
In Silico Study of Potential Small Molecule TIPE2 Inhibitors for the Treatment of Cancer



Abstract
:1. Introduction
2. Methods
2.1. Preparation of Receptor and Ligands
2.2. Preparation of Receptor and Hit Compound Complex
2.3. Molecular Docking
2.4. Fragment Modifications
2.5. Molecular Dynamics
3. Results and Discussion
3.1. Molecular Docking
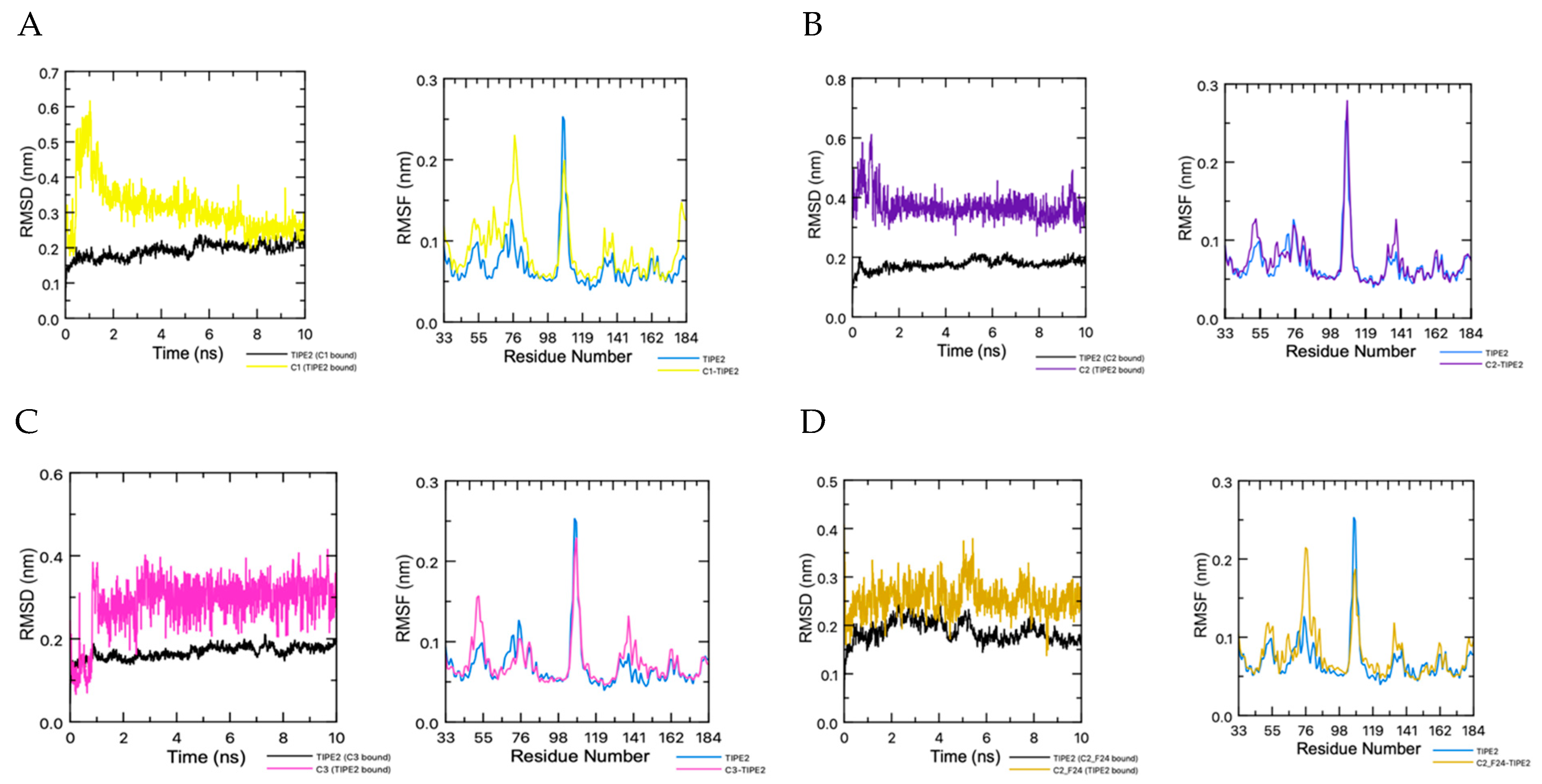
3.2. Molecular Dynamics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Sethi, G.; Shanmugam, M.K.; Ramachandran, L.; Kumar, A.P.; Tergaonkar, V. Multifaceted link between cancer and inflammation. Biosci. Rep. 2012, 32, 1–15. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Gattinoni, L.; Powell, D.J., Jr.; Rosenberg, S.A.; Restifo, N.P. Adoptive immunotherapy for cancer: Building on success. Nat. Rev. Immunol. 2006, 6, 383–393. [Google Scholar] [CrossRef]
- Lin, W.W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Ruegg, C. Leukocytes, inflammation, and angiogenesis in cancer: Fatal attractions. J. Leukoc. Biol. 2006, 80, 682–684. [Google Scholar] [CrossRef]
- Lou, Y.; Liu, S. The TIPE (TNFAIP8) family in inflammation, immunity, and cancer. Mol. Immunol. 2011, 49, 4–7. [Google Scholar] [CrossRef]
- Karin, J.A.D.F.M.M. NF-jB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar]
- Sun, H.; Gong, S.; Carmody, R.J.; Hilliard, A.; Li, L.; Sun, J.; Kong, L.; Xu, L.; Hilliard, B.; Hu, S.; et al. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell 2008, 133, 415–426. [Google Scholar] [CrossRef]
- Oho, M.; Nakano, R.; Nakayama, R.; Sakurai, W.; Miyamoto, A.; Masuhiro, Y.; Hanazawa, S. TIPE2 (Tumor Necrosis Factor α-induced Protein 8-like 2) Is a Novel Negative Regulator of TAK1 Signal. J. Biol. Chem. 2016, 291, 22650–22660. [Google Scholar] [CrossRef]
- Fayngerts, S.A.; Wang, Z.; Zamani, A.; Sun, H.; Boggs, A.E.; Porturas, T.P.; Xie, W.; Lin, M.; Cathopoulis, T.; Goldsmith, J.R.; et al. Direction of leukocyte polarization and migration by the phosphoinositide-transfer protein TIPE2. Nat. Immunol. 2017, 18, 1353–1360. [Google Scholar] [CrossRef]
- Yan, D.; Wang, J.; Sun, H.; Zamani, A.; Zhang, H.; Chen, W.; Tang, A.; Ruan, Q.; Yang, X.; Chen, Y.H.; et al. TIPE2 specifies the functional polarization of myeloid-derived suppressor cells during tumorigenesis. J. Exp. Med. 2020, 217, e20182005. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- The PyMOL Molecular Graphics System; Version 2.0; Schrödinger, LLC.: New York, NY, USA, 2015.
- BIOVIA Dassault Systèmes. Discovery Studio Visualizer, 21.1.0.20298; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Adasme, M.F.; Linnemann, K.L.; Bolz, S.N.; Kaiser, F.; Salentin, S.; Haupt, V.J.; Schroeder, M. PLIP 2021: Expanding the scope of the protein-ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021, 49, W530–W534. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Binding Affinity (kcal mol–1) | Molecular Weight (g mol–1) | Solubility (ESOL) | Partition Coefficient (M logP) |
|---|---|---|---|---|
| C1 | −8.7 | 419.49 | −1.57 | 1.14 |
| C2 | −8.8 | 365.47 | −4.39 | 3.40 |
| C3 | −8.9 | 363.47 | −3.48 | 2.49 |
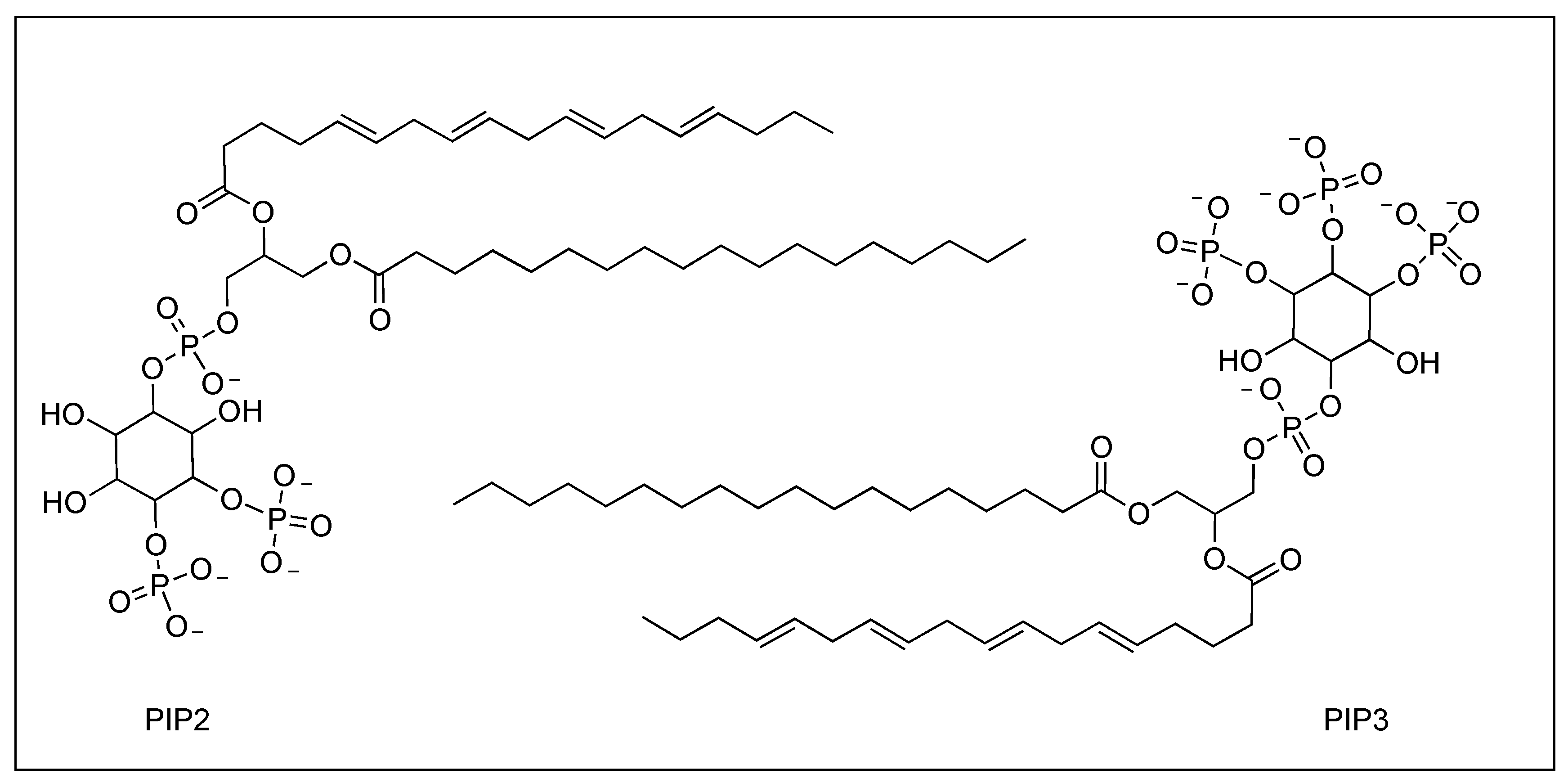
| PIP2 | −7.0 | 1014.01 | −7.90 | 3.07 |
| PIP3 | −7.0 | 1092.98 | −7.57 | 4.63 |
| Compound | Binding Affinity (kcal mol–1) | Molecular Weight (g mol–1) | Solubility (ESOL) | Partition Coefficient (M logP) | TPSA (Å2) |
|---|---|---|---|---|---|
| C1–F1 | −8.0 | 821.97 | −6.34 | 1.08 | 159.28 |
| C1–F2 | −10.1 | 823.02 | −7.50 | 1.58 | 166.73 |
| C1–F3 | −10.2 | 831.98 | −7.38 | 1.82 | 157.79 |
| C1–F4 | −9.0 | 827.95 | −6.89 | 0.47 | 182.52 |
| C1–F5 | −10.3 | 804.90 | −5.77 | 0.53 | 187.23 |
| C1–F6 | −9.5 | 782.89 | −5.58 | 1.54 | 174.81 |
| C1–F7 | −10.9 | 793.96 | −7.34 | 1.82 | 163.93 |
| C1–F8 | −9.0 | 815.92 | −7.27 | 0.58 | 153.14 |
| C1–F9 | −10.9 | 799.37 | −7.54 | 2.86 | 135.82 |
| C1–F10 | −9.1 | 822.88 | −6.72 | 1.63 | 153.49 |
| C2–F11 | −10.8 | 619.80 | −6.68 | 2.79 | 96.07 |
| C2–F12 | −11.1 | 658.77 | −6.83 | 3.15 | 108.96 |
| C2–F13 | −10.0 | 654.80 | −6.80 | 2.97 | 108.96 |
| C2–F14 | −12.7 | 651.71 | −6.73 | 2.62 | 135.93 |
| C2–F15 | −10.7 | 642.79 | −6.34 | 2.87 | 98.1 |
| C2–F16 | −10.7 | 653.82 | −7.41 | 3.37 | 93.17 |
| C2–F17 | −11.4 | 615.77 | −6.56 | 2.95 | 96.07 |
| C2–F18 | −10.3 | 657.80 | −7.05 | 3.04 | 106.31 |
| C2–F19 | −11.0 | 609.74 | −6.23 | 3.64 | 92.31 |
| C2–F20 | −10.6 | 660.85 | −5.91 | 3.29 | 95.88 |
| C3–F21 | −10.7 | 660.82 | −8.59 | 5.18 | 70.08 |
| C3–F22 | −9.3 | 657.75 | −7.31 | 3.51 | 108.41 |
| C3–F23 | −10.6 | 671.82 | −7.69 | 4.35 | 107.97 |
| C3–F24 | −9.4 | 669.85 | −7.79 | 4.50 | 93.87 |
| C3–F25 | −11.3 | 669.85 | −7.73 | 4.50 | 93.87 |
| C3–F26 | −9.9 | 674.76 | −7.94 | 4.43 | 117.79 |
| C3–F27 | −10.6 | 534.60 | −5.49 | 2.95 | 88.84 |
| C3–F28 | −9.8 | 572.71 | −5.68 | 4.29 | 70.08 |
| C3–F29 | −8.6 | 554.72 | −5.51 | 3.94 | 70.08 |
| C3–F30 | −10.6 | 669.85 | −7.95 | 4.5 | 93.87 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, J.; Evangelou, K.; Chen, Y.H.; Ji, H.-F. In Silico Study of Potential Small Molecule TIPE2 Inhibitors for the Treatment of Cancer. Sci 2023, 5, 39. https://doi.org/10.3390/sci5040039
Wilson J, Evangelou K, Chen YH, Ji H-F. In Silico Study of Potential Small Molecule TIPE2 Inhibitors for the Treatment of Cancer. Sci. 2023; 5(4):39. https://doi.org/10.3390/sci5040039
Chicago/Turabian StyleWilson, Jerica, Katerina Evangelou, Youhai H. Chen, and Hai-Feng Ji. 2023. "In Silico Study of Potential Small Molecule TIPE2 Inhibitors for the Treatment of Cancer" Sci 5, no. 4: 39. https://doi.org/10.3390/sci5040039
APA StyleWilson, J., Evangelou, K., Chen, Y. H., & Ji, H. -F. (2023). In Silico Study of Potential Small Molecule TIPE2 Inhibitors for the Treatment of Cancer. Sci, 5(4), 39. https://doi.org/10.3390/sci5040039