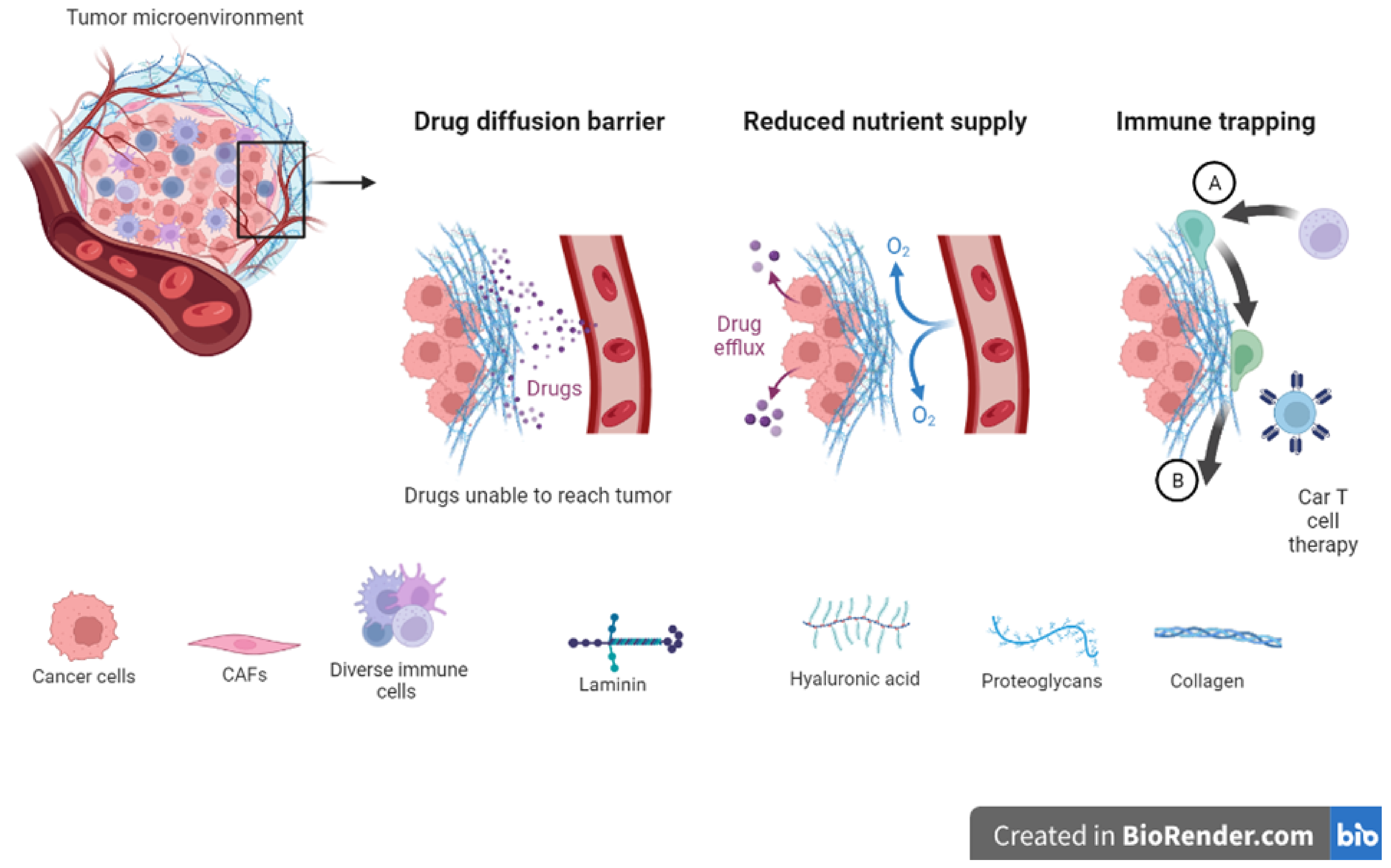
11. Extracellular Matrix in Drug Resistance: The Extracellular Matrix Shields Tumor Cells from Anti-Cancer Drugs
12. Therapeutic Strategies Targeting the Extracellular Matrix
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bissell, M.J.; Hall, H.G.; Parry, G. How does the extracellular matrix direct gene expression? J. Theor. Biol. 1982, 99, 31–68. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.M.; Bissell, M.J. Of extracellular matrix, scaffolds, and signaling: Tissue architecture regulates development, homeostasis, and cancer. Annu. Rev. Cell Dev. Biol. 2006, 22, 287–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.; Mojares, E.; del Río Hernández, A. Role of extracellular matrix in development and cancer progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pupa, S.M.; Ménard, S.; Forti, S.; Tagliabue, E. New insights into the role of extracellular matrix during tumor onset and progression. J. Cell. Physiol. 2002, 192, 259–267. [Google Scholar] [CrossRef]
- Clause, K.C.; Barker, T.H. Extracellular matrix signaling in morphogenesis and repair. Curr. Opin. Biotechnol. 2013, 24, 830–833. [Google Scholar] [CrossRef] [Green Version]
- Dzobo, K.; Vogelsang, M.; Parker, M.I. Wnt/β-catenin and MEK-ERK signaling are required for fibroblast-derived extracellular matrix-mediated endoderm differentiation of embryonic stem cells. Stem Cell Rev. Rep. 2015, 11, 761–773. [Google Scholar] [CrossRef]
- Dzobo, K.; Turnley, T.; Wishart, A.; Rowe, A.; Kallmeyer, K.; Van Vollenstee, F.A.; Thomford, N.E.; Dandara, C.; Chopera, D.; Pepper, M.S. Fibroblast-derived extracellular matrix induces chondrogenic differentiation in human adipose-derived mesenchymal stromal/stem cells in vitro. Int. J. Mol. Sci. 2016, 17, 1259. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Wang, J.; Li, H.; Yu, Y.; Wang, X.; Lu, L.; Lv, C.; Chang, B.; Jin, W.; Guo, W.; et al. Extracellular matrix protein-1 secretory isoform promotes ovarian cancer through increasing alternative mRNA splicing and stemness. Nat. Commun. 2021, 12, 4230. [Google Scholar] [CrossRef]
- Dussoyer, M.; Page, A.; Delolme, F.; Rousselle, P.; Nyström, A.; Moali, C. Comparison of extracellular matrix enrichment protocols for the improved characterization of the skin matrisome by mass spectrometry. J. Proteom. 2022, 251, 104397. [Google Scholar] [CrossRef]
- Mienaltowski, M.J.; Gonzales, N.L.; Beall, J.M.; Pechanec, M.Y. Basic Structure, Physiology, and Biochemistry of Connective Tissues and Extracellular Matrix Collagens. Adv. Exp. Med. Biol. 2021, 1348, 5–43. [Google Scholar] [CrossRef]
- Giblin, S.P.; Schwenzer, A.; Midwood, K.S. Alternative splicing controls cell lineage-specific responses to endogenous innate immune triggers within the extracellular matrix. Matrix Biol. 2020, 93, 95–114. [Google Scholar] [CrossRef]
- Chen, W.; Yuan, Y.; Li, C.; Mao, H.; Liu, B.; Jiang, X. Modulating Tumor Extracellular Matrix by Simultaneous Inhibition of Two Cancer Cell Receptors. Adv. Mater 2021, 34, e2109376. [Google Scholar] [CrossRef]
- Gu, X.; Ge, L.; Ren, B.; Fang, Y.; Li, Y.; Wang, Y.; Xu, H. Glucocorticoids Promote Extracellular Matrix Component Remodeling by Activating YAP in Human Retinal Capillary Endothelial Cells. Front. Cell Dev. Biol. 2021, 9, 738341. [Google Scholar] [CrossRef]
- Laurito, T.L.; França, F.T.; Vieira-Damiani, G.; Pelegati, V.B.; Baratti, M.O.; de Carvalho, H.F.; Cesar, C.L.; de Moraes, A.M.; Cintra, M.L.; Teixeira, F. The texture of collagen in the microenvironments of Merkel cell carcinoma. Medicine 2021, 100, e27925. [Google Scholar] [CrossRef]
- Dzobo, K. Taking a Full Snapshot of Cancer Biology: Deciphering the Tumor Microenvironment for Effective Cancer Therapy in the Oncology Clinic. Omics 2020, 24, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Dzobo, K.; Leaner, V.D.; Parker, M.I. Feedback regulation of the α2(1) collagen gene via the Mek-Erk signaling pathway. IUBMB Life 2012, 64, 87–98. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Jonker, T.; Rowe, A.; Thomford, N.E.; Munro, D.; Dandara, C.; Wonkam, A.; Govender, D.; Calder, B.; Soares, N.C.; et al. The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices. Int. J. Mol. Sci. 2018, 19, 2861. [Google Scholar] [CrossRef] [Green Version]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.R. The matrix in cancer. Nat. Rev. Cancer 2021, 21, 217–238. [Google Scholar] [CrossRef]
- Lukashev, M.E.; Werb, Z. ECM signalling: Orchestrating cell behaviour and misbehaviour. Trends Cell Biol. 1998, 8, 437–441. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Bissell, M.J. Extracellular matrix control of mammary gland morphogenesis and tumorigenesis: Insights from imaging. Histochem. Cell Biol. 2008, 130, 1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzobo, K.; Vogelsang, M.; Thomford, N.E.; Dandara, C.; Kallmeyer, K.; Pepper, M.S.; Parker, M.I. Wharton’s Jelly-Derived Mesenchymal Stromal Cells and Fibroblast-Derived Extracellular Matrix Synergistically Activate Apoptosis in a p21-Dependent Mechanism in WHCO1 and MDA MB 231 Cancer Cells In Vitro. Stem Cells Int. 2016, 2016, 4842134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitale, I.; Manic, G.; Galassi, C.; Galluzzi, L. Stress responses in stromal cells and tumor homeostasis. Pharmacol. Ther. 2019, 200, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.-J. The role of the extracellular matrix in cancer stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef] [Green Version]
- Dzobo, K.; Senthebane, D.A.; Dandara, C. The tumor microenvironment in tumorigenesis and therapy resistance revisited. Cancers 2023, 15, 376. [Google Scholar] [CrossRef]
- Egeblad, M.; Rasch, M.G.; Weaver, V.M. Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 2010, 22, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Deegan, D.B.; Zimmerman, C.; Skardal, A.; Atala, A.; Shupe, T.D. Stiffness of hyaluronic acid gels containing liver extracellular matrix supports human hepatocyte function and alters cell morphology. J. Mech. Behav. Biomed. Mater. 2016, 55, 87–103. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Molecular pathways: Connecting fibrosis and solid tumor metastasis. Clin. Cancer Res. 2014, 20, 3637–3643. [Google Scholar] [CrossRef] [Green Version]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef]
- Cox, T.R.; Bird, D.; Baker, A.M.; Barker, H.E.; Ho, M.W.; Lang, G.; Erler, J.T. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.K.; Kim, H.S.; Jin, T.; Moon, W.K. LOXL4 knockdown enhances tumor growth and lung metastasis through collagen-dependent extracellular matrix changes in triple-negative breast cancer. Oncotarget 2017, 8, 11977–11989. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.H.; Hsia, S.M.; Shieh, T.M. Lysyl Oxidase and the Tumor Microenvironment. Int. J. Mol. Sci. 2016, 18, 62. [Google Scholar] [CrossRef] [Green Version]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Giussani, M.; Triulzi, T.; Sozzi, G.; Tagliabue, E. Tumor extracellular matrix remodeling: New perspectives as a circulating tool in the diagnosis and prognosis of solid tumors. Cells 2019, 8, 81. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.D.; Nakamura, I.; Roberts, L.R. The tumor microenvironment in hepatocellular carcinoma: Current status and therapeutic targets. In Proceedings of the Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2011; pp. 35–43. [Google Scholar]
- Wood, S.L.; Pernemalm, M.; Crosbie, P.A.; Whetton, A.D. The role of the tumor-microenvironment in lung cancer-metastasis and its relationship to potential therapeutic targets. Cancer Treat. Rev. 2014, 40, 558–566. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Cordenonsi, M.; Piccolo, S. YAP/TAZ as therapeutic targets in cancer. Curr. Opin. Pharmacol. 2016, 29, 26–33. [Google Scholar] [CrossRef]
- Fuster, M.M.; Esko, J.D. The sweet and sour of cancer: Glycans as novel therapeutic targets. Nat. Rev. Cancer 2005, 5, 526–542. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A. Overview of the matrisome—An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [Green Version]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, M.; Yuan, J.; Peng, C.; Li, Y. Collagen as a double-edged sword in tumor progression. Tumour Biol. 2014, 35, 2871–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, S.; Vijanto, J.; Raekallio, J.; Penttinen, R. Collagen types in early phases of wound healing in children. Acta Chir. Scand. 1978, 144, 205–211. [Google Scholar]
- Singh, S.; Young, A.; McNaught, C.-E. The physiology of wound healing. Surgery 2017, 35, 473–477. [Google Scholar] [CrossRef]
- Dzobo, K. Matrix-Mediated Regulation of Type 1 Collagen Synthesis and Degradation in Cultured Fibroblasts. Ph.D. Thesis, University of Cape Town, Cape Town, South Africa, 2009. [Google Scholar]
- Scott, I.; Yamauchi, M.; Sricholpech, M. Lysine post-translational modifications of collagen. Essays Biochem. 2012, 52, 113–133. [Google Scholar] [CrossRef] [Green Version]
- Garnero, P.; Borel, O.; Gineyts, E.; Duboeuf, F.; Solberg, H.; Bouxsein, M.L.; Christiansen, C.; Delmas, P.D. Extracellular post-translational modifications of collagen are major determinants of biomechanical properties of fetal bovine cortical bone. Bone 2006, 38, 300–309. [Google Scholar] [CrossRef]
- Sottile, J.; Shi, F.; Rublyevska, I.; Chiang, H.-Y.; Lust, J.; Chandler, J. Fibronectin-dependent collagen I deposition modulates the cell response to fibronectin. Am. J. Physiol. Cell Physiol. 2007, 293, C1934–C1946. [Google Scholar] [CrossRef] [Green Version]
- Kubow, K.E.; Vukmirovic, R.; Zhe, L.; Klotzsch, E.; Smith, M.L.; Gourdon, D.; Luna, S.; Vogel, V. Mechanical forces regulate the interactions of fibronectin and collagen I in extracellular matrix. Nat. Commun. 2015, 6, 8026. [Google Scholar] [CrossRef] [Green Version]
- Dzobo, K.; Leaner, V.D.; Parker, M.I. Absence of feedback regulation in the synthesis of COL1A1. Life Sci. 2014, 103, 25–33. [Google Scholar] [CrossRef]
- Chelyshev, Y.A.; Kabdesh, I.M.; Mukhamedshina, Y.O. Extracellular Matrix in Neural Plasticity and Regeneration. Cell. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Hastings, J.F.; Skhinas, J.N.; Fey, D.; Croucher, D.R.; Cox, T.R. The extracellular matrix as a key regulator of intracellular signalling networks. Br. J. Pharmacol. 2019, 176, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Amatangelo, M.D.; Bassi, D.E.; Klein-Szanto, A.J.; Cukierman, E. Stroma-derived three-dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am. J. Pathol. 2005, 167, 475–488. [Google Scholar] [CrossRef] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.D. Cell Biology of Extracellular Matrix; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar]
- Sonbol, H.S. Extracellular matrix remodeling in human disease. J. Microsc. Ultrastruct. 2018, 6, 123. [Google Scholar] [CrossRef]
- Candiello, J.; Balasubramani, M.; Schreiber, E.M.; Cole, G.J.; Mayer, U.; Halfter, W.; Lin, H. Biomechanical properties of native basement membranes. FEBS J. 2007, 274, 2897–2908. [Google Scholar] [CrossRef]
- Amenta, P.S.; Briggs, K.; Xu, K.; Gamboa, E.; Jukkola, A.F.; Li, D.; Myers, J.C. Type XV collagen in human colonic adenocarcinomas has a different distribution than other basement membrane zone proteins. Hum. Pathol. 2000, 31, 359–366. [Google Scholar] [CrossRef]
- Tosios, K.; Kapranos, N.; Papanicolaou, S. Loss of basement membrane components laminin and type IV collagen parallels the progression of oral epithelial neoplasia. Histopathology 1998, 33, 261–268. [Google Scholar] [CrossRef]
- Spivey, K.A.; Chung, I.; Banyard, J.; Adini, I.; Feldman, H.A.; Zetter, B.R. A role for collagen XXIII in cancer cell adhesion, anchorage-independence and metastasis. Oncogene 2012, 31, 2362–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, C.D. Pancreatic cancer is suppressed by fibroblast-derived collagen I. Cancer Cell 2021, 39, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Kresse, H.; Schönherr, E. Proteoglycans of the extracellular matrix and growth control. J. Cell. Physiol. 2001, 189, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Loganathan, D.; Merchant, Z.; Linhardt, R. Carbohydrate analysis of glycoproteins A review. Appl. Biochem. Biotechnol. 1990, 23, 53–80. [Google Scholar] [CrossRef]
- Hughes, R.C. Membrane Glycoproteins: A Review of Structure and Function; The Butterworth Group: London, UK, 2014. [Google Scholar]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Hardingham, T.E.; Fosang, A.J. Proteoglycans: Many forms and many functions. FASEB J. 1992, 6, 861–870. [Google Scholar] [CrossRef]
- Iozzo, R.V. Matrix proteoglycans: From molecular design to cellular function. Annu. Rev. Biochem. 1998, 67, 609–652. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, J.; Ishihara, A.; Fukunaga, K.; Sasaki, K.; White, M.J.V.; Briquez, P.S.; Hubbell, J.A. Laminin heparin-binding peptides bind to several growth factors and enhance diabetic wound healing. Nat. Commun. 2018, 9, 2163. [Google Scholar] [CrossRef] [Green Version]
- Broekelmann, T.J.; Bodmer, N.K.; Mecham, R.P. Identification of the growth factor—Binding sequence in the extracellular matrix protein MAGP-1. J. Biol. Chem. 2020, 295, 2687–2697. [Google Scholar] [CrossRef]
- Bohaumilitzky, L.; Huber, A.K.; Stork, E.M.; Wengert, S.; Woelfl, F.; Boehm, H. A Trickster in Disguise: Hyaluronan’s Ambivalent Roles in the Matrix. Front. Oncol. 2017, 7, 242. [Google Scholar] [CrossRef] [Green Version]
- Price, Z.K.; Lokman, N.A.; Ricciardelli, C. Differing roles of hyaluronan molecular weight on cancer cell behavior and chemotherapy resistance. Cancers 2018, 10, 482. [Google Scholar] [CrossRef] [Green Version]
- Januchowski, R.; Zawierucha, P.; Ruciński, M.; Nowicki, M.; Zabel, M. Extracellular Matrix Proteins Expression Profiling in Chemoresistant Variants of the A2780 Ovarian Cancer Cell Line. BioMed Res. Int. 2014, 2014, 365867. [Google Scholar] [CrossRef] [Green Version]
- Timpl, R.; Rohde, H.; Robey, P.G.; Rennard, S.I.; Foidart, J.M.; Martin, G.R. Laminin—A glycoprotein from basement membranes. J. Biol. Chem. 1979, 254, 9933–9937. [Google Scholar] [CrossRef]
- Hohenester, E.; Yurchenco, P.D. Laminins in basement membrane assembly. Cell Adhes. Migr. 2013, 7, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Domogatskaya, A.; Rodin, S.; Tryggvason, K. Functional diversity of laminins. Annu. Rev. Cell Dev. Biol. 2012, 28, 523–553. [Google Scholar] [CrossRef]
- Hohenester, E. Structural biology of laminins. Essays Biochem. 2019, 63, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Aumailley, M. The laminin family. Cell Adh. Migr. 2013, 7, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Fukazawa, S.; Shinto, E.; Tsuda, H.; Ueno, H.; Shikina, A.; Kajiwara, Y.; Yamamoto, J.; Hase, K. Laminin β3 expression as a prognostic factor and a predictive marker of chemoresistance in colorectal cancer. Jpn. J. Clin. Oncol. 2015, 45, 533–540. [Google Scholar] [CrossRef] [Green Version]
- Govaere, O.; Wouters, J.; Petz, M.; Vandewynckel, Y.P.; Van den Eynde, K.; Van den Broeck, A.; Verhulst, S.; Dolle, L.; Gremeaux, L.; Ceulemans, A.; et al. Laminin-332 sustains chemoresistance and quiescence as part of the human hepatic cancer stem cell niche. J. Hepatol. 2016, 64, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Hasebe, T.; Oda, T.; Sasaki, S.; Kinoshita, T.; Konishi, M.; Ochiai, T.; Ochiai, A. Cytoplasmic expression of laminin γ2 chain correlates with postoperative hepatic metastasis and poor prognosis in patients with pancreatic ductal adenocarcinoma. Cancer 2002, 94, 1894–1901. [Google Scholar] [CrossRef]
- Shang, M.; Koshikawa, N.; Schenk, S.; Quaranta, V. The LG3 module of laminin-5 harbors a binding site for integrin α3β1 that promotes cell adhesion, spreading, and migration. J. Biol. Chem. 2001, 276, 33045–33053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.-C.; Ziober, B.L.; Squillace, R.M.; Kramer, R.H. α7 integrin mediates cell adhesion and migration on specific laminin isoforms. J. Biol. Chem. 1996, 271, 25598–25603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannelli, G.; Azzariti, A.; Fransvea, E.; Porcelli, L.; Antonaci, S.; Paradiso, A. Laminin-5 offsets the efficacy of gefitinib (‘Iressa’) in hepatocellular carcinoma cells. Br. J. Cancer 2004, 91, 1964–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsurutani, J.; West, K.A.; Sayyah, J.; Gills, J.J.; Dennis, P.A. Inhibition of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway but not the MEK/ERK pathway attenuates laminin-mediated small cell lung cancer cellular survival and resistance to imatinib mesylate or chemotherapy. Cancer Res. 2005, 65, 8423–8432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [Green Version]
- Gopal, S.; Veracini, L.; Grall, D.; Butori, C.; Schaub, S.; Audebert, S.; Camoin, L.; Baudelet, E.; Radwanska, A.; Beghelli-de la Forest Divonne, S.; et al. Fibronectin-guided migration of carcinoma collectives. Nat. Commun. 2017, 8, 14105. [Google Scholar] [CrossRef] [Green Version]
- Rintoul, R.C.; Sethi, T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. Clin. Sci. 2002, 102, 417–424. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Argilagos, R.F.; Emmons, M.; Boulware, D.; Beam, C.A.; Sullivan, D.M.; Dalton, W.S. Cell adhesion to fibronectin (CAM-DR) influences acquired mitoxantrone resistance in U937 cells. Cancer Res. 2006, 66, 2338–2345. [Google Scholar] [CrossRef] [Green Version]
- Kosmehl, H.; Berndt, A.; Strassburger, S.; Borsi, L.; Rousselle, P.; Mandel, U.; Hyckel, P.; Zardi, L.; Katenkamp, D. Distribution of laminin and fibronectin isoforms in oral mucosa and oral squamous cell carcinoma. Br. J. Cancer 1999, 81, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, M.; Zardi, L.; Neri, D. Fibronectin as target for tumor therapy. Int. J. Cancer 2006, 118, 1331–1339. [Google Scholar] [CrossRef]
- Liu, W.; Cheng, S.; Asa, S.L.; Ezzat, S. The melanoma-associated antigen A3 mediates fibronectin-controlled cancer progression and metastasis. Cancer Res. 2008, 68, 8104–8112. [Google Scholar] [CrossRef] [Green Version]
- Bae, Y.K.; Kim, A.; Kim, M.K.; Choi, J.E.; Kang, S.H.; Lee, S.J. Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum. Pathol. 2013, 44, 2028–2037. [Google Scholar] [CrossRef]
- Hu, D.; Ansari, D.; Zhou, Q.; Sasor, A.; Said Hilmersson, K.; Andersson, R. Stromal fibronectin expression in patients with resected pancreatic ductal adenocarcinoma. World J. Surg. Oncol. 2019, 17, 1–8. [Google Scholar] [CrossRef]
- Singh, P.; Reimer, C.L.; Peters, J.H.; Stepp, M.A.; Hynes, R.O.; Van De Water, L. The spatial and temporal expression patterns of integrin α9β1 and one of its ligands, the EIIIA segment of fibronectin, in cutaneous wound healing. J. Investig. Dermatol. 2004, 123, 1176–1181. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Sidell, N.; Roser-Page, S.; Roman, J. Fibronectin stimulates human lung carcinoma cell growth by inducing cyclooxygenase-2 (COX-2) expression. Int. J. Cancer 2004, 111, 322–331. [Google Scholar] [CrossRef]
- Han, S.; Roman, J. Fibronectin induces cell proliferation and inhibits apoptosis in human bronchial epithelial cells: Pro-oncogenic effects mediated by PI3-kinase and NF-κB. Oncogene 2006, 25, 4341–4349. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Sidell, N.; Roman, J. Fibronectin stimulates human lung carcinoma cell proliferation by suppressing p21 gene expression via signals involving Erk and Rho kinase. Cancer Lett. 2005, 219, 71–81. [Google Scholar] [CrossRef]
- Xing, H.; Weng, D.; Chen, G.; Tao, W.; Zhu, T.; Yang, X.; Meng, L.; Wang, S.; Lu, Y.; Ma, D. Activation of fibronectin/PI-3K/Akt2 leads to chemoresistance to docetaxel by regulating survivin protein expression in ovarian and breast cancer cells. Cancer Lett. 2008, 261, 108–119. [Google Scholar] [CrossRef]
- Horiuchi, K.; Amizuka, N.; Takeshita, S.; Takamatsu, H.; Katsuura, M.; Ozawa, H.; Toyama, Y.; Bonewald, L.F.; Kudo, A. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 1999, 14, 1239–1249. [Google Scholar] [CrossRef]
- Moniuszko, T.; Wincewicz, A.; Koda, M.; Domysławska, I.; Sulkowski, S. Role of periostin in esophageal, gastric and colon cancer. Oncol. Lett. 2016, 12, 783–787. [Google Scholar] [CrossRef] [Green Version]
- Gillan, L.; Matei, D.; Fishman, D.A.; Gerbin, C.S.; Karlan, B.Y.; Chang, D.D. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 2002, 62, 5358–5364. [Google Scholar] [PubMed]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.J.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, T.; Tamai, K.; Shibuya, R.; Nakamura, M.; Mochizuki, M.; Yamaguchi, K.; Abe, J.; Takahashi, S.; Sato, I.; Kudo, A. Periostin is a negative prognostic factor and promotes cancer cell proliferation in non-small cell lung cancer. Oncotarget 2018, 9, 31187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, M.; Fejzo, M.S.; Anderson, L.; Dering, J.; Ginther, C.; Ramos, L.; Gasson, J.C.; Karlan, B.Y.; Slamon, D.J. Periostin promotes ovarian cancer angiogenesis and metastasis. Gynecol. Oncol. 2010, 119, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Tumbarello, D.A.; Temple, J.; Brenton, J.D. ß3 integrin modulates transforming growth factor beta induced (TGFBI) function and paclitaxel response in ovarian cancer cells. Mol. Cancer 2012, 11, 36. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.-L.; Jan, Y.-H.; Lin, S.-C.; Huang, C.-C.; Lin, H.; Wen, K.-C.; Chao, K.-C.; Lai, C.-R.; Wang, P.-H.; Chuang, C.-M.; et al. Periostin in tumor microenvironment is associated with poor prognosis and platinum resistance in epithelial ovarian carcinoma. Oncotarget 2016, 7, 4036–4047. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Du, L. Role of pancreatic stellate cells and periostin in pancreatic cancer progression. Tumor Biol. 2015, 36, 3171–3177. [Google Scholar] [CrossRef]
- Tammi, M.I.; Day, A.J.; Turley, E.A. Hyaluronan and homeostasis: A balancing act. J. Biol. Chem. 2002, 277, 4581–4584. [Google Scholar] [CrossRef] [Green Version]
- Fallacara, A.; Baldini, E.; Manfredini, S.; Vertuani, S. Hyaluronic acid in the third millennium. Polymers 2018, 10, 701. [Google Scholar] [CrossRef] [Green Version]
- Kakehi, K.; Kinoshita, M.; Yasueda, S.-i. Hyaluronic acid: Separation and biological implications. J. Chromatogr. B 2003, 797, 347–355. [Google Scholar] [CrossRef]
- Scott, J.E. Supramolecular organization of extracellular matrix glycosaminoglycans, in vitro and in the tissues. FASEB J. 1992, 6, 2639–2645. [Google Scholar] [CrossRef]
- Kupper, S.; Kłosowska-Chomiczewska, I.; Szumała, P. Collagen and hyaluronic acid hydrogel in water-in-oil microemulsion delivery systems. Carbohydr. Polym. 2017, 175, 347–354. [Google Scholar] [CrossRef]
- Pereira, H.; Sousa, D.A.; Cunha, A.; Andrade, R.; Espregueira-Mendes, J.; Oliveira, J.M.; Reis, R.L. Hyaluronic acid. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2018; pp. 137–153. [Google Scholar]
- Henry, C.B.; Duling, B.R. Permeation of the luminal capillary glycocalyx is determined by hyaluronan. Am. J. Physiol. Heart Circ. Physiol. 1999, 277, H508–H514. [Google Scholar] [CrossRef]
- Barbucci, R.; Lamponi, S.; Borzacchiello, A.; Ambrosio, L.; Fini, M.; Torricelli, P.; Giardino, R. Hyaluronic acid hydrogel in the treatment of osteoarthritis. Biomaterials 2002, 23, 4503–4513. [Google Scholar] [CrossRef]
- Luo, Y.; Kirker, K.R.; Prestwich, G.D. Cross-linked hyaluronic acid hydrogel films: New biomaterials for drug delivery. J. Control. Release 2000, 69, 169–184. [Google Scholar] [CrossRef]
- Bhattacharya, D.S.; Svechkarev, D.; Souchek, J.; Hill, T.K.; Taylor, M.; Natarajan, A.; Mohs, A.M. Impact of structurally modifying hyaluronic acid on CD44 interaction. J. Mater. Chem. B 2017, 5, 8183–8192. [Google Scholar] [CrossRef]
- Necas, J.; Bartosikova, L.; Brauner, P.; Kolar, J. Hyaluronic acid (hyaluronan): A review. Vet. Med. 2008, 53, 397–411. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.H. Extracellular matrix in development: Insights from mechanisms conserved between invertebrates and vertebrates. Cold Spring. Harb. Perspect. Biol. 2011, 3, a005082. [Google Scholar] [CrossRef] [Green Version]
- Entchev, E.V.; González-Gaitán, M.A. Morphogen gradient formation and vesicular trafficking. Traffic 2002, 3, 98–109. [Google Scholar] [CrossRef]
- Marois, E.; Mahmoud, A.; Eaton, S. The endocytic pathway and formation of the Wingless morphogen gradient. Development 2006, 133, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Uhler, C.; Shivashankar, G.V. Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol. 2017, 18, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Ilic, D.; Damsky, C.H.; Yamamoto, T. Focal adhesion kinase: At the crossroads of signal transduction. J. Cell Sci. 1997, 110, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Kai, F.; Laklai, H.; Weaver, V.M. Force Matters: Biomechanical Regulation of Cell Invasion and Migration in Disease. Trends Cell Biol. 2016, 26, 486–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kim, D.-H.; Levchenko, A. Topotaxis: A new mechanism of directed cell migration in topographic ECM gradients. Biophys. J. 2018, 114, 1257–1263. [Google Scholar] [CrossRef] [Green Version]
- Doyle, A.D.; Petrie, R.J.; Kutys, M.L.; Yamada, K.M. Dimensions in cell migration. Curr. Opin. Cell Biol. 2013, 25, 642–649. [Google Scholar] [CrossRef] [Green Version]
- Gunawan, R.C.; Silvestre, J.; Gaskins, H.R.; Kenis, P.J.; Leckband, D.E. Cell migration and polarity on microfabricated gradients of extracellular matrix proteins. Langmuir 2006, 22, 4250–4258. [Google Scholar] [CrossRef]
- Wu, J.; Mao, Z.; Tan, H.; Han, L.; Ren, T.; Gao, C. Gradient biomaterials and their influences on cell migration. Interface Focus 2012, 2, 337–355. [Google Scholar] [CrossRef] [Green Version]
- Shellard, A.; Mayor, R. All roads lead to directional cell migration. Trends Cell Biol. 2020, 30, 852–868. [Google Scholar] [CrossRef]
- Palecek, S.P.; Loftus, J.C.; Ginsberg, M.H.; Lauffenburger, D.A.; Horwitz, A.F. Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 1997, 385, 537–540. [Google Scholar] [CrossRef]
- Hartman, C.D.; Isenberg, B.C.; Chua, S.G.; Wong, J.Y. Extracellular matrix type modulates cell migration on mechanical gradients. Exp. Cell Res. 2017, 359, 361–366. [Google Scholar] [CrossRef]
- Plotnikov, S.V.; Waterman, C.M. Guiding cell migration by tugging. Curr. Opin. Cell Biol. 2013, 25, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Pathak, A.; Kumar, S. Independent regulation of tumor cell migration by matrix stiffness and confinement. Proc. Natl. Acad. Sci. USA 2012, 109, 10334–10339. [Google Scholar] [CrossRef] [Green Version]
- Charras, G.; Sahai, E. Physical influences of the extracellular environment on cell migration. Nat. Rev. Mol. Cell Biol. 2014, 15, 813–824. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Rainero, E. Extracellular matrix endocytosis in controlling matrix turnover and beyond: Emerging roles in cancer. Biochem. Soc. Trans. 2016, 44, 1347–1354. [Google Scholar] [CrossRef]
- Hinck, L.; Silberstein, G.B. Key stages in mammary gland development: The mammary end bud as a motile organ. Breast Cancer Res 2005, 7, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Alford, D.; Baeckström, D.; Geyp, M.; Pitha, P.; Taylor-Papadimitriou, J. Integrin-matrix interactions affect the form of the structures developing from human mammary epithelial cells in collagen or fibrin gels. J. Cell. Sci. 1998, 111, 521–532. [Google Scholar] [CrossRef]
- Sakai, T.; Larsen, M.; Yamada, K.M. Fibronectin requirement in branching morphogenesis. Nature 2003, 423, 876–881. [Google Scholar] [CrossRef]
- Ortega, N.; Werb, Z. New functional roles for non-collagenous domains of basement membrane collagens. J. Cell Sci. 2002, 115, 4201–4214. [Google Scholar] [CrossRef] [Green Version]
- Sternlicht, M.D.; Kouros-Mehr, H.; Lu, P.; Werb, Z. Hormonal and local control of mammary branching morphogenesis. Differentiation 2006, 74, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, B. Biology of the extracellular matrix: An overview. J. Glaucoma 2014, 23, S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walma, D.A.C.; Yamada, K.M. The extracellular matrix in development. Development 2020, 147, dev175596. [Google Scholar] [CrossRef] [PubMed]
- Streuli, C.H.; Schmidhauser, C.; Bailey, N.; Yurchenco, P.; Skubitz, A.P.; Roskelley, C.; Bissell, M.J. Laminin mediates tissue-specific gene expression in mammary epithelia. J. Cell Biol. 1995, 129, 591–603. [Google Scholar] [CrossRef]
- Muncie, J.M.; Weaver, V.M. The physical and biochemical properties of the extracellular matrix regulate cell fate. Curr. Top. Dev. Biol. 2018, 130, 1–37. [Google Scholar]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta 2014, 1840, 2506–2519. [Google Scholar] [CrossRef]
- Dityatev, A.; Schachner, M.; Sonderegger, P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neuroscience 2010, 11, 735–746. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Mongiat, M.; Andreuzzi, E.; Tarticchio, G.; Paulitti, A. Extracellular matrix, a hard player in angiogenesis. Int. J. Mol. Sci. 2016, 17, 1822. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Zhou, C.; Wang, Q.; Cai, L.; Du, W.; Li, X.; Zhou, X.; Xie, J. Extracellular matrix elasticity regulates osteocyte gap junction elongation: Involvement of paxillin in intracellular signal transduction. Cell. Physiol. Biochem. 2018, 51, 1013–1026. [Google Scholar] [CrossRef]
- Oh, D.-J.; Kang, M.H.; Ooi, Y.H.; Choi, K.R.; Sage, E.H.; Rhee, D.J. Overexpression of SPARC in human trabecular meshwork increases intraocular pressure and alters extracellular matrix. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3309–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terajima, M.; Taga, Y.; Cabral, W.A.; Liu, Y.; Nagasawa, M.; Sumida, N.; Kayashima, Y.; Chandrasekaran, P.; Han, L.; Maeda, N. Cyclophilin B control of lysine post-translational modifications of skin type I collagen. PLoS Genet. 2019, 15, e1008196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynders, M.; Matsuura, B.S.; Bérouti, M.; Simoneschi, D.; Marzio, A.; Pagano, M.; Trauner, D. PHOTACs enable optical control of protein degradation. Sci. Adv. 2020, 6, eaay5064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Chen, H.; Ma, L.; He, Z.; Wang, D.; Liu, Y.; Lin, Q.; Zhang, T.; Gray, N.; Kaniskan, H.Ü. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020, 6, eaay5154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, A.; Liu, J.; Deiters, A. Targeted protein degradation through fast optogenetic activation and its application to the control of cell signaling. J. Am. Chem. Soc. 2021, 143, 9222–9229. [Google Scholar] [CrossRef]
- Petrie, R.J.; Gavara, N.; Chadwick, R.S.; Yamada, K.M. Nonpolarized signaling reveals two distinct modes of 3D cell migration. J. Cell Biol. 2012, 197, 439–455. [Google Scholar] [CrossRef] [Green Version]
- Viji Babu, P.K.; Rianna, C.; Mirastschijski, U.; Radmacher, M. Nano-mechanical mapping of interdependent cell and ECM mechanics by AFM force spectroscopy. Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Maller, O.; Martinson, H.; Schedin, P. Extracellular matrix composition reveals complex and dynamic stromal-epithelial interactions in the mammary gland. J. Mammary Gland Biol. Neoplasia 2010, 15, 301–318. [Google Scholar] [CrossRef]
- Naba, A.; Clauser, K.R.; Hynes, R.O. Enrichment of extracellular matrix proteins from tissues and digestion into peptides for mass spectrometry analysis. JoVE 2015, 101, e53057. [Google Scholar]
- Robertson, J.; Humphries, J.D.; Paul, N.R.; Warwood, S.; Knight, D.; Byron, A.; Humphries, M.J. Characterization of the phospho-adhesome by mass spectrometry-based proteomics. Kinase Signal. Netw. 2017, 1636, 235–251. [Google Scholar]
- De Castro Brás, L.E.; Ramirez, T.A.; DeLeon-Pennell, K.Y.; Chiao, Y.A.; Ma, Y.; Dai, Q.; Halade, G.V.; Hakala, K.; Weintraub, S.T.; Lindsey, M.L. Texas 3-step decellularization protocol: Looking at the cardiac extracellular matrix. J. Proteom. 2013, 86, 43–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera, J.A.; Mallikarjun, V.; Rosini, S.; Montero, M.A.; Lawless, C.; Warwood, S.; O’Cualain, R.; Knight, D.; Schwartz, M.A.; Swift, J. Laser capture microdissection coupled mass spectrometry (LCM-MS) for spatially resolved analysis of formalin-fixed and stained human lung tissues. Clin. Proteom. 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Coronado, R.E.; Somaraki-Cormier, M.; Natesan, S.; Christy, R.J.; Ong, J.L.; Halff, G.A. Decellularization and solubilization of porcine liver for use as a substrate for porcine hepatocyte culture: Method optimization and comparison. Cell Transplant. 2017, 26, 1840–1854. [Google Scholar] [CrossRef] [Green Version]
- Alevra Sarika, N.; Payen, V.L.; Fléron, M.; Ravau, J.; Brusa, D.; Najimi, M.; De Pauw, E.; Eppe, G.; Mazzucchelli, G.; Sokal, E.M. Human liver-derived extracellular matrix for the culture of distinct human primary liver cells. Cells 2020, 9, 1357. [Google Scholar] [CrossRef]
- Byron, A.; Humphries, J.D.; Humphries, M.J. Defining the extracellular matrix using proteomics. Int. J. Exp. Pathol. 2013, 94, 75–92. [Google Scholar] [CrossRef]
- Goddard, E.T.; Hill, R.C.; Barrett, A.; Betts, C.; Guo, Q.; Maller, O.; Borges, V.F.; Hansen, K.C.; Schedin, P. Quantitative extracellular matrix proteomics to study mammary and liver tissue microenvironments. Int. J. Biochem. Cell Biol. 2016, 81, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Krasny, L.; Bland, P.; Kogata, N.; Wai, P.; Howard, B.A.; Natrajan, R.C.; Huang, P.H. SWATH mass spectrometry as a tool for quantitative profiling of the matrisome. J. Proteom. 2018, 189, 11–22. [Google Scholar] [CrossRef]
- Nguyen, E.V.; Pereira, B.A.; Lawrence, M.G.; Ma, X.; Rebello, R.J.; Chan, H.; Niranjan, B.; Wu, Y.; Ellem, S.; Guan, X. Proteomic Profiling of Human Prostate Cancer-associated Fibroblasts (CAF) Reveals LOXL2-dependent Regulation of the Tumor Microenvironment*[S]. Mol. Cell. Proteom. 2019, 18, 1410–1427. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.-Y.; Schepmoes, A.A.; Zhang, X.; Moore, R.J.; Monroe, M.E.; Lee, J.H.; Camp, D.G.; Smith, R.D.; Qian, W.-J. Improved LC− MS/MS spectral counting statistics by recovering low-scoring spectra matched to confidently identified peptide sequences. J. Proteome Res. 2010, 9, 5698–5704. [Google Scholar] [CrossRef] [Green Version]
- Toghi Eshghi, S.; Shah, P.; Yang, W.; Li, X.; Zhang, H. GPQuest: A spectral library matching algorithm for site-specific assignment of tandem mass spectra to intact N-glycopeptides. Anal. Chem. 2015, 87, 5181–5188. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Herrmann, C.J.; Simonovic, M.; Szklarczyk, D.; von Mering, C. Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 2015, 15, 3163–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roycik, M.D.; Fang, X.; Sang, Q.X. A fresh prospect of extracellular matrix hydrolytic enzymes and their substrates. Curr. Pharm. Des. 2009, 15, 1295–1308. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J. Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis. Matrix Biol. 2003, 22, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Xu, R. Roles of PLODs in Collagen Synthesis and Cancer Progression. Front. Cell Dev. Biol. 2018, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Bajpai, S.; Wong, C.C.; Chaturvedi, P.; Hubbi, M.E.; Wirtz, D.; Semenza, G.L. Procollagen Lysyl Hydroxylase 2 Is Essential for Hypoxia-Induced Breast Cancer MetastasisPLOD2 Is Essential for Hypoxia-Induced Metastasis. Mol. Cancer Res. 2013, 11, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Eddy, A.A. Molecular basis of renal fibrosis. Pediatr. Nephrol. 2000, 15, 290–301. [Google Scholar] [CrossRef]
- Alcolado, R.; Arthur, M.; Iredale, J. Pathogenesis of liver fibrosis. Clin. Sci. 1997, 92, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Libring, S.; Shinde, A.; Chanda, M.K.; Nuru, M.; George, H.; Saleh, A.M.; Abdullah, A.; Kinzer-Ursem, T.L.; Calve, S.; Wendt, M.K. The dynamic relationship of breast cancer cells and fibroblasts in fibronectin accumulation at primary and metastatic tumor sites. Cancers 2020, 12, 1270. [Google Scholar] [CrossRef]
- Comoglio, P.M.; Trusolino, L. Cancer: The matrix is now in control. Nat. Med. 2005, 11, 1156–1158. [Google Scholar] [CrossRef]
- Pires, A.; Greenshields-Watson, A.; Jones, E.; Smart, K.; Lauder, S.N.; Somerville, M.; Milutinovic, S.; Kendrick, H.; Hindley, J.P.; French, R.; et al. Immune Remodeling of the Extracellular Matrix Drives Loss of Cancer Stem Cells and Tumor Rejection. Cancer Immunol. Res. 2020, 8, 1520–1531. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Wang, C.Y.; Werb, Z. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol. 2015, 44-46, 184–190. [Google Scholar] [CrossRef]
- Fonović, M.; Turk, B. Cysteine cathepsins and extracellular matrix degradation. Biochim. Biophys. Acta 2014, 1840, 2560–2570. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Fibrosis and Cancer: Partners in Crime or Opposing Forces? Trends Cancer 2016, 2, 279–282. [Google Scholar] [CrossRef]
- Filipe, E.C.; Chitty, J.L.; Cox, T.R. Charting the unexplored extracellular matrix in cancer. Int. J. Exp. Pathol. 2018, 99, 58–76. [Google Scholar] [CrossRef] [Green Version]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [Green Version]
- Nicolas-Boluda, A.; Vaquero, J.; Vimeux, L.; Guilbert, T.; Barrin, S.; Kantari-Mimoun, C.; Ponzo, M.; Renault, G.; Deptula, P.; Pogoda, K.; et al. Tumor stiffening reversion through collagen crosslinking inhibition improves T cell migration and anti-PD-1 treatment. Elife 2021, 10, e58688. [Google Scholar] [CrossRef]
- Kench, J.A.; Russell, D.M.; Fadok, V.A.; Young, S.K.; Worthen, G.S.; Jones-Carson, J.; Henson, J.E.; Henson, P.M.; Nemazee, D. Aberrant wound healing and TGF-β production in the autoimmune-prone MRL/+ mouse. Clin. Immunol. 1999, 92, 300–310. [Google Scholar] [CrossRef]
- Zhao, Y.; Bao, L.; Chan, L.S.; DiPietro, L.A.; Chen, L. Aberrant wound healing in an epidermal interleukin-4 transgenic mouse model of atopic dermatitis. PLoS ONE 2016, 11, e0146451. [Google Scholar] [CrossRef]
- Hertle, M.D.; Kubler, M.-D.; Leigh, I.M.; Watt, F. Aberrant integrin expression during epidermal wound healing and in psoriatic epidermis. J. Clin. Investig. 1992, 89, 1892–1901. [Google Scholar] [CrossRef]
- Stephens, P.; Grenard, P.; Aeschlimann, P.; Langley, M.; Blain, E.; Errington, R.; Kipling, D.; Thomas, D.; Aeschlimann, D. Crosslinking and G-protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. J. Cell Sci. 2004, 117, 3389–3403. [Google Scholar] [CrossRef] [Green Version]
- Chitty, J.L.; Setargew, Y.F.I.; Cox, T.R. Targeting the lysyl oxidases in tumour desmoplasia. Biochem. Soc. Trans. 2019, 47, 1661–1678. [Google Scholar] [CrossRef] [PubMed]
- Ponce, I.; Garrido, N.; Tobar, N.; Melo, F.; Smith, P.C.; Martínez, J. Matrix stiffness modulates metabolic interaction between human stromal and breast cancer cells to stimulate epithelial motility. Metabolites 2021, 11, 432. [Google Scholar] [CrossRef] [PubMed]
- DuFort, C.C.; DelGiorno, K.E.; Hingorani, S.R. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology 2016, 150, 1545–1557.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpellini, A.; Germack, R.; Lortat-Jacob, H.; Muramatsu, T.; Billett, E.; Johnson, T.; Verderio, E.A. Heparan sulfate proteoglycans are receptors for the cell-surface trafficking and biological activity of transglutaminase-2. J. Biol. Chem. 2009, 284, 18411–18423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, I.; Stamnaes, J.; Andersen, J.T.; Melino, G.; Iversen, R.; Sollid, L.M. Transglutaminase 2 interactions with extracellular matrix proteins as probed with celiac disease autoantibodies. FEBS J. 2015, 282, 2063–2075. [Google Scholar] [CrossRef]
- Akimov, S.S.; Krylov, D.; Fleischman, L.F.; Belkin, A.M. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J. Cell Biol. 2000, 148, 825–838. [Google Scholar] [CrossRef] [Green Version]
- Richter, P.; Junker, K.; Franz, M.; Berndt, A.; Geyer, C.; Gajda, M.; Kosmehl, H.; Berndt, A.; Wunderlich, H. IIICS de novo glycosylated fibronectin as a marker for invasiveness in urothelial carcinoma of the urinary bladder (UBC). J. Cancer Res. Clin. Oncol. 2008, 134, 1059–1065. [Google Scholar] [CrossRef]
- Freire-de-Lima, L.; Gelfenbeyn, K.; Ding, Y.; Mandel, U.; Clausen, H.; Handa, K.; Hakomori, S.-i. Involvement of O-glycosylation defining oncofetal fibronectin in epithelial-mesenchymal transition process. Proc. Natl. Acad. Sci. USA 2011, 108, 17690–17695. [Google Scholar] [CrossRef] [Green Version]
- SUZUkI, O.; Abe, M.; Hashimoto, Y. Sialylation and glycosylation modulate cell adhesion and invasion to extracellular matrix in human malignant lymphoma: Dependency on integrin and the Rho GTPase family. Int. J. Oncol. 2015, 47, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Singh, C.; Shyanti, R.K.; Singh, V.; Kale, R.K.; Mishra, J.P.; Singh, R.P. Integrin expression and glycosylation patterns regulate cell-matrix adhesion and alter with breast cancer progression. Biochem. Biophys. Res. Commun. 2018, 499, 374–380. [Google Scholar] [CrossRef]
- Rathinam, R.; Alahari, S.K. Important role of integrins in the cancer biology. Cancer Metastasis Rev. 2010, 29, 223–237. [Google Scholar] [CrossRef]
- Yalak, G.; Vogel, V. Ectokinases as novel cancer markers and drug targets in cancer therapy. Cancer Med. 2015, 4, 404–414. [Google Scholar] [CrossRef]
- Yalak, G.; Shiu, J.-Y.; Schoen, I.; Mitsi, M.; Vogel, V. Phosphorylated fibronectin enhances cell attachment and upregulates mechanical cell functions. PLoS ONE 2019, 14, e0218893. [Google Scholar] [CrossRef] [Green Version]
- Wolanska, K.I.; Morgan, M.R. Fibronectin remodelling: Cell-mediated regulation of the microenvironment. Biochem. Soc. Trans. 2015, 43, 122–128. [Google Scholar] [CrossRef]
- Soares Da Costa, D.; Reis, R.L.; Pashkuleva, I. Sulfation of glycosaminoglycans and its implications in human health and disorders. Annu. Rev. Biomed. Eng. 2017, 19, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Escobar Galvis, M.L.; Jia, J.; Zhang, X.; Jastrebova, N.; Spillmann, D.; Gottfridsson, E.; Van Kuppevelt, T.H.; Zcharia, E.; Vlodavsky, I.; Lindahl, U. Transgenic or tumor-induced expression of heparanase upregulates sulfation of heparan sulfate. Nat. Chem. Biol. 2007, 3, 773–778. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Gross-Cohen, M.; Weissmann, M.; Ilan, N.; Sanderson, R.D. Opposing functions of heparanase-1 and heparanase-2 in cancer progression. Trends Biochem. Sci. 2018, 43, 18–31. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Hamacher, S.; Matern, S.; Roeb, E. Extracellular matrix—From basic research to clinical significance. An overview with special consideration of matrix metalloproteinases. Dtsch. Med. Wochenschr. 2004, 129, 1976–1980. [Google Scholar]
- Toth, M.; Fridman, R. Assessment of gelatinases (MMP-2 and MMP-9 by gelatin zymography. In Metastasis Research Protocols; Springer: Berlin/Heidelberg, Germany, 2001; Volume 57, pp. 163–174. [Google Scholar]
- Zhou, Z.; Liu, Y.; Gao, S.; Zhou, M.; Qi, F.; Ding, N.; Zhang, J.; Li, R.; Wang, J.; Shi, J. Excessive DNA damage mediates ECM degradation via the RBBP8/NOTCH1 pathway in sporadic aortic dissection. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166303. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef] [PubMed]
- Stetler-Stevenson, W.G.; Liotta, L.A.; Kleiner, D.E., Jr. Extracellular matrix 6: Role of matrix metalloproteinases in tumor invasion and metastasis. FASEB J. 1993, 7, 1434–1441. [Google Scholar] [CrossRef] [PubMed]
- Yuzhalin, A.E.; Lim, S.Y.; Kutikhin, A.G.; Gordon-Weeks, A.N. Dynamic matrisome: ECM remodeling factors licensing cancer progression and metastasis. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 207–228. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Cena, J.; Schulz, R. Acute actions and novel targets of matrix metalloproteinases in the heart and vasculature. Br. J. Pharmacol. 2007, 152, 189–205. [Google Scholar] [CrossRef] [Green Version]
- Mott, J.D.; Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 2004, 16, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Siqueira, A.S.; Carvalho, M.R.; Monteiro, A.C.; Freitas, V.M.; Jaeger, R.G.; Pinheiro, J.J. Matrix metalloproteinases, TIMPs and growth factors regulating ameloblastoma behaviour. Histopathology 2010, 57, 128–137. [Google Scholar] [CrossRef]
- Rodríguez, D.; Morrison, C.J.; Overall, C.M. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Nagase, H.; Visse, R.; Murphy, G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Shimoda, M.; Ohtsuka, T.; Okada, Y.; Kanai, Y. Stromal metalloproteinases: Crucial contributors to the tumor microenvironment. Pathol. Int. 2021, 71, 1–14. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Mittal, R.; Patel, A.P.; Debs, L.H.; Nguyen, D.; Patel, K.; Grati, M.h.; Mittal, J.; Yan, D.; Chapagain, P.; Liu, X.Z. Intricate functions of matrix metalloproteinases in physiological and pathological conditions. J. Cell. Physiol. 2016, 231, 2599–2621. [Google Scholar] [CrossRef]
- Jacob, M.P. Extracellular matrix remodeling and matrix metalloproteinases in the vascular wall during aging and in pathological conditions. Biomed. Pharmacother. 2003, 57, 195–202. [Google Scholar] [CrossRef]
- Stadlmann, S.; Pollheimer, J.; Moser, P.L.; Raggi, A.; Amberger, A.; Margreiter, R.; Offner, F.A.; Mikuz, G.; Dirnhofer, S.; Moch, H. Cytokine-regulated expression of collagenase-2 (MMP-8) is involved in the progression of ovarian cancer. Eur. J. Cancer 2003, 39, 2499–2505. [Google Scholar] [CrossRef]
- Åström, P.; Juurikka, K.; Hadler-Olsen, E.S.; Svineng, G.; Cervigne, N.K.; Coletta, R.D.; Risteli, J.; Kauppila, J.H.; Skarp, S.; Kuttner, S.; et al. The interplay of matrix metalloproteinase-8, transforming growth factor-β1 and vascular endothelial growth factor-C cooperatively contributes to the aggressiveness of oral tongue squamous cell carcinoma. Br. J. Cancer 2017, 117, 1007–1016. [Google Scholar] [CrossRef]
- Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and their inhibitors: Potential for the development of new therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef]
- Markland, F.S. Snake venoms and the hemostatic system. Toxicon 1998, 36, 1749–1800. [Google Scholar] [CrossRef]
- Rossello, A.; Nuti, E.; Ferrini, S.; Fabbi, M. Targeting ADAM17 Sheddase Activity in Cancer. Curr. Drug. Targets 2016, 17, 1908–1927. [Google Scholar] [CrossRef]
- Van Goor, H.; Melenhorst, W.B.; Turner, A.J.; Holgate, S.T. Adamalysins in biology and disease. J. Pathol. A J. Pathol. Soc. Great Br. Irel. 2009, 219, 277–286. [Google Scholar] [CrossRef]
- Killar, L.; White, J.; Black, R.; Peschon, J. Adamalysins: A family of metzincins including TNF-α converting enzyme (TACE). Ann. N. Y. Acad. Sci. 1999, 878, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta Proteins Proteom. 2012, 1824, 68–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conus, S. Cathepsins and their involvement in immune responses. Swiss Med. Wkly. 2010, 140, w13042. [Google Scholar] [CrossRef] [PubMed]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The ins and outs of cathepsins: Physiological function and role in disease management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef]
- Ishidoh, K.; Kominami, E. Procathepsin L degrades extracellular matrix proteins in the presence of glycosaminoglycans in vitro. Biochem. Biophys. Res. Commun. 1995, 217, 624–631. [Google Scholar] [CrossRef]
- Taleb, S.; Cancello, R.; Clément, K.; Lacasa, D. Cathepsin s promotes human preadipocyte differentiation: Possible involvement of fibronectin degradation. Endocrinology 2006, 147, 4950–4959. [Google Scholar] [CrossRef]
- Nomura, T.; Katunuma, N. Involvement of cathepsins in the invasion, metastasis and proliferation of cancer cells. J. Med. Investig. 2005, 52, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Tan, G.-J.; Peng, Z.-K.; Lu, J.-P.; Tang, F.-Q. Cathepsins mediate tumor metastasis. World J. Biol. Chem. 2013, 4, 91. [Google Scholar] [CrossRef]
- Rudzińska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A. The role of cysteine cathepsins in cancer progression and drug resistance. Int. J. Mol. Sci. 2019, 20, 3602. [Google Scholar] [CrossRef]
- Llorens, F.; Thüne, K.; Sikorska, B.; Schmitz, M.; Tahir, W.; Fernández-Borges, N.; Cramm, M.; Gotzmann, N.; Carmona, M.; Streichenberger, N. Altered Ca2+ homeostasis induces Calpain-Cathepsin axis activation in sporadic Creutzfeldt-Jakob disease. Acta Neuropathol. Commun. 2017, 5, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Büth, H.; Buttigieg, P.L.; Ostafe, R.; Rehders, M.; Dannenmann, S.R.; Schaschke, N.; Stark, H.-J.; Boukamp, P.; Brix, K. Cathepsin B is essential for regeneration of scratch-wounded normal human epidermal keratinocytes. Eur. J. Cell Biol. 2007, 86, 747–761. [Google Scholar] [CrossRef]
- Hopkins, D.R.; Keles, S.; Greenspan, D.S. The bone morphogenetic protein 1/Tolloid-like metalloproteinases. Matrix Biol. 2007, 26, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Steiglitz, B.M.; Ayala, M.; Narayanan, K.; George, A.; Greenspan, D.S. Bone morphogenetic protein-1/Tolloid-like proteinases process dentin matrix protein-1. J. Biol. Chem. 2004, 279, 980–986. [Google Scholar] [CrossRef] [Green Version]
- Vadon-Le Goff, S.; Hulmes, D.J.; Moali, C. BMP-1/tolloid-like proteinases synchronize matrix assembly with growth factor activation to promote morphogenesis and tissue remodeling. Matrix Biol. 2015, 44-46, 14–23. [Google Scholar] [CrossRef]
- Ge, G.; Greenspan, D.S. Developmental roles of the BMP1/TLD metalloproteinases. Birth Defects Res. C Embryo Today 2006, 78, 47–68. [Google Scholar] [CrossRef]
- Malecaze, F.; Massoudi, D.; Fournié, P.; Tricoire, C.; Cassagne, M.; Malbouyres, M.; Hulmes, D.J.; Moali, C.; Galiacy, S.D. Upregulation of bone morphogenetic protein-1/mammalian tolloid and procollagen C-proteinase enhancer-1 in corneal scarring. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6712–6721. [Google Scholar] [CrossRef]
- Muir, A.M.; Massoudi, D.; Nguyen, N.; Keene, D.R.; Lee, S.-J.; Birk, D.E.; Davidson, J.M.; Marinkovich, M.P.; Greenspan, D.S. BMP1-like proteinases are essential to the structure and wound healing of skin. Matrix Biol. 2016, 56, 114–131. [Google Scholar] [CrossRef] [Green Version]
- Stern, R. Hyaluronidases in cancer biology. Semin Cancer Biol 2008, 18, 275–280. [Google Scholar] [CrossRef]
- Liu, M.; Tolg, C.; Turley, E. Dissecting the Dual Nature of Hyaluronan in the Tumor Microenvironment. Front. Immunol. 2019, 10, 947. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Yamamoto, H.; Tobisawa, Y.; Irie, F. TMEM2: A missing link in hyaluronan catabolism identified? Matrix Biol. 2019, 78-79, 139–146. [Google Scholar] [CrossRef]
- Tammi, M.I.; Oikari, S.; Pasonen-Seppänen, S.; Rilla, K.; Auvinen, P.; Tammi, R.H. Activated hyaluronan metabolism in the tumor matrix—Causes and consequences. Matrix Biol. 2019, 78–79, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Stern, R. Hyaluronan metabolism: A major paradox in cancer biology. Pathol. Biol. 2005, 53, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Girish, K.; Kemparaju, K. The magic glue hyaluronan and its eraser hyaluronidase: A biological overview. Life Sci. 2007, 80, 1921–1943. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.; Asari, A.A.; Sugahara, K.N. Hyaluronan fragments: An information-rich system. Eur. J. Cell Biol. 2006, 85, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan in Tissue Injury and Repair. Annu. Rev. Cell Dev. Biol. 2007, 23, 435–461. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Chanmee, T.; Itano, N. Hyaluronan: Metabolism and function. Biomolecules 2020, 10, 1525. [Google Scholar] [CrossRef]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan as an immune regulator in human diseases. Physiol. Rev. 2011, 91, 221–264. [Google Scholar] [CrossRef] [Green Version]
- Noble, P.W. Hyaluronan and its catabolic products in tissue injury and repair. Matrix Biol. 2002, 21, 25–29. [Google Scholar] [CrossRef]
- Heldin, P.; Basu, K.; Olofsson, B.; Porsch, H.; Kozlova, I.; Kahata, K. Deregulation of hyaluronan synthesis, degradation and binding promotes breast cancer. J. Biochem. 2013, 154, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Bame, K.J. Heparanases: Endoglycosidases that degrade heparan sulfate proteoglycans. Glycobiology 2001, 11, 91R–98R. [Google Scholar] [CrossRef] [Green Version]
- Zcharia, E.; Zilka, R.; Yaar, A.; Yacoby-Zeevi, O.; Zetser, A.; Metzger, S.; Sarid, R.; Naggi, A.; Casu, B.; Ilan, N. Heparanase accelerates wound angiogenesis and wound healing in mouse and rat models. FASEB J. 2005, 19, 211–221. [Google Scholar] [CrossRef]
- Crispel, Y.; Ghanem, S.; Attias, J.; Kogan, I.; Brenner, B.; Nadir, Y. Involvement of the heparanase procoagulant domain in bleeding and wound healing. J. Thromb. Haemost. 2017, 15, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, R.D.; Elkin, M.; Rapraeger, A.C.; Ilan, N.; Vlodavsky, I. Heparanase regulation of cancer, autophagy and inflammation: New mechanisms and targets for therapy. FEBS J. 2017, 284, 42–55. [Google Scholar] [CrossRef] [Green Version]
- Khanna, M.; Parish, C.R. Heparanase: Historical Aspects and Future Perspectives. Adv. Exp. Med. Biol. 2020, 1221, 71–96. [Google Scholar] [CrossRef]
- Itoh, Y. Membrane-type matrix metalloproteinases: Their functions and regulations. Matrix Biol. 2015, 44, 207–223. [Google Scholar] [CrossRef]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Brew, K.; Nagase, H. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim. Biophys. Acta Mol. Cell Res. 2010, 1803, 55–71. [Google Scholar] [CrossRef] [Green Version]
- Baker, E.A.; Leaper, D.J. Profiles of matrix metalloproteinases and their tissue inhibitors in intraperitoneal drainage fluid: Relationship to wound healing. Wound Repair Regen. 2003, 11, 268–274. [Google Scholar] [CrossRef]
- Xue, M.; Jackson, C.J. Extracellular Matrix Reorganization During Wound Healing and Its Impact on Abnormal Scarring. Adv. Wound Care 2015, 4, 119–136. [Google Scholar] [CrossRef] [Green Version]
- Breznik, B.; Mitrović, A.; Lah, T.T.; Kos, J. Cystatins in cancer progression: More than just cathepsin inhibitors. Biochimie 2019, 166, 233–250. [Google Scholar] [CrossRef]
- Van Gent, D.; Sharp, P.; Morgan, K.; Kalsheker, N. Serpins: Structure, function and molecular evolution. Int. J. Biochem. Cell Biol. 2003, 35, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Rau, J.; Beaulieu, L.; Huntington, J.; CHURCH, F.C. Serpins in thrombosis, hemostasis and fibrinolysis. J. Thromb. Haemost. 2007, 5, 102–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrell, R.W.; Evans, D.L.; Stein, P.E. Mobile reactive centre of serpins and the control of thrombosis. Nature 1991, 353, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S.; Vallet, S.D. Proteases decode the extracellular matrix cryptome. Biochimie 2016, 122, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Isayeva, T.; Larson, M.R.; Sawant, A.; Cha, H.R.; Chanda, D.; Chesnokov, I.N.; Ponnazhagan, S. Endostatin: A novel inhibitor of androgen receptor function in prostate cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 1392–1397. [Google Scholar] [CrossRef] [Green Version]
- Magnon, C.; Galaup, A.; Mullan, B.; Rouffiac, V.; Bouquet, C.; Bidart, J.M.; Griscelli, F.; Opolon, P.; Perricaudet, M. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with alphavbeta3 and alphavbeta5 integrins. Cancer Res. 2005, 65, 4353–4361. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Lu, X.A.; Liu, P.; Fu, Y.; Jia, L.; Zhan, S.; Luo, Y. Endostatin has ATPase activity, which mediates its antiangiogenic and antitumor activities. Mol. Cancer 2015, 14, 1192–1201. [Google Scholar] [CrossRef] [Green Version]
- Colorado, P.C.; Torre, A.; Kamphaus, G.; Maeshima, Y.; Hopfer, H.; Takahashi, K.; Volk, R.; Zamborsky, E.D.; Herman, S.; Sarkar, P.K.; et al. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 2000, 60, 2520–2526. [Google Scholar]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [Green Version]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Schönherr, E.; Hausser, H.-J. Extracellular Matrix and Cytokines: A Functional Unit. Dev. Immunol. 2000, 7, 031748. [Google Scholar] [CrossRef]
- Foster, D.S.; Jones, R.E.; Ransom, R.C.; Longaker, M.T.; Norton, J.A. The evolving relationship of wound healing and tumor stroma. JCI Insight 2018, 3, e99911. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal—A historical perspective with a focus on the fundamental roles of increased vascular permeability and clotting. In Proceedings of the Seminars in Thrombosis and Hemostasis; Thieme: Teningen, Germany, 2019; pp. 576–592. [Google Scholar]
- Dzobo, K.; Dandara, C. Architecture of Cancer-Associated Fibroblasts in Tumor Microenvironment: Mapping Their Origins, Heterogeneity, and Role in Cancer Therapy Resistance. Omics 2020, 24, 314–339. [Google Scholar] [CrossRef]
- Dzobo, K.; Dandara, C. Broadening Drug Design and Targets to Tumor Microenvironment? Cancer-Associated Fibroblast Marker Expression in Cancers and Relevance for Survival Outcomes. Omics 2020, 24, 340–351. [Google Scholar] [CrossRef]
- Bochet, L.; Lehuédé, C.; Dauvillier, S.; Wang, Y.Y.; Dirat, B.; Laurent, V.; Dray, C.; Guiet, R.; Maridonneau-Parini, I.; Le Gonidec, S.; et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013, 73, 5657–5668. [Google Scholar] [CrossRef] [Green Version]
- Mu, W.; Rana, S.; Zöller, M. Host matrix modulation by tumor exosomes promotes motility and invasiveness. Neoplasia 2013, 15, 875–887. [Google Scholar] [CrossRef] [Green Version]
- Webber, J.P.; Spary, L.K.; Sanders, A.J.; Chowdhury, R.; Jiang, W.G.; Steadman, R.; Wymant, J.; Jones, A.T.; Kynaston, H.; Mason, M.D.; et al. Differentiation of tumour-promoting stromal myofibroblasts by cancer exosomes. Oncogene 2015, 34, 290–302. [Google Scholar] [CrossRef]
- Liu, L.; Liu, L.; Yao, H.H.; Zhu, Z.Q.; Ning, Z.L.; Huang, Q. Stromal Myofibroblasts Are Associated with Poor Prognosis in Solid Cancers: A Meta-Analysis of Published Studies. PLoS ONE 2016, 11, e0159947. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, M.; Ogawa, T.; Zhang, X.; Hanamura, N.; Kashikura, Y.; Takamura, M.; Yoneda, M.; Shiraishi, T. Role of stromal myofibroblasts in invasive breast cancer: Stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer 2012, 19, 170–176. [Google Scholar] [CrossRef]
- Tsujino, T.; Seshimo, I.; Yamamoto, H.; Ngan, C.Y.; Ezumi, K.; Takemasa, I.; Ikeda, M.; Sekimoto, M.; Matsuura, N.; Monden, M. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin. Cancer Res. 2007, 13, 2082–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Mak, M. Fibroblast-mediated uncaging of cancer cells and dynamic evolution of the physical microenvironment. Sci. Rep. 2022, 12, 791. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Öhlund, D.; Rickelt, S.; Lidström, T.; Huang, Y.; Hao, L.; Zhao, R.T.; Franklin, O.; Bhatia, S.N.; Tuveson, D.A.; et al. Cancer Cell–Derived Matrisome Proteins Promote Metastasis in Pancreatic Ductal Adenocarcinoma. Cancer Res. 2020, 80, 1461–1474. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.B.; Chua, M.L.K.; Yeong, J.P.S.; Tan, S.J.; Lim, W.-T.; Lim, C.T. Pan-cancer analysis connects tumor matrisome to immune response. NPJ Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Rafaeva, M.; Erler, J.T. Framing cancer progression: Influence of the organ-and tumour-specific matrisome. FEBS J. 2020, 287, 1454–1477. [Google Scholar] [CrossRef] [Green Version]
- Le, C.P.; Nowell, C.J.; Kim-Fuchs, C.; Botteri, E.; Hiller, J.G.; Ismail, H.; Pimentel, M.A.; Chai, M.G.; Karnezis, T.; Rotmensz, N.; et al. Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nat. Commun. 2016, 7, 10634. [Google Scholar] [CrossRef] [Green Version]
- Tacconi, C.; Correale, C.; Gandelli, A.; Spinelli, A.; Dejana, E.; D’Alessio, S.; Danese, S. Vascular endothelial growth factor C disrupts the endothelial lymphatic barrier to promote colorectal cancer invasion. Gastroenterology 2015, 148, 1438–1451.e8. [Google Scholar] [CrossRef] [Green Version]
- Le, C.P.; Karnezis, T.; Achen, M.G.; Stacker, S.A.; Sloan, E.K. Lymphovascular and neural regulation of metastasis: Shared tumour signalling pathways and novel therapeutic approaches. Best Pract. Res. Clin. Anaesthesiol 2013, 27, 409–425. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja, A.S.; Dood, R.L.; Armaiz-Pena, G.; Kang, Y.; Wu, S.Y.; Allen, J.K.; Jennings, N.B.; Mangala, L.S.; Pradeep, S.; Lyons, Y.; et al. Adrenergic-mediated increases in INHBA drive CAF phenotype and collagens. JCI Insight 2017, 2, e93076. [Google Scholar] [CrossRef]
- Insua-Rodríguez, J.; Pein, M.; Hongu, T.; Meier, J.; Descot, A.; Lowy, C.M.; De Braekeleer, E.; Sinn, H.P.; Spaich, S.; Sütterlin, M.; et al. Stress signaling in breast cancer cells induces matrix components that promote chemoresistant metastasis. EMBO Mol. Med. 2018, 10, e9003. [Google Scholar] [CrossRef]
- Afasizheva, A.; Devine, A.; Tillman, H.; Fung, K.L.; Vieira, W.D.; Blehm, B.H.; Kotobuki, Y.; Busby, B.; Chen, E.I.; Tanner, K. Mitogen-activated protein kinase signaling causes malignant melanoma cells to differentially alter extracellular matrix biosynthesis to promote cell survival. BMC Cancer 2016, 16, 186. [Google Scholar] [CrossRef] [Green Version]
- Steins, A.; van Mackelenbergh, M.G.; van der Zalm, A.P.; Klaassen, R.; Serrels, B.; Goris, S.G.; Kocher, H.M.; Waasdorp, C.; de Jong, J.H.; Tekin, C.; et al. High-grade mesenchymal pancreatic ductal adenocarcinoma drives stromal deactivation through CSF-1. EMBO Rep. 2020, 21, e48780. [Google Scholar] [CrossRef]
- Vera, R.E.; Fernandez-Zapico, M.E. Stromal deactivation by CSF1: A new feature of the aggressive pancreatic cancer microenvironment. EMBO Rep. 2020, 21, e50468. [Google Scholar] [CrossRef]
- Fu, Y.; Liu, S.; Zeng, S.; Shen, H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol. Cancer 2018, 17, 62. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2018, 9, 1065. [Google Scholar] [CrossRef] [Green Version]
- Palumbo Jr, A.; Ferreira, L.B.; de Souza, P.A.R.; de Oliveira, F.L.; Pontes, B.; Viana, N.B.; Machado, D.E.; Palmero, C.Y.; Alves, L.M.; Gimba, E.R. Extracellular matrix secreted by reactive stroma is a main inducer of pro-tumorigenic features on LNCaP prostate cancer cells. Cancer Lett. 2012, 321, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Barcus, C.E.; Holt, E.C.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Dense collagen-I matrices enhance pro-tumorigenic estrogen-prolactin crosstalk in MCF-7 and T47D breast cancer cells. PLoS ONE 2015, 10, e0116891. [Google Scholar] [CrossRef]
- Brown, Y.; Hua, S.; Tanwar, P.S. Extracellular matrix-mediated regulation of cancer stem cells and chemoresistance. Int. J. Biochem. Cell Biol. 2019, 109, 90–104. [Google Scholar] [CrossRef]
- Yeldag, G.; Rice, A.; del Río Hernández, A. Chemoresistance and the self-maintaining tumor microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietilä, E.A.; Gonzalez-Molina, J.; Moyano-Galceran, L.; Jamalzadeh, S.; Zhang, K.; Lehtinen, L.; Turunen, S.P.; Martins, T.A.; Gultekin, O.; Lamminen, T. Co-evolution of matrisome and adaptive adhesion dynamics drives ovarian cancer chemoresistance. Nat. Commun. 2021, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Keeratichamroen, S.; Lirdprapamongkol, K.; Svasti, J. Mechanism of ECM-induced dormancy and chemoresistance in A549 human lung carcinoma cells. Oncol. Rep. 2018, 39, 1765–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocanu, M.M.; Fazekas, Z.; Petrás, M.; Nagy, P.; Sebestyén, Z.; Isola, J.; Tímár, J.; Park, J.W.; Vereb, G.; Szöllosi, J. Associations of ErbB2, beta1-integrin and lipid rafts on Herceptin (Trastuzumab) resistant and sensitive tumor cell lines. Cancer Lett 2005, 227, 201–212. [Google Scholar] [CrossRef]
- Guo, W.; Pylayeva, Y.; Pepe, A.; Yoshioka, T.; Muller, W.J.; Inghirami, G.; Giancotti, F.G. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 2006, 126, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.V.; Sleiman, M.; Moriarty, T.; Herrick, W.G.; Peyton, S.R. Sorafenib resistance and JNK signaling in carcinoma during extracellular matrix stiffening. Biomaterials 2014, 35, 5749–5759. [Google Scholar] [CrossRef] [Green Version]
- Keely, P.J. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J. Mammary Gland Biol. Neoplasia 2011, 16, 205–219. [Google Scholar] [CrossRef]
- Weigelt, B.; Lo, A.T.; Park, C.C.; Gray, J.W.; Bissell, M.J. HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Res. Treat. 2010, 122, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Dzobo, K. Integrins Within the Tumor Microenvironment: Biological Functions, Importance for Molecular Targeting, and Cancer Therapeutics Innovation. Omics 2021, 25, 417–430. [Google Scholar] [CrossRef]
- Guerrero, P.A.; McCarty, J.H. Integrins in Vascular Development and Pathology. Adv. Pharm. 2018, 81, 129–153. [Google Scholar] [CrossRef]
- Conway, J.R.W.; Jacquemet, G. Cell matrix adhesion in cell migration. Essays Biochem. 2019, 63, 535–551. [Google Scholar] [CrossRef]
- Zhang, Y.; Reif, G.; Wallace, D.P. Extracellular matrix, integrins, and focal adhesion signaling in polycystic kidney disease. Cell Signal. 2020, 72, 109646. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [Green Version]
- Samaržija, I.; Dekanić, A.; Humphries, J.D.; Paradžik, M.; Stojanović, N.; Humphries, M.J.; Ambriović-Ristov, A. Integrin crosstalk contributes to the complexity of signalling and unpredictable cancer cell fates. Cancers 2020, 12, 1910. [Google Scholar] [CrossRef]
- Varner, J.A.; Emerson, D.A.; Juliano, R.L. Integrin alpha 5 beta 1 expression negatively regulates cell growth: Reversal by attachment to fibronectin. Mol. Biol. Cell 1995, 6, 725–740. [Google Scholar] [CrossRef] [Green Version]
- Stoeltzing, O.; Liu, W.; Reinmuth, N.; Fan, F.; Parry, G.C.; Parikh, A.A.; McCarty, M.F.; Bucana, C.D.; Mazar, A.P.; Ellis, L.M. Inhibition of integrin α5β1 function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int. J. Cancer 2003, 104, 496–503. [Google Scholar] [CrossRef]
- Jin, H.; Varner, J. Integrins: Roles in cancer development and as treatment targets. Br. J. Cancer 2004, 90, 561–565. [Google Scholar] [CrossRef]
- Varner, J.A.; Cheresh, D.A. Integrins and cancer. Curr. Opin. Cell Biol. 1996, 8, 724–730. [Google Scholar] [CrossRef]
- Weis, S.M.; Cheresh, D.A. αV integrins in angiogenesis and cancer. Cold Spring Harb. Perspect. Med. 2011, 1, a006478. [Google Scholar] [CrossRef] [Green Version]
- Janiszewska, M.; Primi, M.C.; Izard, T. Cell adhesion in cancer: Beyond the migration of single cells. J. Biol. Chem. 2020, 295, 2495–2505. [Google Scholar] [CrossRef] [Green Version]
- Truong, H.; Danen, E.H. Integrin switching modulates adhesion dynamics and cell migration. Cell Adhes. Migr. 2009, 3, 179–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuidema, A.; Wang, W.; Sonnenberg, A. Crosstalk between cell adhesion complexes in regulation of mechanotransduction. Bioessays 2020, 42, 2000119. [Google Scholar] [CrossRef] [PubMed]
- Kerrisk, M.E.; Cingolani, L.A.; Koleske, A.J. ECM receptors in neuronal structure, synaptic plasticity, and behavior. Prog. Brain Res. 2014, 214, 101–131. [Google Scholar] [PubMed] [Green Version]
- Boraschi-Diaz, I.; Wang, J.; Mort, J.S.; Komarova, S.V. Collagen type I as a ligand for receptor-mediated signaling. Front. Phys. 2017, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Takai, K.; Drain, A.P.; Lawson, D.A.; Littlepage, L.E.; Karpuj, M.; Kessenbrock, K.; Le, A.; Inoue, K.; Weaver, V.M.; Werb, Z. Discoidin domain receptor 1 (DDR1) ablation promotes tissue fibrosis and hypoxia to induce aggressive basal-like breast cancers. Genes Dev. 2018, 32, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.E.; Martin, E.E.; Anwar, T.; Arellano-Garcia, C.; Medhora, N.; Lama, A.; Chen, Y.-C.; Tanager, K.S.; Yoon, E.; Kidwell, K.M. Mesenchymal stem cell-induced DDR2 mediates stromal-breast cancer interactions and metastasis growth. Cell Rep. 2017, 18, 1215–1228. [Google Scholar] [CrossRef]
- Bayer, S.V.; Grither, W.R.; Brenot, A.; Hwang, P.Y.; Barcus, C.E.; Ernst, M.; Pence, P.; Walter, C.; Pathak, A.; Longmore, G.D. DDR2 controls breast tumor stiffness and metastasis by regulating integrin mediated mechanotransduction in CAFs. Elife 2019, 8, e45508. [Google Scholar] [CrossRef]
- Liao, W.-C.; Yen, H.-R.; Chen, C.-H.; Chu, Y.-H.; Song, Y.-C.; Tseng, T.-J.; Liu, C.-H. CHPF promotes malignancy of breast cancer cells by modifying syndecan-4 and the tumor microenvironment. Am. J. Cancer Res. 2021, 11, 812. [Google Scholar]
- Leblanc, R.; Sahay, D.; Houssin, A.; Machuca-Gayet, I.; Peyruchaud, O. Autotaxin-β interaction with the cell surface via syndecan-4 impacts on cancer cell proliferation and metastasis. Oncotarget 2018, 9, 33170. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Liu, T.; Li, R.; Sy, M.-S. Mechanisms regulating the binding activity of CD44 to hyaluronic acid. Front. Biosci. Landmark 1998, 3, 631–636. [Google Scholar]
- Naor, D. interaction between hyaluronic acid and its receptors (CD44, RHAMM) regulates the activity of inflammation and cancer. Front. Immunol. 2016, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Misra, S.; Heldin, P.; Hascall, V.C.; Karamanos, N.K.; Skandalis, S.S.; Markwald, R.R.; Ghatak, S. Hyaluronan–CD44 interactions as potential targets for cancer therapy. FEBS J. 2011, 278, 1429–1443. [Google Scholar] [CrossRef] [Green Version]
- Toole, B.P. Hyaluronan-CD44 interactions in cancer: Paradoxes and possibilities. Clin. Cancer Res. 2009, 15, 7462–7468. [Google Scholar] [CrossRef] [Green Version]
- Jalkanen, S.; Jalkanen, M. Lymphocyte CD44 binds the COOH-terminal heparin-binding domain of fibronectin. J. Cell Biol. 1992, 116, 817–825. [Google Scholar] [CrossRef]
- Dzobo, K.; Sinkala, M. Cancer Stem Cell Marker CD44 Plays Multiple Key Roles in Human Cancers: Immune Suppression/Evasion, Drug Resistance, Epithelial–Mesenchymal Transition, and Metastasis. OMICS A J. Integr. Biol. 2021, 25, 313–332. [Google Scholar] [CrossRef]
- Thapa, R.; Wilson, G.D. The importance of CD44 as a stem cell biomarker and therapeutic target in cancer. Stem Cells Int. 2016, 2016, 2087204. [Google Scholar] [CrossRef] [Green Version]
- Hirata, K.; Suzuki, H.; Imaeda, H.; Matsuzaki, J.; Tsugawa, H.; Nagano, O.; Asakura, K.; Saya, H.; Hibi, T. CD44 variant 9 expression in primary early gastric cancer as a predictive marker for recurrence. Br. J. Cancer 2013, 109, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Palapattu, G.S.; Wu, C.; Silvers, C.R.; Martin, H.B.; Williams, K.; Salamone, L.; Bushnell, T.; Huang, L.S.; Yang, Q.; Huang, J. Selective expression of CD44, a putative prostate cancer stem cell marker, in neuroendocrine tumor cells of human prostate cancer. Prostate 2009, 69, 787–798. [Google Scholar] [CrossRef]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.J.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun. 2012, 3, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Leung, E.L.-H.; Fiscus, R.R.; Tung, J.W.; Tin, V.P.-C.; Cheng, L.C.; Sihoe, A.D.-L.; Fink, L.M.; Ma, Y.; Wong, M.P. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS ONE 2010, 5, e14062. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.R.; Cho, S.; Discher, D.E. Stem cell differentiation is regulated by extracellular matrix mechanics. Physiology 2018, 33, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, E.J.; Ponik, S.M. Biomechanical contributions to macrophage activation in the tumor microenvironment. Front. Oncol. 2020, 10, 787. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, O.; Cooper-White, J.; Janmey, P.A.; Mooney, D.J.; Shenoy, V.B. Effects of extracellular matrix viscoelasticity on cellular behaviour. Nature 2020, 584, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Charrier, E.E.; Pogoda, K.; Wells, R.G.; Janmey, P.A. Control of cell morphology and differentiation by substrates with independently tunable elasticity and viscous dissipation. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elosegui-Artola, A. The extracellular matrix viscoelasticity as a regulator of cell and tissue dynamics. Curr. Opin. Cell Biol. 2021, 72, 10–18. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Lagares, D. Mechano-therapeutics: Targeting mechanical signaling in fibrosis and tumor stroma. Pharmacol. Ther. 2020, 212, 107575. [Google Scholar] [CrossRef]
- Pratt, S.J.; Lee, R.M.; Martin, S.S. The mechanical microenvironment in breast cancer. Cancers 2020, 12, 1452. [Google Scholar] [CrossRef]
- Nazemi, M.; Rainero, E. Cross-talk between the tumor microenvironment, extracellular matrix, and cell metabolism in cancer. Front. Oncol. 2020, 10, 239. [Google Scholar] [CrossRef] [Green Version]
- Pizzo, A.M.; Kokini, K.; Vaughn, L.C.; Waisner, B.Z.; Voytik-Harbin, S.L. Extracellular matrix (ECM) microstructural composition regulates local cell-ECM biomechanics and fundamental fibroblast behavior: A multidimensional perspective. J. Appl. Physiol. 2005, 98, 1909–1921. [Google Scholar] [CrossRef] [Green Version]
- Wala, J.; Das, S. Mapping of biomechanical properties of cell lines on altered matrix stiffness using atomic force microscopy. Biomech. Model. Mechanobiol. 2020, 19, 1523–1536. [Google Scholar] [CrossRef]
- Souza, S.T.; Agra, L.C.; Santos, C.E.; Barreto, E.; Hickmann, J.M.; Fonseca, E.J. Macrophage adhesion on fibronectin evokes an increase in the elastic property of the cell membrane and cytoskeleton: An atomic force microscopy study. Eur. Biophys. J. 2014, 43, 573–579. [Google Scholar] [CrossRef]
- Park, J.S.; Burckhardt, C.J.; Lazcano, R.; Solis, L.M.; Isogai, T.; Li, L.; Chen, C.S.; Gao, B.; Minna, J.D.; Bachoo, R. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 2020, 578, 621–626. [Google Scholar] [CrossRef]
- Vitillo, L.; Kimber, S.J. Integrin and FAK regulation of human pluripotent stem cells. Curr. Stem Cell Rep. 2017, 3, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Dedhar, S. Cell–substrate interactions and signaling through ILK. Curr. Opin. Cell Biol. 2000, 12, 250–256. [Google Scholar] [CrossRef]
- Schlie-Wolter, S.; Ngezahayo, A.; Chichkov, B.N. The selective role of ECM components on cell adhesion, morphology, proliferation and communication in vitro. Exp. Cell Res. 2013, 319, 1553–1561. [Google Scholar] [CrossRef]
- Nallanthighal, S.; Rada, M.; Heiserman, J.P.; Cha, J.; Sage, J.; Zhou, B.; Yang, W.; Hu, Y.; Korgaonkar, C.; Hanos, C.T. Inhibition of collagen XI alpha 1-induced fatty acid oxidation triggers apoptotic cell death in cisplatin-resistant ovarian cancer. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Even-Ram, S.; Yamada, K.M. Cell migration in 3D matrix. Curr. Opin. Cell Biol. 2005, 17, 524–532. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the hallmarks of metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G.; Lehembre, F. Distinct mechanisms of tumor invasion and metastasis. Trends Mol. Med. 2007, 13, 535–541. [Google Scholar] [CrossRef]
- Conklin, M.W.; Gangnon, R.E.; Sprague, B.L.; Van Gemert, L.; Hampton, J.M.; Eliceiri, K.W.; Bredfeldt, J.S.; Liu, Y.; Surachaicharn, N.; Newcomb, P.A. Collagen Alignment as a Predictor of Recurrence after Ductal Carcinoma In SituCollagen Alignment and Recurrence of DCIS. Cancer Epidemiol. Biomark. Prev. 2018, 27, 138–145. [Google Scholar] [CrossRef] [Green Version]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Drifka, C.R.; Loeffler, A.G.; Mathewson, K.; Keikhosravi, A.; Eickhoff, J.C.; Liu, Y.; Weber, S.M.; Kao, W.J.; Eliceiri, K.W. Highly aligned stromal collagen is a negative prognostic factor following pancreatic ductal adenocarcinoma resection. Oncotarget 2016, 7, 76197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Friedl, A. Syndecan-1-induced ECM fiber alignment requires integrin αvβ3 and syndecan-1 ectodomain and heparan sulfate chains. PLoS ONE 2016, 11, e0150132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdogan, B.; Ao, M.; White, L.M.; Means, A.L.; Brewer, B.M.; Yang, L.; Washington, M.K.; Shi, C.; Franco, O.E.; Weaver, A.M. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J. Cell Biol. 2017, 216, 3799–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ao, M.; Brewer, B.M.; Yang, L.; Franco Coronel, O.E.; Hayward, S.W.; Webb, D.J.; Li, D. Stretching fibroblasts remodels fibronectin and alters cancer cell migration. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, T.Y.; Zheng, H.; Liu, R.; Wicha, M.S.; Yu, S.M.; Weiss, S.J. Divergent matrix-remodeling strategies distinguish developmental from neoplastic mammary epithelial cell invasion programs. Dev. Cell 2018, 47, 145–160.e6. [Google Scholar] [CrossRef] [Green Version]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Werb, Z. The many faces of metalloproteases: Cell growth, invasion, angiogenesis and metastasis. Trends Cell Biol. 2001, 11, S37–S43. [Google Scholar] [CrossRef]
- Elia, I.; Rossi, M.; Stegen, S.; Broekaert, D.; Doglioni, G.; van Gorsel, M.; Boon, R.; Escalona-Noguero, C.; Torrekens, S.; Verfaillie, C. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 2019, 568, 117–121. [Google Scholar] [CrossRef]
- Ge, J.; Cui, H.; Xie, N.; Banerjee, S.; Guo, S.; Dubey, S.; Barnes, S.; Liu, G. Glutaminolysis promotes collagen translation and stability via α-ketoglutarate–mediated mTOR activation and proline hydroxylation. Am. J. Respir. Cell Mol. Biol. 2018, 58, 378–390. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Gao, Y.; Bado, I.; Wang, H.; Zhang, W.; Rosen, J.M.; Zhang, X.H.-F. Metastasis organotropism: Redefining the congenial soil. Dev. Cell 2019, 49, 375–391. [Google Scholar] [CrossRef]
- Haemmerle, M.; Taylor, M.L.; Gutschner, T.; Pradeep, S.; Cho, M.S.; Sheng, J.; Lyons, Y.M.; Nagaraja, A.S.; Dood, R.L.; Wen, Y. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Ting, D.T.; Stott, S.L.; Wittner, B.S.; Ozsolak, F.; Paul, S.; Ciciliano, J.C.; Smas, M.E.; Winokur, D.; Gilman, A.J. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature 2012, 487, 510–513. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Alix-Panabières, C.; Riethdorf, S. Cancer micrometastases. Nat. Rev. Clin. Oncol. 2009, 6, 339–351. [Google Scholar] [CrossRef]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant tumour cells, their niches and the influence of immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef]
- Yeh, A.C.; Ramaswamy, S. Mechanisms of cancer cell dormancy—Another hallmark of cancer? Cancer Res. 2015, 75, 5014–5022. [Google Scholar] [CrossRef] [Green Version]
- Boire, A.; Coffelt, S.B.; Quezada, S.A.; Vander Heiden, M.G.; Weeraratna, A.T. Tumour dormancy and reawakening: Opportunities and challenges. Trends Cancer 2019, 5, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef]
- Gay, L.J.; Malanchi, I. The sleeping ugly: Tumour microenvironment’s act to make or break the spell of dormancy. Biochim. Et Biophys. Acta Rev. Cancer 2017, 1868, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Mancini, M.L.; Sonis, S.T. Mechanisms of cellular fibrosis associated with cancer regimen-related toxicities. Front. Pharmacol. 2014, 5, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Principe, D.R.; Narbutis, M.; Kumar, S.; Park, A.; Viswakarma, N.; Dorman, M.J.; Kamath, S.D.; Grippo, P.J.; Fishel, M.L.; Hwang, R.F. Long-Term Gemcitabine Treatment Reshapes the Pancreatic Tumor Microenvironment and Sensitizes Murine Carcinoma to Combination ImmunotherapyGemcitabine Primes Pancreatic Cancer for Immunotherapy. Cancer Res. 2020, 80, 3101–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.J.; Sharma, A.; Vuong, D.-V.; Erler, J.T.; Pruschy, M.; Broggini-Tenzer, A. Ionizing radiation induces tumor cell lysyl oxidase secretion. BMC Cancer 2014, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Roy, D.M.; Walsh, L.A. Candidate prognostic markers in breast cancer: Focus on extracellular proteases and their inhibitors. Breast Cancer Targets Ther. 2014, 6, 81. [Google Scholar]
- Kanayama, H.o.; Yokota, K.y.; Kurokawa, Y.; Murakami, Y.; Nishitani, M.; Kagawa, S. Prognostic values of matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-2 expression in bladder cancer. Cancer Interdiscip. Int. J. Am. Cancer Soc. 1998, 82, 1359–1366. [Google Scholar]
- Bergamaschi, A.; Tagliabue, E.; Sørlie, T.; Naume, B.; Triulzi, T.; Orlandi, R.; Russnes, H.; Nesland, J.; Tammi, R.; Auvinen, P. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. J. Pathol. 2008, 214, 357–367. [Google Scholar] [CrossRef]
- Valdembri, D.; Serini, G. The roles of integrins in cancer. Fac. Rev. 2021, 10, 45. [Google Scholar] [CrossRef]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr. Biol. 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [Green Version]
- Bordeleau, F.; Mason, B.N.; Lollis, E.M.; Mazzola, M.; Zanotelli, M.R.; Somasegar, S.; Califano, J.P.; Montague, C.; LaValley, D.J.; Huynh, J.; et al. Matrix stiffening promotes a tumor vasculature phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 492–497. [Google Scholar] [CrossRef] [Green Version]
- Hui, L.; Zhang, J.; Ding, X.; Guo, X.; Jiang, X. Matrix stiffness regulates the proliferation, stemness and chemoresistance of laryngeal squamous cancer cells. Int. J. Oncol. 2017, 50, 1439–1447. [Google Scholar] [CrossRef] [Green Version]
- McLane, J.S.; Ligon, L.A. Stiffened Extracellular Matrix and Signaling from Stromal Fibroblasts via Osteoprotegerin Regulate Tumor Cell Invasion in a 3-D Tumor in Situ Model. Cancer Microenviron. 2016, 9, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Tadeo, I.; Berbegall, A.P.; Navarro, S.; Castel, V.; Noguera, R. A stiff extracellular matrix is associated with malignancy in peripheral neuroblastic tumors. Pediatr. Blood Cancer 2017, 64, e26449. [Google Scholar] [CrossRef]
- Qin, X.; Lv, X.; Li, P.; Yang, R.; Xia, Q.; Chen, Y.; Peng, Y.; Li, L.; Li, S.; Li, T. Matrix stiffness modulates ILK-mediated YAP activation to control the drug resistance of breast cancer cells. Biochim. Et Biophys. Acta Mol. Basis Dis. 2020, 1866, 165625. [Google Scholar] [CrossRef]
- Wang, D.; Li, Y.; Ge, H.; Ghadban, T.; Reeh, M.; Güngör, C. The Extracellular Matrix: A Key Accomplice of Cancer Stem Cell Migration, Metastasis Formation, and Drug Resistance in PDAC. Cancers 2022, 14, 3998. [Google Scholar] [CrossRef]
- Grantab, R.H.; Tannock, I.F. Penetration of anticancer drugs through tumour tissue as a function of cellular packing density and interstitial fluid pressure and its modification by bortezomib. BMC Cancer 2012, 12, 214. [Google Scholar] [CrossRef] [Green Version]
- Harisi, R.; Jeney, A. Extracellular matrix as target for antitumor therapy. OncoTargets Ther. 2015, 8, 1387–1398. [Google Scholar] [CrossRef] [Green Version]
- Holle, A.W.; Young, J.L.; Spatz, J.P. In vitro cancer cell-ECM interactions inform in vivo cancer treatment. Adv. Drug Deliv. Rev. 2016, 97, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Miroshnikova, Y.A.; Mouw, J.K.; Barnes, J.M.; Pickup, M.W.; Lakins, J.N.; Kim, Y.; Lobo, K.; Persson, A.I.; Reis, G.F.; McKnight, T.R.; et al. Tissue mechanics promote IDH1-dependent HIF1alpha-tenascin C feedback to regulate glioblastoma aggression. Nat. Cell Biol. 2016, 18, 1336–1345. [Google Scholar] [CrossRef]
- Mittal, V.; El Rayes, T.; Narula, N.; McGraw, T.E.; Altorki, N.K.; Barcellos-Hoff, M.H. The Microenvironment of Lung Cancer and Therapeutic Implications. Adv. Exp. Med. Biol. 2016, 890, 75–110. [Google Scholar] [CrossRef] [PubMed]
- Mumenthaler, S.M.; Foo, J.; Choi, N.C.; Heise, N.; Leder, K.; Agus, D.B.; Pao, W.; Michor, F.; Mallick, P. The Impact of Microenvironmental Heterogeneity on the Evolution of Drug Resistance in Cancer Cells. Cancer Inf. 2015, 14, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Nieponice, A.; McGrath, K.; Qureshi, I.; Beckman, E.J.; Luketich, J.D.; Gilbert, T.W.; Badylak, S.F. An extracellular matrix scaffold for esophageal stricture prevention after circumferential EMR. Gastrointest. Endosc. 2009, 69, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Barcus, C.E.; O’Leary, K.A.; Brockman, J.L.; Rugowski, D.E.; Liu, Y.; Garcia, N.; Yu, M.; Keely, P.J.; Eliceiri, K.W.; Schuler, L.A. Elevated collagen-I augments tumor progressive signals, intravasation and metastasis of prolactin-induced estrogen receptor alpha positive mammary tumor cells. Breast Cancer Res. 2017, 19, 9. [Google Scholar] [CrossRef] [Green Version]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.; Gillen, A.; Cittelly, D.M.; Tan, A.C.; Sams, S.B.; Pillai, M.; Elias, A.; Robinson, W.A.; et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 23, 1710–1721. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Tse, K.; Zahedi, P.; Harding, S.M.; Zafarana, G.; Jaffray, D.A.; Bristow, R.G.; Allen, C. Hypoxia and cellular localization influence the radiosensitizing effect of gold nanoparticles (AuNPs) in breast cancer cells. Radiat. Res. 2014, 182, 475–488. [Google Scholar] [CrossRef]
- Cun, X.; Ruan, S.; Chen, J.; Zhang, L.; Li, J.; He, Q.; Gao, H. A dual strategy to improve the penetration and treatment of breast cancer by combining shrinking nanoparticles with collagen depletion by losartan. Acta Biomater. 2016, 31, 186–196. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Sung, S.Y.; Hsieh, C.L.; Wu, D.; Chung, L.W.; Johnstone, P.A. Tumor microenvironment promotes cancer progression, metastasis, and therapeutic resistance. Curr. Probl. Cancer 2007, 31, 36–100. [Google Scholar] [CrossRef]
- Whatcott, C.J.; Han, H.; Posner, R.G.; Hostetter, G.; Von Hoff, D.D. Targeting the tumor microenvironment in cancer: Why hyaluronidase deserves a second look. Cancer Discov. 2011, 1, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Mbeunkui, F.; Johann, D.J. Cancer and the tumor microenvironment: A review of an essential relationship. Cancer Chemother. Pharmacol. 2009, 63, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Q.; Ge, G. Lysyl oxidase, extracellular matrix remodeling and cancer metastasis. Cancer Microenviron. 2012, 5, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Xiao, Q.; Ma, H.; Li, L.; Liu, J.; Feng, Y.; Fang, Z.; Wu, J.; Han, X.; Zhang, J. LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proc. Natl. Acad. Sci. USA 2010, 107, 18892–18897. [Google Scholar] [CrossRef] [Green Version]
- Vallet, S.D.; Ricard-Blum, S. Lysyl oxidases: From enzyme activity to extracellular matrix cross-links. Essays Biochem. 2019, 63, 349–364. [Google Scholar]
- Seewaldt, V. ECM stiffness paves the way for tumor cells. Nat. Med. 2014, 20, 332–333. [Google Scholar] [CrossRef]
- Sato, S.; Chong, S.G.; Upagupta, C.; Yanagihara, T.; Saito, T.; Shimbori, C.; Bellaye, P.-S.; Nishioka, Y.; Kolb, M.R. Fibrotic extracellular matrix induces release of extracellular vesicles with pro-fibrotic miRNA from fibrocytes. Thorax 2021, 76, 895–906. [Google Scholar] [CrossRef]
- Netti, P.A.; Baxter, L.T.; Boucher, Y.; Skalak, R.; Jain, R.K. Time-dependent behavior of interstitial fluid pressure in solid tumors: Implications for drug delivery. Cancer Res. 1995, 55, 5451–5458. [Google Scholar]
- Morin, P.J. Drug resistance and the microenvironment: Nature and nurture. Drug Resist. Updat. 2003, 6, 169–172. [Google Scholar] [CrossRef]
- Gouarderes, S.; Mingotaud, A.-F.; Vicendo, P.; Gibot, L. Vascular and extracellular matrix remodeling by physical approaches to improve drug delivery at the tumor site. Expert Opin. Drug Deliv. 2020, 17, 1703–1726. [Google Scholar] [CrossRef]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the physical microenvironment of tumors to improve drug delivery and efficacy: From mathematical modeling to bench to bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef] [Green Version]
- Sercu, S.; Zhang, L.; Merregaert, E.J. The extracellular matrix protein 1: Its molecular interaction and implication in tumor progression. Cancer Investig. 2008, 26, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troup, S.; Njue, C.; Kliewer, E.V.; Parisien, M.; Roskelley, C.; Chakravarti, S.; Roughley, P.J.; Murphy, L.C.; Watson, P.H. Reduced expression of the small leucine-rich proteoglycans, lumican, and decorin is associated with poor outcome in node-negative invasive breast cancer. Clin. Cancer Res. 2003, 9, 207–214. [Google Scholar] [PubMed]
- Biaoxue, R.; Xiguang, C.; Hua, L.; Hui, M.; Shuanying, Y.; Wei, Z.; Wenli, S.; Jie, D. Decreased expression of decorin and p57 (KIP2) correlates with poor survival and lymphatic metastasis in lung cancer patients. Int. J. Biol. Mrk. 2011, 26, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Matsumine, A.; Shintani, K.; Kusuzaki, K.; Matsubara, T.; Satonaka, H.; Wakabayashi, T.; Iino, T.; Uchida, A. Expression of decorin, a small leucine-rich proteoglycan, as a prognostic factor in soft tissue tumors. J. Surg. Oncol. 2007, 96, 411–418. [Google Scholar] [CrossRef]
- Järveläinen, H.; Sainio, A.; Koulu, M.; Wight, T.N.; Penttinen, R. Extracellular matrix molecules: Potential targets in pharmacotherapy. Pharmacol. Rev. 2009, 61, 198–223. [Google Scholar] [CrossRef] [Green Version]
- Erkan, M.; Michalski, C.W.; Rieder, S.; Reiser–Erkan, C.; Abiatari, I.; Kolb, A.; Giese, N.A.; Esposito, I.; Friess, H.; Kleeff, J. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin. Gastroenterol. Hepatol. 2008, 6, 1155–1161. [Google Scholar] [CrossRef]
- Niland, S.; Eble, J.A. Hold on or cut? Integrin-and MMP-mediated cell–matrix interactions in the tumor microenvironment. Int. J. Mol. Sci. 2020, 22, 238. [Google Scholar] [CrossRef]
- Henke, E.; Nandigama, R.; Ergün, S. Extracellular matrix in the tumor microenvironment and its impact on cancer therapy. Front. Mol. Biosci. 2020, 6, 160. [Google Scholar] [CrossRef] [Green Version]
- Hanker, A.B.; Estrada, M.V.; Bianchini, G.; Moore, P.D.; Zhao, J.; Cheng, F.; Koch, J.P.; Gianni, L.; Tyson, D.R.; Sánchez, V. Extracellular Matrix/Integrin Signaling Promotes Resistance to Combined Inhibition of HER2 and PI3K in HER2+ Breast CancerECM Promotes Resistance to HER2/PI3K Inhibition. Cancer Res. 2017, 77, 3280–3292. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Zheng, L.; Bullock, A.J.; Seery, T.E.; Harris, W.P.; Sigal, D.S.; Braiteh, F.; Ritch, P.S.; Zalupski, M.M.; Bahary, N. HALO 202: Randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J. Clin. Oncol. 2018, 36, 359–366. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Philip, P.A.; Hingorani, S.R.; Lacy, J.; Kortmansky, J.S.; Thumar, J.; Chiorean, E.G.; Shields, A.F.; Behl, D. Phase IB/II randomized study of FOLFIRINOX plus pegylated recombinant human hyaluronidase versus FOLFIRINOX alone in patients with metastatic pancreatic adenocarcinoma: SWOG S1313. J. Clin. Oncol. 2019, 37, 1062. [Google Scholar] [CrossRef]
- Lokeshwar, B.L. MMP inhibition in prostate cancer. Ann. N. Y. Acad. Sci. 1999, 878, 271–289. [Google Scholar] [CrossRef]
- Turpeenniemi-Hujanen, T. Gelatinases (MMP-2 and-9) and their natural inhibitors as prognostic indicators in solid cancers. Biochimie 2005, 87, 287–297. [Google Scholar] [CrossRef]
- Gomes, L.R.; Terra, L.F.; Wailemann, R.A.; Labriola, L.; Sogayar, M.C. TGF-β1 modulates the homeostasis between MMPs and MMP inhibitors through p38 MAPK and ERK1/2 in highly invasive breast cancer cells. BMC Cancer 2012, 12, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Matter, H.; Schudok, M. Recent advances in the design of matrix metalloprotease inhibitors. Curr. Opin. Drug Discov. Dev. 2004, 7, 513–535. [Google Scholar]
- Nuti, E.; Tuccinardi, T.; Rossello, A. Matrix metalloproteinase inhibitors: New challenges in the era of post broad-spectrum inhibitors. Curr. Pharm. Des. 2007, 13, 2087–2100. [Google Scholar] [CrossRef]
- Mannello, F.; Tonti, G.; Papa, S. Matrix metalloproteinase inhibitors as anticancer therapeutics. Curr. Cancer Drug Targets 2005, 5, 285–298. [Google Scholar] [CrossRef]
- Radisky, E.S.; Raeeszadeh-Sarmazdeh, M.; Radisky, D.C. Therapeutic potential of matrix metalloproteinase inhibition in breast cancer. J. Cell. Biochem. 2017, 118, 3531–3548. [Google Scholar] [CrossRef] [Green Version]
- Devel, L.; Czarny, B.; Beau, F.; Georgiadis, D.; Stura, E.; Dive, V. Third generation of matrix metalloprotease inhibitors: Gain in selectivity by targeting the depth of the S1’ cavity. Biochimie 2010, 92, 1501–1508. [Google Scholar] [CrossRef]
- Broekgaarden, M.; Weijer, R.; van Gulik, T.M.; Hamblin, M.R.; Heger, M. Tumor cell survival pathways activated by photodynamic therapy: A molecular basis for pharmacological inhibition strategies. Cancer Metastasis Rev. 2015, 34, 643–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, S. Cotargeting survival signaling pathways in cancer. J. Clin. Investig. 2008, 118, 3003–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhou, J.-Y.; Wei, W.-Z.; Wu, G.S. Activation of the Akt survival pathway contributes to TRAIL resistance in cancer cells. PLoS ONE 2010, 5, e10226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy-Luzarraga, M.; Hodivala-Dilke, K. Molecular Pathways: Endothelial Cell FAK—A Target for Cancer TreatmentEndothelial Cell FAK in Cancer Treatment. Clin. Cancer Res. 2016, 22, 3718–3724. [Google Scholar] [CrossRef] [Green Version]
- McCormick, F. Small-molecule inhibitors of cell signaling. Curr. Opin. Biotechnol. 2000, 11, 593–597. [Google Scholar] [CrossRef]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Lin, R.; Yi, S.; Gong, L.; Liu, W.; Wang, P.; Liu, N.; Zhao, L.; Wang, P. Inhibition of TGF-β signaling with halofuginone can enhance the antitumor effect of irradiation in Lewis lung cancer. OncoTargets Ther. 2015, 8, 3549. [Google Scholar] [CrossRef] [Green Version]
- Juárez, P.; Mohammad, K.S.; Yin, J.J.; Fournier, P.G.; McKenna, R.C.; Davis, H.W.; Peng, X.H.; Niewolna, M.; Javelaud, D.; Chirgwin, J.M. Halofuginone Inhibits the Establishment and Progression of Melanoma Bone MetastasesHalofuginone Decreases Melanoma Bone Metastases. Cancer Res. 2012, 72, 6247–6256. [Google Scholar] [CrossRef] [Green Version]
- Miyazono, K. Transforming growth factor-β signaling in epithelial-mesenchymal transition and progression of cancer. Proc. Jpn. Acad. Ser. B 2009, 85, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Shaty, M.H.; Arif, I.S.; Al-Ezzi, M.I.; Hanna, D.B. Metformin attenuate fibrosis in both acute and chronic doxorubicin cardiotoxicity in rabbits. J. Pharm. Sci. Res. 2018, 10, 1559–1565. [Google Scholar]
- Lampi, M.C.; Reinhart-King, C.A. Targeting extracellular matrix stiffness to attenuate disease: From molecular mechanisms to clinical trials. Sci. Transl. Med. 2018, 10, eaao0475. [Google Scholar] [CrossRef] [Green Version]
- Sporn, M.B.; Roberts, A.B.; Wakefield, L.M.; Assoian, R.K. Transforming growth factor-β: Biological function and chemical structure. Science 1986, 233, 532–534. [Google Scholar] [CrossRef]
- Krane, S.M. Collagenases and collagen degradation. J. Investig. Dermatol. 1982, 79, 83–86. [Google Scholar] [CrossRef]
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 2509–2513. [Google Scholar] [CrossRef] [Green Version]
- Parks, W.C.; Wilson, C.L.; López-Boado, Y.S. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat. Rev. Immunol. 2004, 4, 617–629. [Google Scholar] [CrossRef]
- Khokha, R.; Murthy, A.; Weiss, A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 649–665. [Google Scholar] [CrossRef]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer—Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef]
- Castellani, P.; Viale, G.; Dorcaratto, A.; Nicolo, G.; Kaczmarek, J.; Querze, G.; Zardi, L. The fibronectin isoform containing the ED-B oncofetal domain: A marker of angiogenesis. Int. J. Cancer 1994, 59, 612–618. [Google Scholar] [CrossRef]
- Glukhova, M.A.; Frid, M.G.; Shekhonin, B.V.; Balabanov, Y.V.; Koteliansky, V.E. Expression of fibronectin variants in vascular and visceral smooth muscle cells in development. Dev. Biol. 1990, 141, 193–202. [Google Scholar] [CrossRef]
- Han, Z.; Lu, Z.-R. Targeting fibronectin for cancer imaging and therapy. J. Mater. Chem. B 2017, 5, 639–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnemolla, B.; Leprini, A.; Allemanni, G.; Saginati, M.; Zardi, L. The inclusion of the type III repeat ED-B in the fibronectin molecule generates conformational modifications that unmask a cryptic sequence. J. Biol. Chem. 1992, 267, 24689–24692. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.-M.; Lan, Y.; Lauder, S.; Zhang, J.; Brunkhorst, B.; Qin, G.; Verma, R.; Courtenay-Luck, N.; Gillies, S.D. huBC1-IL12, an immunocytokine which targets EDB-containing oncofetal fibronectin in tumors and tumor vasculature, shows potent anti-tumor activity in human tumor models. Cancer Immunol. Immunother. 2007, 56, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Kim, S.; Lee, I.-h.; Park, J.; Yu, M.; Lee, J.; Kim, J.-I.; Jon, S. Aptide-conjugated liposome targeting tumor-associated fibronectin for glioma therapy. J. Mater. Chem. B 2013, 1, 4723–4726. [Google Scholar] [CrossRef]
- Lokman, N.A.; Price, Z.K.; Hawkins, E.K.; Macpherson, A.M.; Oehler, M.K.; Ricciardelli, C. 4-Methylumbelliferone inhibits cancer stem cell activation and overcomes chemoresistance in ovarian cancer. Cancers 2019, 11, 1187. [Google Scholar] [CrossRef] [Green Version]
- Kohli, A.G.; Kivimäe, S.; Tiffany, M.R.; Szoka, F.C. Improving the distribution of Doxil® in the tumor matrix by depletion of tumor hyaluronan. J. Control. Release 2014, 191, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Doherty, G.J.; Tempero, M.; Corrie, P.G. HALO-109–301: A Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Future Oncol. 2018, 14, 13–22. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Storm, G.; Prakash, J. Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Adv. Drug Deliv. Rev. 2018, 129, 37–53. [Google Scholar] [CrossRef]
- Gutheil, J.C.; Campbell, T.N.; Pierce, P.R.; Watkins, J.D.; Huse, W.D.; Bodkin, D.J.; Cheresh, D.A. Targeted antiangiogenic therapy for cancer using Vitaxin: A humanized monoclonal antibody to the integrin ανβ3. Clin. Cancer Res. 2000, 6, 3056–3061. [Google Scholar]
- Raguse, J.-D.; Gath, H.J.; Bier, J.; Riess, H.; Oettle, H. Cilengitide (EMD 121974) arrests the growth of a heavily pretreated highly vascularised head and neck tumour. Oral Oncol. 2004, 40, 228–230. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Anti-Cancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [Green Version]
- Tijink, B.M.; Buter, J.; De Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; Van Dongen, G.A. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 2006, 12, 6064–6072. [Google Scholar] [CrossRef] [Green Version]
- Riechelmann, H.; Sauter, A.; Golze, W.; Hanft, G.; Schroen, C.; Hoermann, K.; Erhardt, T.; Gronau, S. Phase I trial with the CD44v6-targeting immunoconjugate bivatuzumab mertansine in head and neck squamous cell carcinoma. Oral Oncol. 2008, 44, 823–829. [Google Scholar] [CrossRef]
- Liu, S.; Qin, S.; He, M.; Zhou, D.; Qin, Q.; Wang, H. Current applications of poly (lactic acid) composites in tissue engineering and drug delivery. Compos. Part B Eng. 2020, 199, 108238. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, J.; Yang, Y.; Liang, H.; Jia, H.; Li, D. Current trends of targeted drug delivery for oral cancer therapy. Front. Bioeng. Biotechnol. 2020, 8, 618931. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Liu, Y.; Xu, K.; Sun, X.; Yu, Q.; Wu, Z.; Zhao, Z. Biomimetic polydopamine-modified silk fibroin/curcumin nanofibrous scaffolds for chemo-photothermal therapy of bone tumor. ACS Omega 2021, 6, 22213–22223. [Google Scholar] [CrossRef]
- Ozcelikkale, A.; Moon, H.r.; Linnes, M.; Han, B. In vitro microfluidic models of tumor microenvironment to screen transport of drugs and nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1460. [Google Scholar] [CrossRef]
- Wang, L.; Huo, M.; Chen, Y.; Shi, J. Tumor microenvironment-enabled nanotherapy. Adv. Healthc. Mater. 2018, 7, 1701156. [Google Scholar] [CrossRef]
- Sheikhpour, M.; Barani, L.; Kasaeian, A. Biomimetics in drug delivery systems: A critical review. J. Control. Release 2017, 253, 97–109. [Google Scholar] [CrossRef]
- Frith, J.E.; Thomson, B.; Genever, P.G. Dynamic three-dimensional culture methods enhance mesenchymal stem cell properties and increase therapeutic potential. Tissue Eng. Part C Methods 2010, 16, 735–749. [Google Scholar] [CrossRef]
- Martino, M.M.; Briquez, P.S.; Güç, E.; Tortelli, F.; Kilarski, W.W.; Metzger, S.; Rice, J.J.; Kuhn, G.A.; Müller, R.; Swartz, M.A. Growth factors engineered for super-affinity to the extracellular matrix enhance tissue healing. Science 2014, 343, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Gjorgieva, D.; Zaidman, N.; Bosnakovski, D. Mesenchymal stem cells for anti-cancer drug delivery. Recent Pat. Anti-Cancer Drug Discov. 2013, 8, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Studeny, M.; Marini, F.C.; Dembinski, J.L.; Zompetta, C.; Cabreira-Hansen, M.; Bekele, B.N.; Champlin, R.E.; Andreeff, M. Mesenchymal stem cells: Potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J. Natl. Cancer Inst. 2004, 96, 1593–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzobo, K. Recent trends in multipotent human mesenchymal stem/stromal cells: Learning from history and advancing clinical applications. OMICS A J. Integr. Biol. 2021, 25, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, L.H.; Heyn, G.S.; Magalhaes, K.G. The impact of the adipose organ plasticity on inflammation and cancer progression. Cells 2019, 8, 662. [Google Scholar] [CrossRef] [Green Version]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of mesenchymal stem cells for therapeutic agent delivery in anti-tumor treatment. Front. Pharmacol. 2018, 9, 259. [Google Scholar] [CrossRef]
- Gao, P.; Ding, Q.; Wu, Z.; Jiang, H.; Fang, Z. Therapeutic potential of human mesenchymal stem cells producing IL-12 in a mouse xenograft model of renal cell carcinoma. Cancer Lett. 2010, 290, 157–166. [Google Scholar] [CrossRef]
- Bu, S.; Wang, Q.; Zhang, Q.; Sun, J.; He, B.; Xiang, C.; Liu, Z.; Lai, D. Human endometrial mesenchymal stem cells exhibit intrinsic anti-tumor properties on human epithelial ovarian cancer cells. Sci. Rep. 2016, 6, 37019. [Google Scholar] [CrossRef] [Green Version]
- Rasti Boroojeni, F.; Mashayekhan, S.; Abbaszadeh, H.-A.; Ansarizadeh, M.; Khoramgah, M.-S.; Rahimi Movaghar, V. Bioinspired nanofiber scaffold for differentiating bone marrow-derived neural stem cells to oligodendrocyte-like cells: Design, fabrication, and characterization. Int. J. Nanomed. 2020, 15, 3903–3920. [Google Scholar] [CrossRef]
- Chen, C.; Tang, J.; Gu, Y.; Liu, L.; Liu, X.; Deng, L.; Martins, C.; Sarmento, B.; Cui, W.; Chen, L. Bioinspired hydrogel electrospun fibers for spinal cord regeneration. Adv. Funct. Mater. 2019, 29, 1806899. [Google Scholar] [CrossRef]
- Ahmadi, S.; Nasiri, M.; Pourrajab-miandoab, A.; Abbasi, F.; Hassanpour, F.; Sabzi, E. Photo-and thermo-responsive extracellular matrix mimicking nano-coatings prepared from poly (N-isopropylacrylamide)-spiropyran copolymer for effective cell sheet harvesting. Prog. Org. Coat. 2022, 167, 106847. [Google Scholar] [CrossRef]
- Valente, K.P.; Thind, S.S.; Akbari, M.; Suleman, A.; Brolo, A.G. Collagen Type I–Gelatin Methacryloyl Composites: Mimicking the Tumor Microenvironment. ACS Biomater. Sci. Eng. 2019, 5, 2887–2898. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Fan, Z.; Deng, J.; Lemons, P.K.; Arhontoulis, D.C.; Bowne, W.B.; Cheng, H. Hyaluronidase embedded in nanocarrier PEG shell for enhanced tumor penetration and highly efficient antitumor efficacy. Nano Lett. 2016, 16, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Şen, Ö.; Emanet, M.; Ciofani, G. Nanotechnology-Based Strategies to Evaluate and Counteract Cancer Metastasis and Neoangiogenesis. Adv. Healthc. Mater. 2021, 10, 2002163. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Prabhakaran, M.P.; Ramakrishna, S. Advances in drug delivery via electrospun and electrosprayed nanomaterials. Int. J. Nanomed. 2013, 8, 2997–3017. [Google Scholar]
- Fu, Y.; Li, X.; Ren, Z.; Mao, C.; Han, G. Multifunctional electrospun nanofibers for enhancing localized cancer treatment. Small 2018, 14, 1801183. [Google Scholar] [CrossRef]
- Lee, H.J.; Mun, S.; Pham, D.M.; Kim, P. Extracellular matrix-based hydrogels to tailoring tumor organoids. ACS Biomater. Sci. Eng. 2021, 7, 4128–4135. [Google Scholar] [CrossRef]
- Votanopoulos, K.I.; Forsythe, S.; Sivakumar, H.; Mazzocchi, A.; Aleman, J.; Miller, L.; Levine, E.; Triozzi, P.; Skardal, A. Model of patient-specific immune-enhanced organoids for immunotherapy screening: Feasibility study. Ann. Surg. Oncol. 2020, 27, 1956–1967. [Google Scholar] [CrossRef]
- Stirling, E.; Willey-Shelkey, E.; Wilson, A.; Skardal, A.; Triozzi, P.; Terabe, M.; Miller, L.; Soker, S.; Soto-Pantoja, D. 206 An immune-competent tumor organoid platform to test novel immune checkpoint combinations targeting the receptor CD47 in triple negative breast cancer. J. Immunother. Cancer 2020, 8, A121–A122. [Google Scholar]
- Tang, R.-Z.; Liu, Z.-Z.; Gu, S.-S.; Liu, X.-Q. Multiple local therapeutics based on nano-hydrogel composites in breast cancer treatment. J. Mater. Chem. B 2021, 9, 1521–1535. [Google Scholar] [CrossRef]
- Li, A.; Zhao, J.; Fu, J.; Cai, J.; Zhang, P. Recent advances of biomimetic nano-systems in the diagnosis and treatment of tumor. Asian J. Pharm. Sci. 2021, 16, 161–174. [Google Scholar] [CrossRef]
- Oliveira, M.; Conceição, P.; Kant, K.; Ainla, A.; Diéguez, L. Electrochemical sensing in 3D cell culture models: New Tools for developing better cancer diagnostics and treatments. Cancers 2021, 13, 1381. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dzobo, K.; Dandara, C. The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis. Biomimetics 2023, 8, 146. https://doi.org/10.3390/biomimetics8020146
Dzobo K, Dandara C. The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis. Biomimetics. 2023; 8(2):146. https://doi.org/10.3390/biomimetics8020146
Chicago/Turabian StyleDzobo, Kevin, and Collet Dandara. 2023. "The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis" Biomimetics 8, no. 2: 146. https://doi.org/10.3390/biomimetics8020146
APA StyleDzobo, K., & Dandara, C. (2023). The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis. Biomimetics, 8(2), 146. https://doi.org/10.3390/biomimetics8020146