

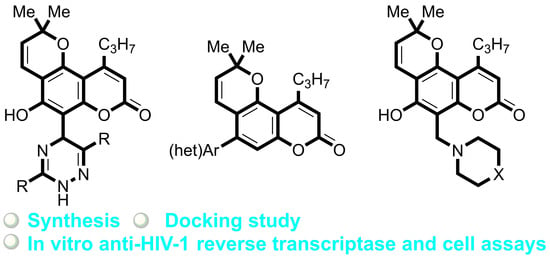
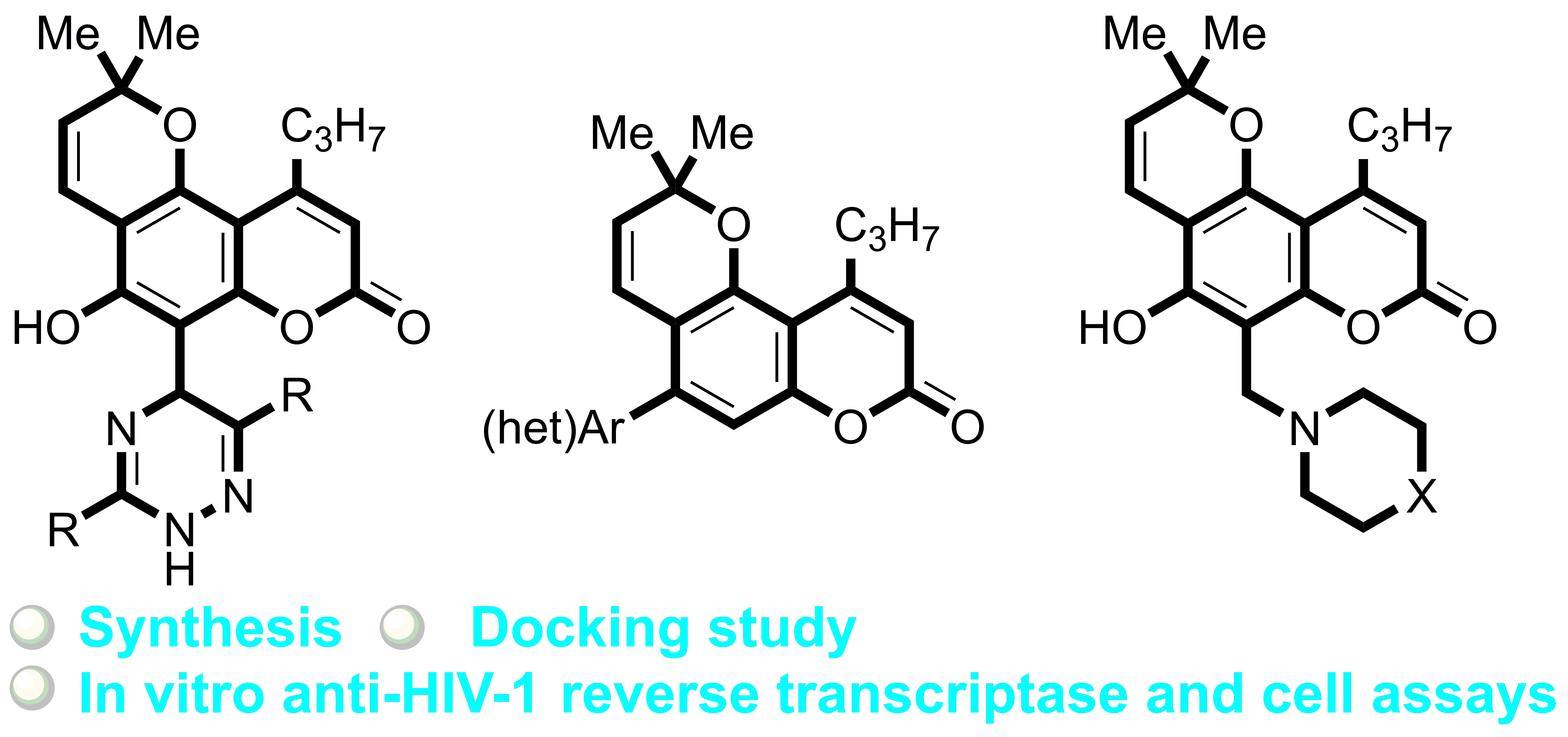
Bioinspired Pyrano[2,3-f]chromen-8-ones: Ring C-Opened Analogues of Calanolide A: Synthesis and Anti-HIV-1 Evaluation

 , , , ,
, , , , 
Abstract
:
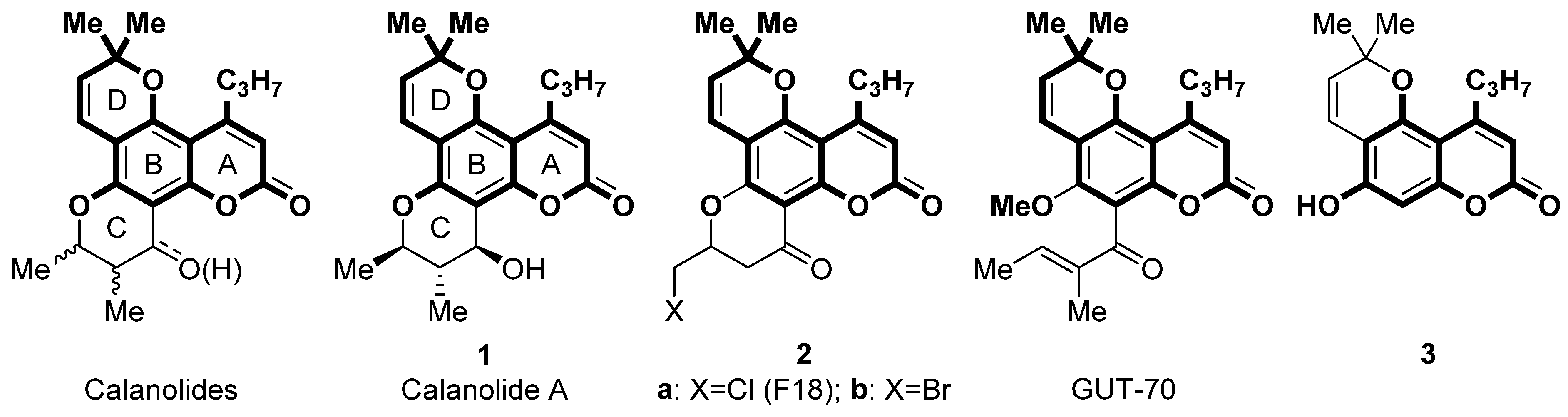
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. Synthesis of 6-Triazyninyl Derivatives of 5-Hydroxy-2,2-dimethyl-10-propyl-2H,8H-pyrano[2,3-f]chromen-8-ones 4a–h and 5a–f
General Procedure for the Synthesis of Dihydrotriazines 4a–h
General Procedure for Aromatization of Dihydrotriazines 4a–d and 4f to triazines 5a–d and 5f
2.1.2. Synthesis of 5-(het)Aryl-2,2-dimethyl-10-propyl-2H,8H-pyrano[2,3-f]chromen-8-ones 7a–l
General Procedure for the Suzuki Cross-Coupling Reaction
2.1.3. Synthesis of Aminomethyl Derivatives of 5-Hydroxy-2,2-dimethyl-10-propyl-2H,8H-pyrano[2,3-f]chromen-8-one 9a–c
General Procedure for the Mannich Reaction
2.2. Anti-HIV Assay
2.2.1. Antiviral Activity on the Model HIV-1 Reverse Transcriptase
2.2.2. Antiviral Activity in a Model of Human Cells Infected with HIV-1
- Cells.
- Viruses.
- Assay Design.
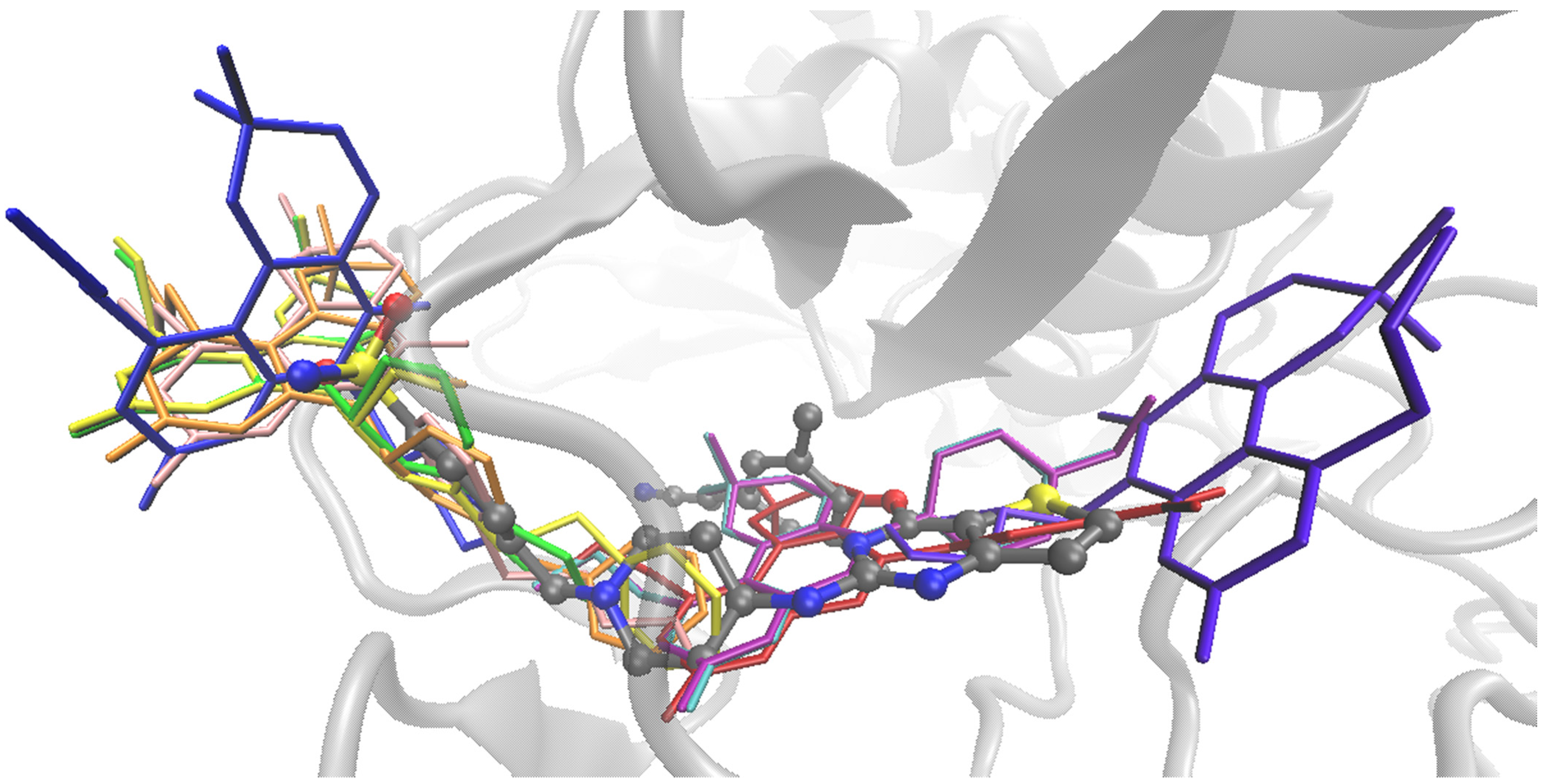
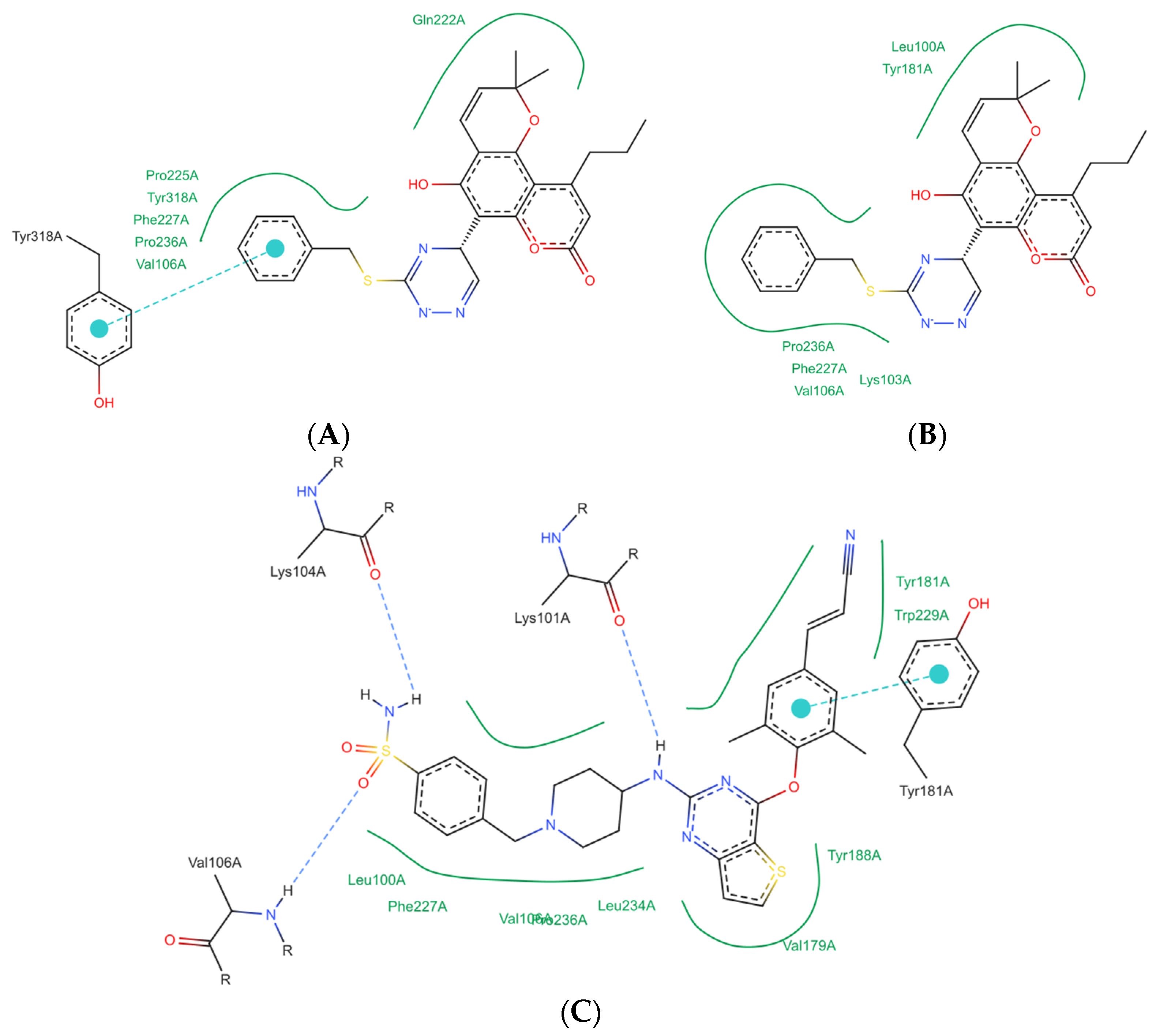
2.3. Computational Study
- Ligand preparation.
- Docking protocol
- Validation.
3. Results and Discussion
3.1. Chemistry
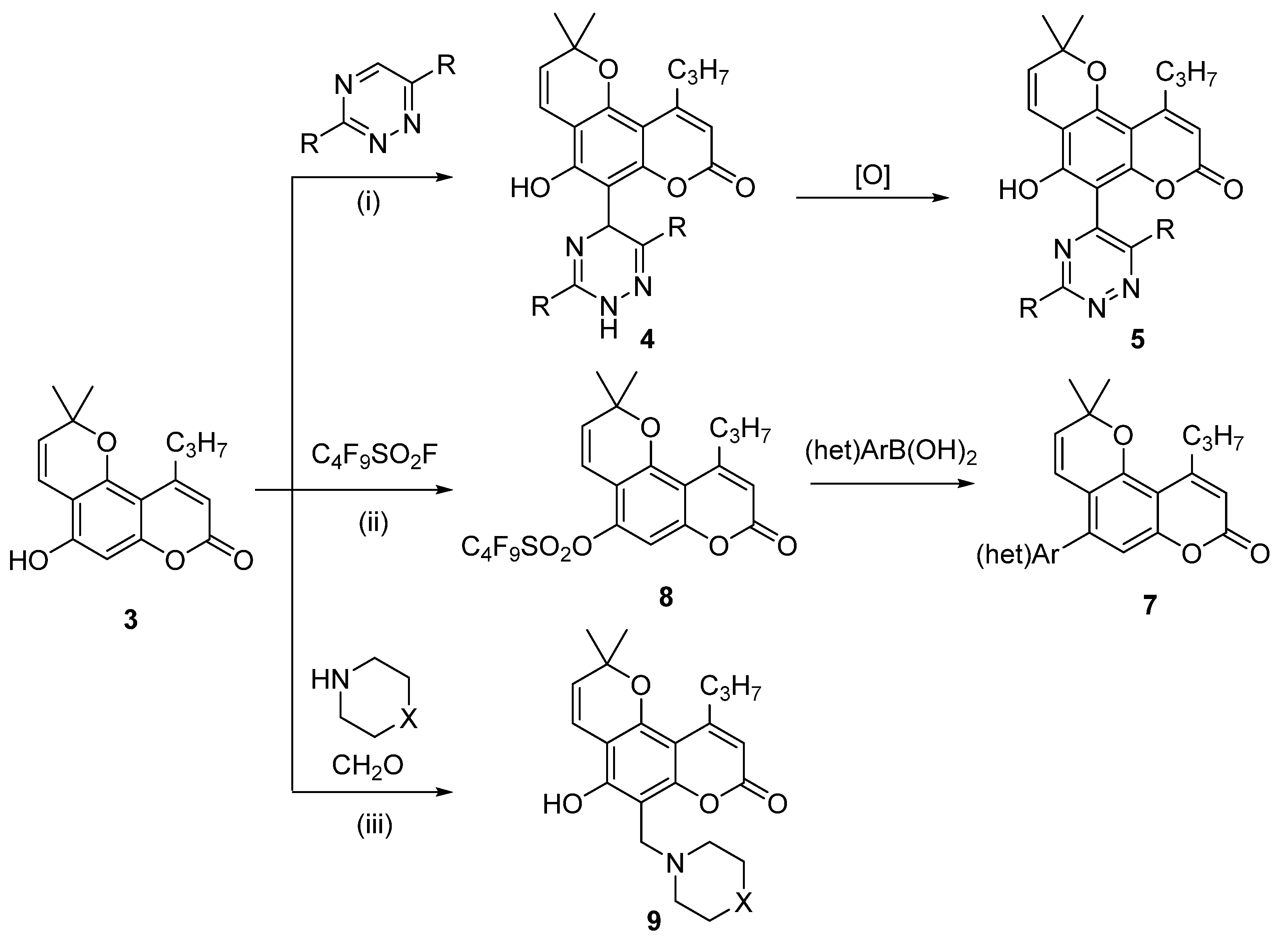
3.1.1. Synthesis of Calanolide Analogues with 1,2,4-Triazine Moiety
3.1.2. Synthesis of Calanolide Analogues with (het)Aryl Moiety via Suzuki Cross-Coupling Reaction
3.1.3. Synthesis of Calanolide Analogues with Aminomethyl Moiety
3.2. Anti-HIV Assay
3.2.1. Antiviral Activity on the Model HIV-1 Reverse Transcriptase
3.2.2. Antiviral Activity in the Model of Human Cells Infected with HIV-1
3.3. Computational Study
4. Conclusions
- Reaction of compound 3 with 1,2,4-triazines in the presence of methanesulfonic acid afforded dihydrotriazines 4. The dihydrotriazine ring was aromatized by refluxing with tetrachloro-1,4-benzoquinone to obtain compound 5.
- Sulfonylation of compound 3 followed by Suzuki cross-coupling reaction with (het)aryl boronic acids.
- Aminomethylation of 3 by Mannich reaction with cyclic secondary amines.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- HIV and AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 22 December 2023).
- Andrade, H.B.; Shinotsuka, C.R.; da Silva, I.R.F.; Donini, C.S.; Yeh Li, H.; de Carvalho, F.B.; do Brasil, P.E.A.A.; Bozza, F.A.; Japiassu, A.M. Highly active antiretroviral therapy for critically ill HIV patients: A systematic review and metaanalysis. PLoS ONE 2017, 12, e0186968. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Pannecouque, C.; De Clercq, E.; Liu, X. Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem. 2016, 59, 2849–2878. [Google Scholar] [CrossRef]
- Maeda, K.; Das, D.; Kobayakawa, T.; Tamamura, H.; Takeuchi, H. Discovery and Development of Anti-HIV Therapeutic Agents: Progress Towards Improved HIV Medication. Curr. Top. Med. Chem. 2019, 19, 1621–1649. [Google Scholar] [CrossRef]
- Hu, S.; Chen, J.; Cao, J.-X.; Zhang, S.-S.; Gu, S.-X.; Chen, F.-E. Quinolines and isoquinolines as HIV-1 inhibitors: Chemical structures, action targets, and biological activities. Bioorg. Chem. 2023, 136, 106549. [Google Scholar] [CrossRef] [PubMed]
- Romeo, R.; Iannazzo, D.; Veltri, L.; Gabriele, B.; Macchi, B.; Frezza, C.; Marino-Merlo, F.; Giofrè, S.V. Pyrimidine 2,4-Diones in the Design of New HIV RT Inhibitors. Molecules 2019, 24, 1718. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, G.C.; Martins, L.M.; Bregadiolli, B.A.; Moreno, V.F.; da Silva-Filho, L.C.; da Silva, B.H.S.T. Heterocyclic compounds as antiviral drugs: Synthesis, structure–activity relationship and traditional applications. J. Heterocyclic. Chem. 2021, 58, 2226–2260. [Google Scholar] [CrossRef]
- Mousavi, H. A comprehensive survey upon diverse and prolific applications of chitosan-based catalytic systems in one-pot multi-component synthesis of heterocyclic rings. Int. J. Biol. Macromol. 2021, 186, 1003–1166. [Google Scholar] [CrossRef]
- Viktorova, V.V.; Steparuk, E.V.; Obydennov, D.L.; Sosnovskikh, V.Y. The Construction of Polycyclic Pyridones via Ring-Opening Transformations of 3-hydroxy-3,4-dihydropyrido[2,1-c][1,4]oxazine-1,8-diones. Molecules 2023, 28, 1285. [Google Scholar] [CrossRef]
- Rotella, D.P. Heterocycles in drug discovery: Properties and preparation. In Advances in Heterocyclic Chemistry; Meanwell, N.A., Lolli, M.L., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 134, pp. 149–183. [Google Scholar]
- Nahar, L.; Talukdar, A.D.; Nath, D.; Nath, S.; Mehan, A.; Ismail, F.M.D.; Sarker, S.D. Naturally Occurring Calanolides: Occurrence, Biosynthesis, and Pharmacological Properties Including Therapeutic Potential. Molecules 2020, 25, 4983. [Google Scholar] [CrossRef]
- Sharapov, A.D.; Fatykhov, R.F.; Khalymbadzha, I.A.; Zyryanov, G.V.; Chupakhin, O.N.; Tsurkan, M.V. Plant Coumarins with Anti-HIV Activity: Isolation and Mechanisms of Action. Int. J. Mol. Sci. 2023, 24, 2839. [Google Scholar] [CrossRef]
- Creagh, T.; Ruckle, J.L.; Tolbert, D.T.; Giltner, J.; Eiznhamer, D.A.; Dutta, B.; Flavin, M.T.; Xu, Z.-Q. Safety and Pharmacokinetics of Single Doses of (+)-Calanolide A, a Novel, Naturally Occurring Nonnucleoside Reverse Transcriptase Inhibitor, in Healthy, Human Immunodeficiency Virus-Negative Human Subjects. Antimicrob. Agents Chemother. 2001, 45, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Flavin, M.T.; Rizzo, J.D.; Khilevich, A.; Kucherenko, A.; Sheinkman, A.K.; Vilaychack, V.; Lin, L.; Chen, W.; Greenwood, E.M.; Pengsuparp, T.; et al. Synthesis, Chromatographic Resolution, and Anti-Human Immunodeficiency Virus Activity of (±)-Calanolide A and Its Enantiomers. J. Med. Chem. 1996, 39, 1303–1313. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.P.; Tagliaferri, F.; Victory, S.F.; Yan, S.; Baker, D.C. Synthesis of Optically Active Calanolides A and B. J. Org. Chem. 1995, 60, 2964–2965. [Google Scholar] [CrossRef]
- Zembower, D.E.; Liao, S.; Flavin, M.T.; Xu, Z.-Q.; Stup, T.L.; Buckheit, R.W.; Khilevich, A.; Mar, A.A.; Sheinkman, A.K. Structural Analogues of the Calanolide Anti-HIV Agents. Modification of the trans-10,11-Dimethyldihydropyran-12-ol Ring (Ring C). J. Med. Chem. 1997, 40, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Lu, X.; Zheng, P.; Liu, L.; Han, C.; Hu, J.; Liu, Z.; Ma, T.; Li, Y.; Wang, L.; et al. Highly Suppressing Wild-Type HIV-1 and Y181C Mutant HIV-1 Strains by 10-Chloromethyl-11-demethyl-12-oxo-calanolide A with Druggable Profile. J. Med. Chem. 2010, 53, 1397–1401. [Google Scholar] [CrossRef]
- Ma, T.; Liu, L.; Xue, H.; Li, L.; Han, C.; Wang, L.; Chen, Z.; Liu, G. Chemical Library and Structure–Activity Relationships of 11-Demethyl-12-oxo Calanolide A Analogues as Anti-HIV-1 Agents. J. Med. Chem. 2008, 51, 1432–1446. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Hattori, S.; Kariya, R.; Komizu, Y.; Kudo, E.; Goto, H.; Taura, M.; Ueoka, R.; Kimura, S.; Okada, S. Inhibition of HIV-1 entry by the tricyclic coumarin GUT-70 through the modification of membrane fluidity. Biochem. Biophys. Res. Commun. 2015, 457, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Kudo, E.; Taura, M.; Matsuda, K.; Shimamoto, M.; Kariya, R.; Goto, H.; Hattori, S.; Kimura, S.; Okada, S. Inhibition of HIV-1 replication by a tricyclic coumarin GUT-70 in acutely and chronically infected cells. Bioorg. Med. Chem. Lett. 2013, 23, 606–609. [Google Scholar] [CrossRef]
- Fatykhov, R.F.; Khalymbadzha, I.A.; Chupakhin, O.N.; Charushin, V.N.; Inyutina, A.K.; Slepukhin, P.A.; Kartsev, V.G. Nicotinoylbenzotriazole: A Convenient Tool for Site-Selective Protection of 5,7-Dihydroxycoumarins. Synthesis 2019, 51, 3617–3624. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Fatykhov, R.F.; Khalymbadzha, I.A.; Sharapov, A.D.; Potapova, A.P.; Mochulskaya, N.N.; Tsmokalyuk, A.N.; Ivoilova, A.V.; Mozharovskaia, P.N.; Santra, S.; Chupakhin, O.N. MnO2-Mediated Oxidative Cyclization of “Formal” Schiff’s Bases: Easy Access to Diverse Naphthofuro-Annulated Triazines. Molecules 2022, 27, 7105. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Reverse Transcriptase Assay, Colorimetric. Available online: https://www.sigmaaldrich.com/RU/en/product/roche/11468120910 (accessed on 22 December 2023).
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program for Chemistry Aware Data Visualization and Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model. 2019, 59, 895–913. [Google Scholar] [CrossRef]
- Bedoya, L.M.; Beltrán, M.; Sancho, R.; Olmedo, D.A.; Sánchez-Palomino, S.; del Olmo, E.; López-Pérez, J.L.; Muñoz, E.; Feliciano, A.S.; Alcamí, J. 4-Phenylcoumarins as HIV transcription inhibitors. Bioorganic Med. Chem. Lett. 2005, 15, 4447–4450. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Wang, Z.; Zhang, H.; Wu, G.; Zhao, T.; Zhou, Z.; Huo, Z.; Huang, B.; Feng, D.; Ding, X.; et al. Further Exploring Solvent-Exposed Tolerant Regions of Allosteric Binding Pocket for Novel HIV-1 NNRTIs Discovery. ACS Med. Chem. Lett. 2018, 9, 370–375. [Google Scholar] [CrossRef]
- Ippolito, J.A.; Niu, H.; Bertoletti, N.; Carter, Z.J.; Jin, S.; Spasov, K.A.; Cisneros, J.A.; Valhondo, M.; Cutrona, K.J.; Anderson, K.S.; et al. Covalent Inhibition of Wild-Type HIV-1 Reverse Transcriptase Using a Fluorosulfate Warhead. ACS Med. Chem. Lett. 2021, 12, 249–255. [Google Scholar] [CrossRef]
- Famiglini, V.; La Regina, G.; Coluccia, A.; Pelliccia, S.; Brancale, A.; Maga, G.; Crespan, E.; Badia, R.; Clotet, B.; Esté, J.A.; et al. New indolylarylsulfones as highly potent and broad spectrum HIV-1 non-nucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem. 2014, 80, 101–111. [Google Scholar] [CrossRef]
- Artico, M.; Di Santo, R.; Costi, R.; Massa, S.; Scintu, F.; Loi, A.G.; De Montis, A.; La Colla, P. 1-Arylsulfonyl-3-(α-hydroxybenzyl)-1H-pyrroles, a novel class of anti-HIV-1 reverse transcriptase inhibitors. Bioorganic Med. Chem. Lett. 1997, 7, 1931–1936. [Google Scholar] [CrossRef]
- Wishka, D.G.; Graber, D.R.; Kopta, L.A.; Olmsted, R.A.; Friis, J.M.; Hosley, J.D.; Adams, W.J.; Seest, E.P.; Castle, T.M.; Dolak, L.A.; et al. (−)-6-Chloro-2-[(1-furo[2,3-c]pyridin-5-ylethyl)thio]-4-pyrimidinamine, PNU-142721, a New Broad Spectrum HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitor. J. Med. Chem. 1998, 41, 1357–1360. [Google Scholar] [CrossRef]
- Tillekeratne, L.M.V.; Sherette, A.; Fulmer, J.A.; Hupe, L.; Hupe, D.; Gabbara, S.; Peliska, J.A.; Hudson, R.A. Differential Inhibition of polymerase and Strand-Transfer Activities of HIV-1 Reverse Transcriptase. Bioorganic Med. Chem. Lett. 2002, 12, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, K.; Krause, B.; Berg, O.; Rarey, M. PoseEdit: Enhanced Ligand Binding Mode Communication by Interactive 2D Diagrams. J. Comput. Aided Mol. Des. 2023, 37, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Fatykhov, R.F.; Savchuk, M.I.; Starnovskaya, E.S.; Bobkina, M.V.; Kopchuk, D.S.; Nosova, E.V.; Zyryanov, G.V.; Khalymbadzha, I.A.; Chupakhin, O.N.; Charushin, V.N.; et al. Nucleophilic substitution of hydrogen–the Boger reaction sequence as an approach towards 8-(pyridin-2-yl)coumarins. Mendeleev Commun. 2019, 29, 299–300. [Google Scholar] [CrossRef]
- Khalymbadzha, I.A.; Chupakhin, O.N.; Fatykhov, R.F.; Charushin, V.N.; Schepochkin, A.V.; Kartsev, V.G. Transition-Metal-Free Cross-Dehydrogenative Coupling of Triazines with 5,7-Dihydroxycoumarins. Synlett 2016, 27, 2606–2610. [Google Scholar] [CrossRef]
- Yang, Y.; Kang, D.; Nguyen, L.A.; Smithline, Z.B.; Pannecouque, C.; Zhan, P.; Liu, X.; Steitz, T.A. Structural basis for potent and broad inhibition of HIV-1 RT by thiophene[3,2-d]pyrimidine nonnucleoside inhibitors. eLife 2018, 7, e36340. [Google Scholar] [CrossRef]
- Schellhammer, I.; Rarey, M. TrixX: Structure-based molecule indexing for large-scale virtual screening in sublinear time. J. Comput. Aided Mol. Des. 2007, 21, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Henzler, A.M.; Urbaczek, S.; Hilbig, M.; Rarey, M. An integrated approach to knowledge-driven structure-based virtual screening. J. Comput. Aided Mol. Des. 2014, 28, 927–939. [Google Scholar] [CrossRef]
- Flachsenberg, F.; Meyder, A.; Sommer, K.; Penner, P.; Rarey, M. A Consistent Scheme for Gradient-Based Optimization of Protein-Ligand Poses. J. Chem. Inf. Model. 2020, 60, 6502–6522. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Griewel, A.; Grombacher, T.; Rarey, M. Analyzing the topology of active sites: On the prediction of pockets and subpockets. J. Chem. Inf. Model. 2010, 50, 2041–2052. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://proteins.plus/ (accessed on 22 December 2023).
- Schöning-Stierand, K.; Diedrich, K.; Ehrt, C.; Flachsenberg, F.; Graef, J.; Sieg, J.; Penner, P.; Poppinga, M.; Ungethüm, A.; Rarey, M. ProteinsPlus: A Comprehensive Collection of Web-Based Molecular Modeling Tools. Nucleic Acids Res. 2022, 50, W611–W615. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Concentration 1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 100 μM | 75 μM | 50 μM | 25 μM | 5–10 μM | 1 μM | 0.5 μM | 0.1 μM | 0.01 μM | |
| 4a | T | T | T | 25% (T) | 42.5–95% | 45–95% | 70–98% | ||
| 4e | T | T | T | T | 62.5–95% | 62.5–95% | 70–98% | ||
| 4h | T | 95% | 95% | 95% | 42.5–95% | 45–98% | 100% | ||
| 7l 2 | T | T | T | 0% (T) | 25.0–80% | 62.5–80% | 70–98% | ||
| Zidovudine | 0% | 0% | 0% | 37.5–50% | |||||
| Ligand | Inhibition at 50 µM, % | Jamda Score | SwissADME Characteristics | ||||||
|---|---|---|---|---|---|---|---|---|---|
| TPSA | WlogP | ESOL logS | GI Absorption | BBB Permeant | CYP1A2 Inhibitor | CYP3A4 Inhibitor | |||
| 4g | 43 | −2.47 | 124.66 | 3.2 | −5.24 | High | No | Yes | Yes |
| 4d | 32 | −2.31 | 121.72 | 3.83 | −5.82 | High | No | Yes | Yes |
| 7h | 25 | −2.20 | 65.46 | 5.08 | −5.26 | High | Yes | Yes | Yes |
| 7e | 25 | −1.93 | 48.67 | 5.5 | −5.68 | High | Yes | Yes | No |
| 4e | 23 | −2.05 | 121.72 | 3.84 | −5.85 | High | No | Yes | Yes |
| 5b | 16 | −2.25 | 123.64 | 5.27 | −5.33 | Low | No | No | Yes |
| 9c | 14 | −1.88 | 62.91 | 3.59 | −4.47 | High | Yes | Yes | No |
| 7k | 13 | −1.97 | 84.75 | 5.75 | −5.62 | High | No | Yes | Yes |
| 9a | 10 | −1.80 | 62.91 | 3.98 | −4.77 | High | Yes | No | No |
| Pearson coefficient | –0.81 | ||||||||
| Spearman coefficient | –0.78 | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalymbadzha, I.A.; Fatykhov, R.F.; Butorin, I.I.; Sharapov, A.D.; Potapova, A.P.; Muthipeedika, N.J.; Zyryanov, G.V.; Melekhin, V.V.; Tokhtueva, M.D.; Deev, S.L.; et al. Bioinspired Pyrano[2,3-f]chromen-8-ones: Ring C-Opened Analogues of Calanolide A: Synthesis and Anti-HIV-1 Evaluation. Biomimetics 2024, 9, 44. https://doi.org/10.3390/biomimetics9010044
Khalymbadzha IA, Fatykhov RF, Butorin II, Sharapov AD, Potapova AP, Muthipeedika NJ, Zyryanov GV, Melekhin VV, Tokhtueva MD, Deev SL, et al. Bioinspired Pyrano[2,3-f]chromen-8-ones: Ring C-Opened Analogues of Calanolide A: Synthesis and Anti-HIV-1 Evaluation. Biomimetics. 2024; 9(1):44. https://doi.org/10.3390/biomimetics9010044
Chicago/Turabian StyleKhalymbadzha, Igor A., Ramil F. Fatykhov, Ilya I. Butorin, Ainur D. Sharapov, Anastasia P. Potapova, Nibin Joy Muthipeedika, Grigory V. Zyryanov, Vsevolod V. Melekhin, Maria D. Tokhtueva, Sergey L. Deev, and et al. 2024. "Bioinspired Pyrano[2,3-f]chromen-8-ones: Ring C-Opened Analogues of Calanolide A: Synthesis and Anti-HIV-1 Evaluation" Biomimetics 9, no. 1: 44. https://doi.org/10.3390/biomimetics9010044
APA StyleKhalymbadzha, I. A., Fatykhov, R. F., Butorin, I. I., Sharapov, A. D., Potapova, A. P., Muthipeedika, N. J., Zyryanov, G. V., Melekhin, V. V., Tokhtueva, M. D., Deev, S. L., Kukhanova, M. K., Mochulskaya, N. N., & Tsurkan, M. V. (2024). Bioinspired Pyrano[2,3-f]chromen-8-ones: Ring C-Opened Analogues of Calanolide A: Synthesis and Anti-HIV-1 Evaluation. Biomimetics, 9(1), 44. https://doi.org/10.3390/biomimetics9010044