
Essential Electronic Properties of Stage-1 Li/Li+-Graphite-Intercalation Compounds for Different Concentrations


Abstract
:1. Introduction
2. Theoretical Calculations
3. Results and Discussions
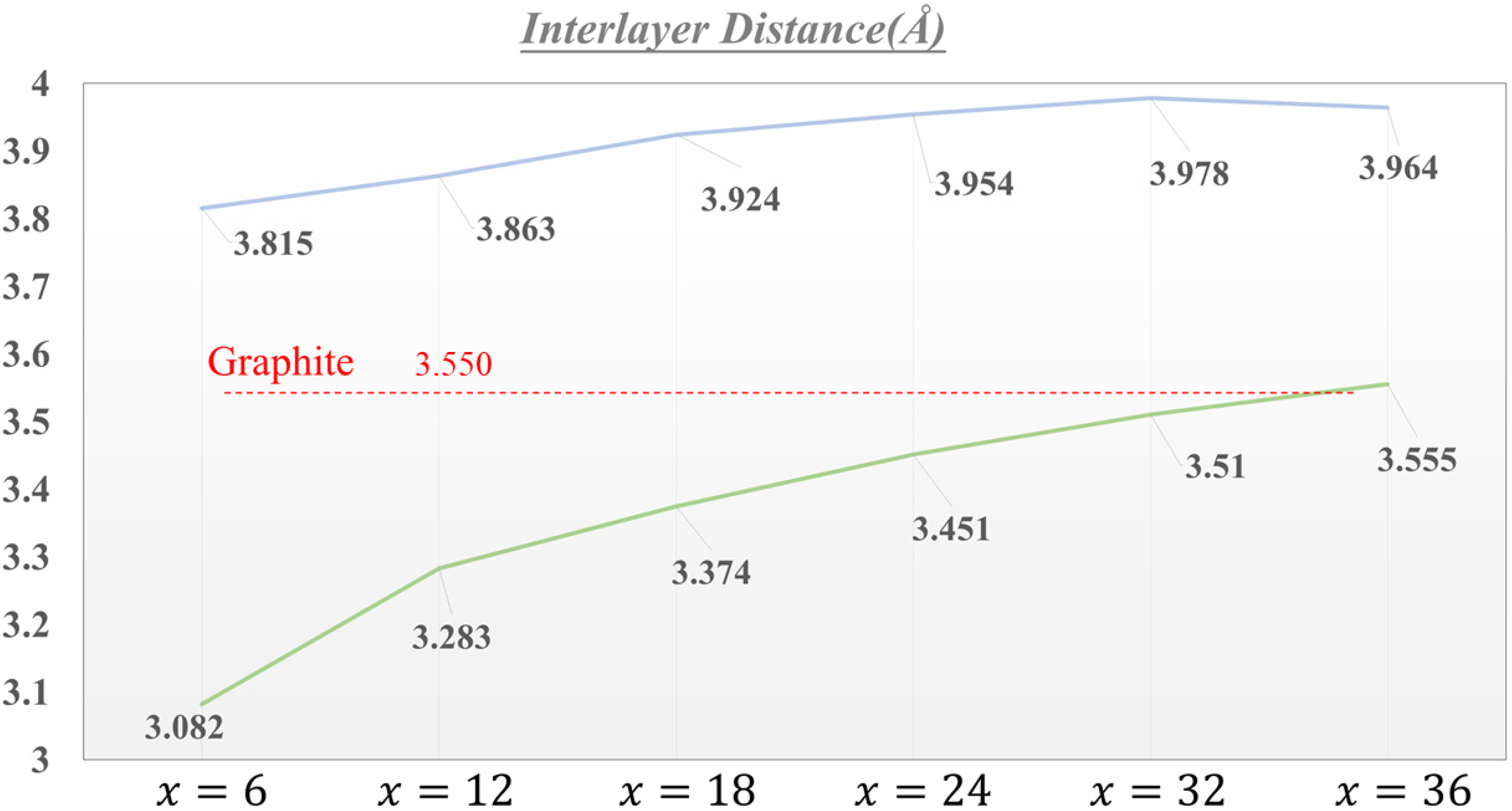
3.1. Geometric Structures
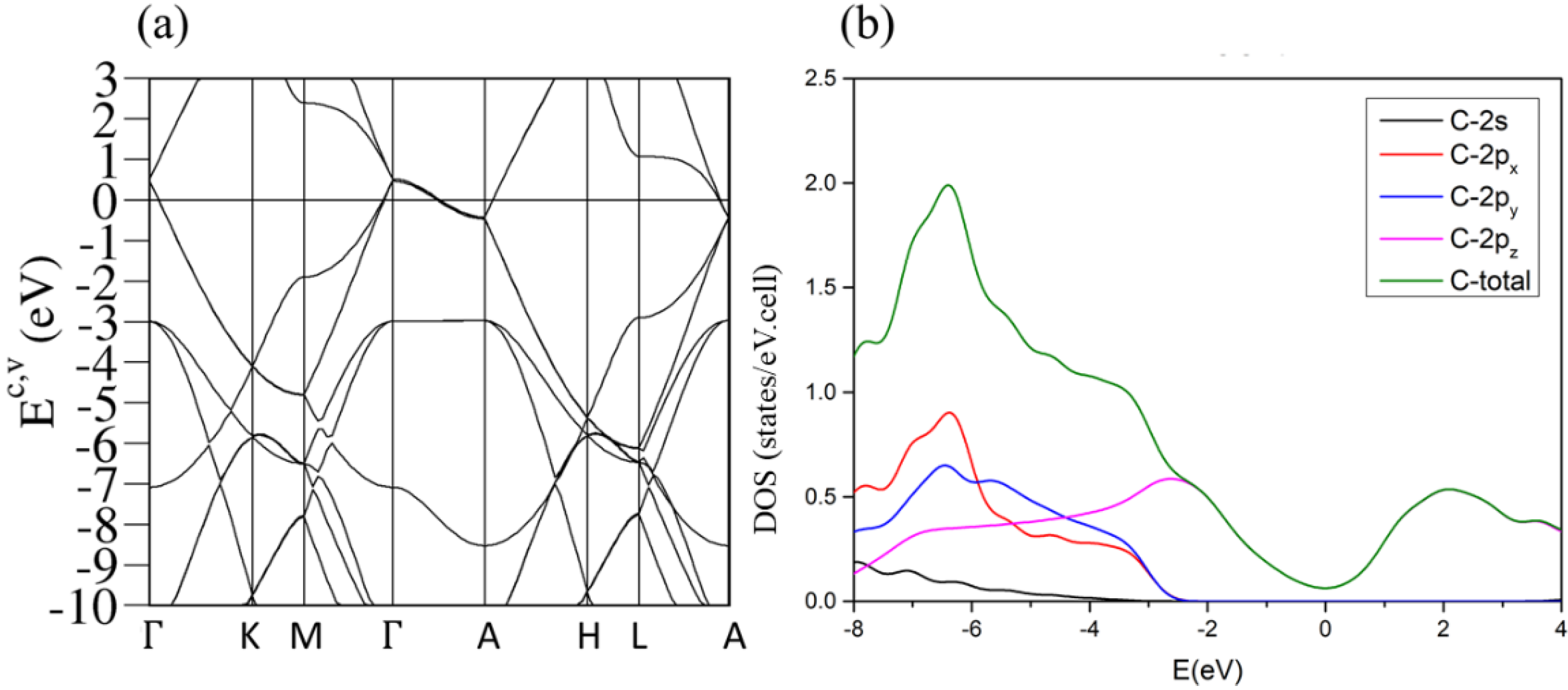
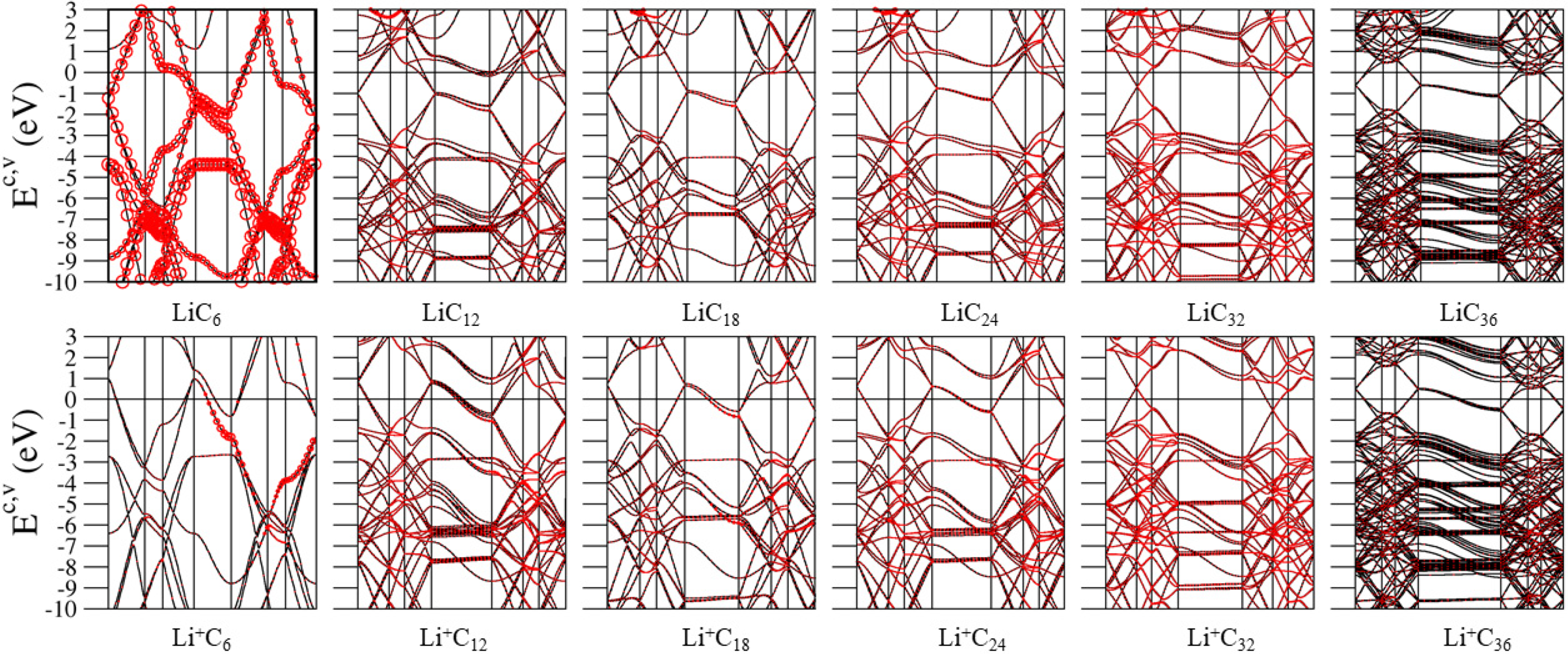
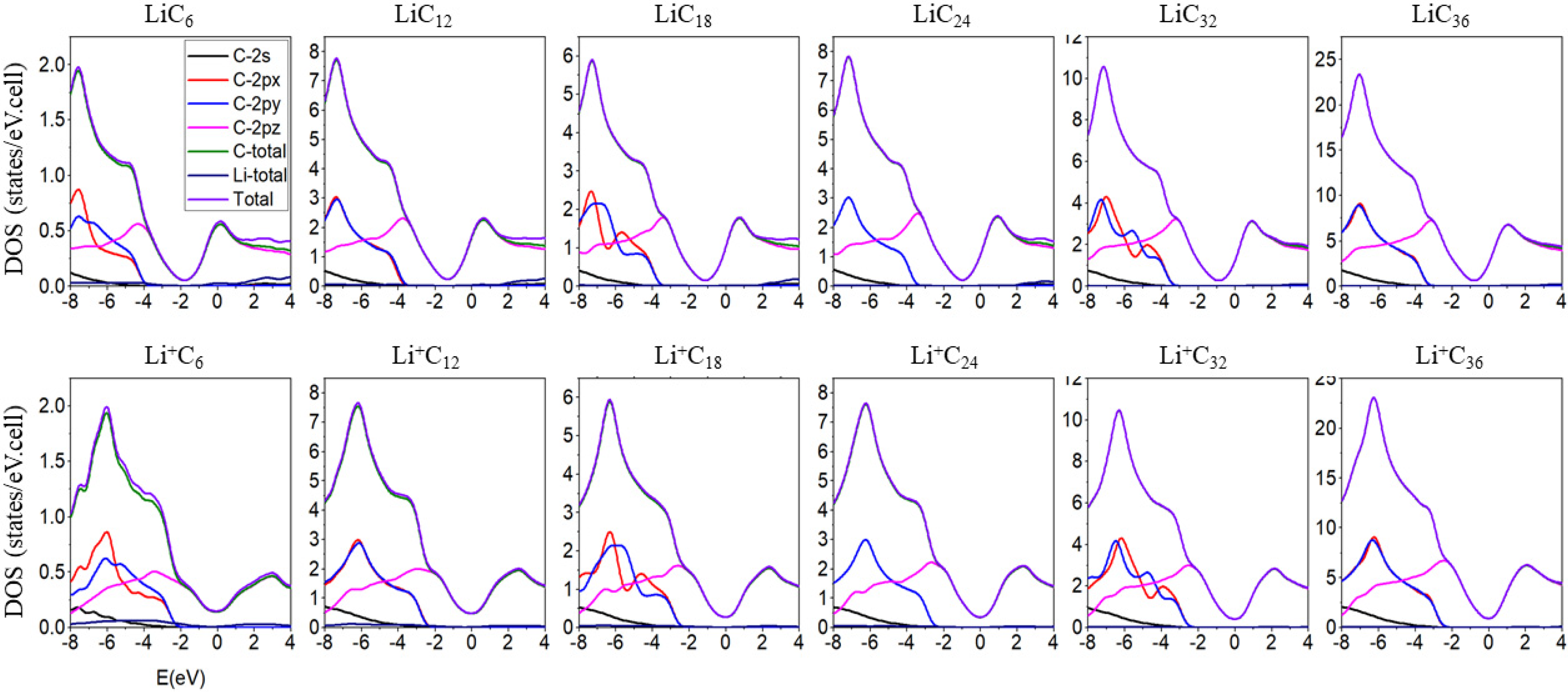
3.2. Band Structures and Density of States
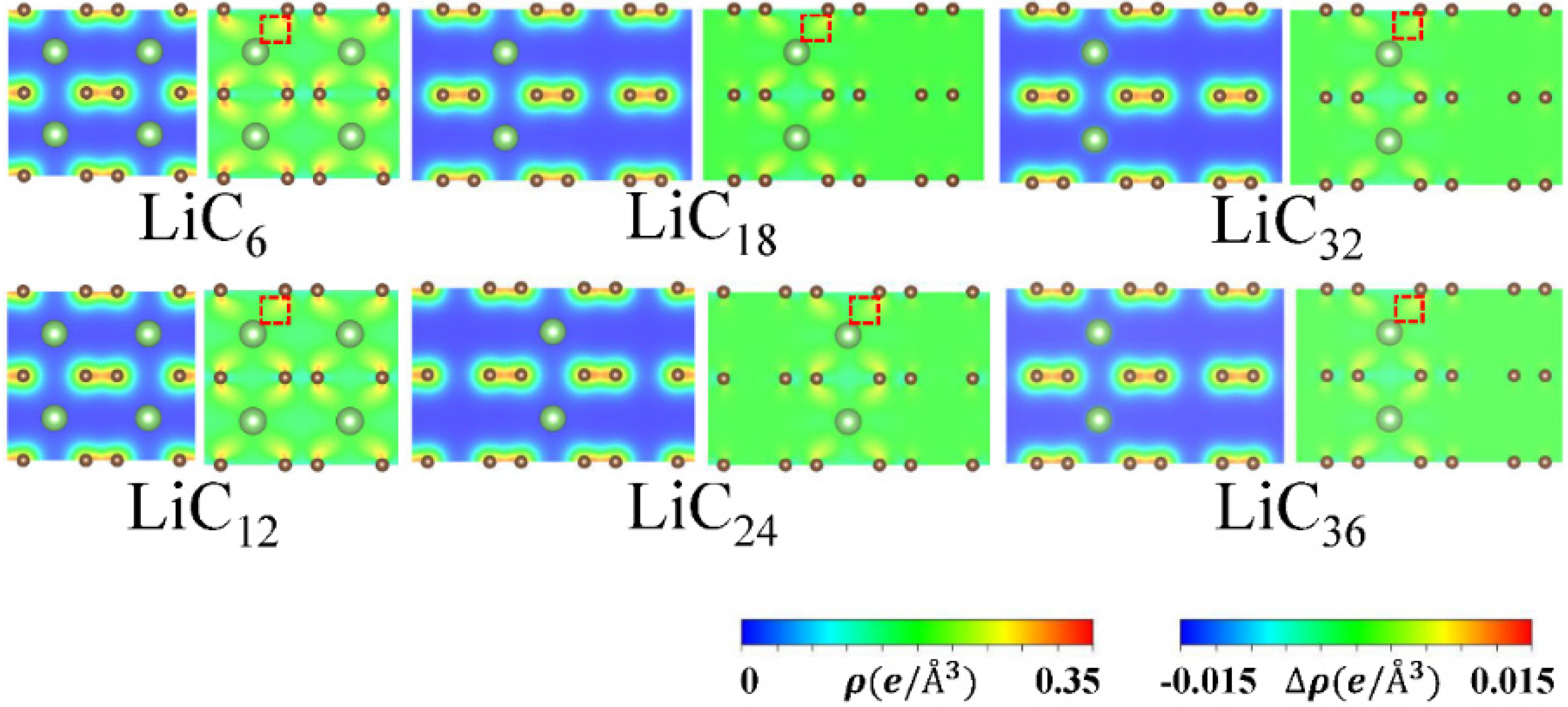
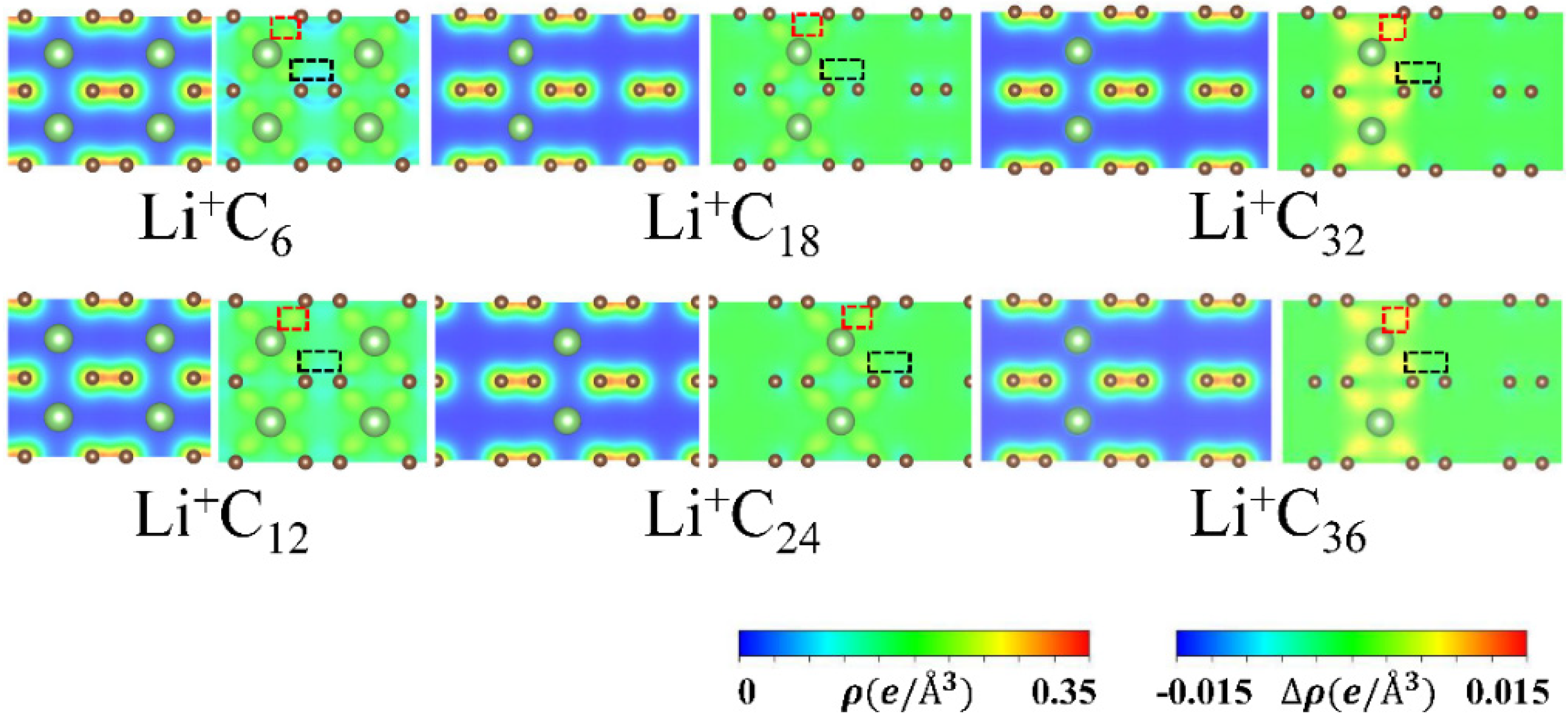
3.3. Charge Distributions and Charge Transfer
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, J.K.; Lee, S.C.; Ahn, J.P.; Kim, S.C.; Wilson, J.I.; John, P. The growth of AA graphite on (111) diamond. J. Chem. Phys. 2008, 129, 234709. [Google Scholar] [CrossRef] [PubMed]
- Yankowitz, M.; Wang, J.I.J.; Birdwell, A.G.; Chen, Y.A.; Watanabe, K.; Taniguchi, T.; Jacquod, P.; San-Jose, P.; Jarillo-Herrero, P.; LeRoy, B.J. Electric field control of soliton motion and stacking in trilayer graphene. Nat. Mater. 2014, 13, 786789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coletti, C.; Forti, S.; Principi, A.; Emtsev, K.V.; Zakharov, A.A.; Daniels, K.M.; Daas, B.K.; Chandrashekhar, M.V.S.; Ouisse, T.; Chaussende, D.; et al. Revealing the electronic band structure of trilayer graphene on SiC: An angle-resolved photoemission study. Phys. Rev. B 2013, 88, 155439. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, T.; Saito, R. First-principles study on interlayer state in alkali and alkaline earth metal atoms intercalated bilayer graphene. Surf. Sci. 2017, 665, 1–9. [Google Scholar] [CrossRef]
- Rydberg, H. Van der Waals Density Functional for Layered Structures. Phys. Rev. Lett. 2003, 91, 126402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethuraman, V.A.; Hardwick, L.J.; Srinivasan, V.; Kostecki, R. Surface structural disordering in graphite upon lithium intercalation/deintercalation. J. Power Sources 2010, 195, 3655–3660. [Google Scholar] [CrossRef] [Green Version]
- Park, T.R.; Rhee, S.S. Multilayer model of interlayer spacing in graphite intercalation compounds. Appl. Phys. A 2001, 72, 367–372. [Google Scholar] [CrossRef]
- Chan, K.T.; Neaton, J.B.; Cohen, M.L. First-principles study of metal adatom adsorption on graphene. Phys. Rev. B 2008, 77, 235430. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.H.; Choi, S.M.; Jhi, S.H. Crossover in the adsorption properties of alkali metals on graphene. Phys. Rev. B 2010, 82, 033414. [Google Scholar] [CrossRef]
- Praveen, C.S.; Piccinin, S.; Fabris, S. Adsorption of alkali adatoms on graphene supported by the Au/Ni (111) surface. Phys. Rev. B 2015, 92, 075403. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.Y.; Lin, Y.T.; Tran, N.T.T.; Su, W.P.; Lin, M.F. Feature-rich electronic properties of aluminum-doped graphenes. arXiv 2016, arXiv:1606.01624. [Google Scholar]
- Aoki, M.; Amawashi, H. Dependence of band structures on stacking and field in layered graphene. Solid State Commun. 2007, 142, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Sahu, B.; Min, H.; MacDonald, A.H. Band structure of A B C-stacked graphene trilayers. Phys. Rev. B 2010, 82, 035409. [Google Scholar] [CrossRef] [Green Version]
- Avramov, P.V.; Sakai, S.; Entani, S.; Matsumoto, Y.; Naramoto, H. Ab initio LC-DFT study of graphene, multilayer graphenes and graphite. Chem. Phys. Lett. 2011, 508, 86–89. [Google Scholar]
- Menezes, M.G.; Capaz, R.B.; Louie, S.G. Ab initio quasiparticle band structure of ABA and ABC-stacked graphene trilayers. Phys. Rev. B 2014, 89, 035431. [Google Scholar] [CrossRef] [Green Version]
- Persson, K.; Hinuma, Y.; Meng, Y.S.; Van der Ven, A.; Ceder, G. Thermodynamic and kinetic properties of the Li-graphite system from first-principles calculations. Phys. Rev. B 2010, 82, 125416. [Google Scholar] [CrossRef]
- Wang, Z.; Selbach, S.M.; Grande, T. Van der Waals density functional study of the energetics of alkali metal intercalation in graphite. RSC Adv. 2014, 4, 4069. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.Y.; Chang, S.L.; Tran, N.T.T.; Yang, P.H.; Lin, M.F. H–Si bonding-induced unusual electronic properties of silicene: A method to identify hydrogen concentration. Phys. Chem. Chem. Phys. 2015, 39, 26443–26450. [Google Scholar] [CrossRef] [Green Version]
- Rafique, M.; Unar, M.A.; Ahmed, I.; Chachar, A.R.; Shuai, Y. Ab-initio investigations on physisorption of alkaline earth metal atoms on monolayer hexagonal boron nitride (h-BN). J. Phys. Chem. Solids 2018, 118, 114–125. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Blchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dresselhaus, M.S.; Dresselhaus, G. Intercalation compounds of graphite. Adv. Phys. 2002, 51, 1–186. [Google Scholar] [CrossRef]
- Okamoto, Y. Density functional theory calculations of alkali metal (Li, Na, and K) graphite intercalation compounds. J. Phys. Chem. C 2013, 118, 16–19. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interlayer Distance (Å) | |
|---|---|
| LiC6 | 3.815 |
| LiC12 | 3.863 |
| LiC18 | 3.924 |
| LiC24 | 3.954 |
| LiC32 | 3.978 |
| LiC36 | 3.964 |
| Li+C6 | 3.082 |
| Li+C12 | 3.283 |
| Li+C18 | 3.374 |
| Li+C24 | 3.451 |
| Li+C32 | 3.510 |
| Li+C36 | 3.555 |
| Graphite | 3.550 |
| Charge Transfer (e/atom) | ||
|---|---|---|
| C | Li | |
| LiC6 | 0.144 | −0.865 |
| LiC12 | 0.073 | −0.871 |
| LiC18 | 0.049 | −0.878 |
| LiC24 | 0.037 | −0.880 |
| LiC32 | 0.028 | −0.884 |
| LiC36 | 0.025 | −0.883 |
| Li+C6 | −0.030 | 0.191 |
| Li+C12 | −0.015 | 0.176 |
| Li+C18 | −0.009 | 0.173 |
| Li+C24 | −0.007 | 0.163 |
| Li+C32 | −0.005 | 0.159 |
| Li+C36 | −0.004 | 0.155 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.-B.; Lin, S.-Y.; Lin, M.-F.; Lin, K.-I. Essential Electronic Properties of Stage-1 Li/Li+-Graphite-Intercalation Compounds for Different Concentrations. Condens. Matter 2022, 7, 35. https://doi.org/10.3390/condmat7020035
Li W-B, Lin S-Y, Lin M-F, Lin K-I. Essential Electronic Properties of Stage-1 Li/Li+-Graphite-Intercalation Compounds for Different Concentrations. Condensed Matter. 2022; 7(2):35. https://doi.org/10.3390/condmat7020035
Chicago/Turabian StyleLi, Wei-Bang, Shih-Yang Lin, Ming-Fa Lin, and Kuang-I Lin. 2022. "Essential Electronic Properties of Stage-1 Li/Li+-Graphite-Intercalation Compounds for Different Concentrations" Condensed Matter 7, no. 2: 35. https://doi.org/10.3390/condmat7020035
APA StyleLi, W. -B., Lin, S. -Y., Lin, M. -F., & Lin, K. -I. (2022). Essential Electronic Properties of Stage-1 Li/Li+-Graphite-Intercalation Compounds for Different Concentrations. Condensed Matter, 7(2), 35. https://doi.org/10.3390/condmat7020035