1. Introduction
Polyaniline (PANI) is among a series of unique polymers studied in recent decades. The growing attractiveness, such as ease of preparation, low cost, and its multitude of applications owes to its remarkable properties. PANI-based materials are widely used in catalysis, adsorption, solar cells, battery production, gas and vapour sensors, etc. [
1,
2]. Although PANI has been known for well over 150 years, comprehensive studies span less than 50 years [
3]. The conductive properties of PANI have potential utility for fabrication of electronic devices [
4,
5], batteries [
6,
7] and sensors [
8,
9,
10]. PANI can be prepared in various product morphologies [
11], where it can be combined with other polymers to prepare composite materials [
12]. Such composites may have tunable properties, depending on the type of components and their composition. This study reports on three-component (ternary) polyaniline-based composite materials.
PANI is insoluble in common solvents, including aqueous media across a wide range of pH [
13]. It is a thermoset polymer and, therefore, infusible since it will decompose at elevated temperature before it melts [
14]. To achieve the required properties, PANI can be modified by alteration of its functional groups by various methods of functionalization, such as cross-linking [
15], grafting [
16] and chemical modification [
17]. A key feature related to PANI is that it retains its intrinsic properties (conductivity, thermal properties, and chemical stability) even after modification. In some cases, modification of PANI upon incorporation into composites results in enhanced textural properties (pore structure and surface area) relative to its pristine form [
18]. PANI-based composites find utility in sensor devices, where such materials often display enhanced detection of rapid chemical fluctuations in the environment [
19]. Such PANI composites with unique properties are known upon blending with cellulose and/or chitosan biopolymers [
20,
21].
Chitosan (CHT) is an abundant marine biopolymer derived from the hydrolysis of chitin [
22]. Alkaline hydrolysis of chitin yields chitosan [
23] with variable deacetylation, where typical commercial CHT samples have deacetylation levels that range from 70% to 95% [
24]. The abundant functional groups (–NH
2 and –OH) of CHT are amenable for modification (
Figure 1) by physical and chemical means. Cross-linking is a versatile method for modification of chitosan, where the degree of cross-linking in composite materials affects the range of swelling, mechanical, and adsorption properties [
25]. In the case of biopolymer composites that contain chitosan and PANI, there is limited covalent cross-linking between polyaniline and chitosan, whereas hydrogen bond formation occurs between the –OH or –NH
2 groups of chitosan and the imine/amine (=N– or –NH–) groups of PANI (
Figure 1b).
A report on cellulose/PANI nanocomposites [
21] indicated a similar structure to that shown in
Figure 1b, where the extent of “hydrogen-bond mediated cross-linking” depends on the relative ratio of the precursors. In addition, the properties of such polymer composites are affected by composition such as the mechanical properties, electrical conductivity, dye adsorption and textural properties.
Although binary CHT-PANI composites possess enhanced physical properties relative to the single precursor components, their mechanical properties are not sufficiently high. These properties become modified upon physical blending with a third component, such as polyvinyl alcohol (PVA). PVA is a water-soluble synthetic polymer that possesses high hydrophilicity and plasticity suitable for production of thin films. Its water solubility is rather moderate and requires elevated temperatures to achieve higher solubility. PVA-based composite films possess enhanced mechanical properties, such as tensile strength [
27]; and greater swelling (with respect to pristine PANI) due to its high propensity to absorb water. The film-forming ability of PVA allows for the formation of flexible and elastic samples; thereby widening their field of application and versatility.
Ternary PANI–CHT–PVA composites are unique materials with promising potential in the field of composite science. Although binary PANI–biopolymer composites are known [
28], there are sparse studies on ternary composite systems that contain PANI. This work details a systematic study on ternary composites (PANI–CHT–PVA) with a focus on the correlation between their adsorptive and electromechanical properties. An overall goal of this study compares the physico-chemical and physico-mechanical properties amongst composites with variable composition, and how the structure and properties are governed by their composition.
2. Materials and Methods
2.1. Materials
Aniline (density = 1.02 g/cm3) was an ACS reagent grade (99.5%) and chitosan flakes (medium molecular weight) with a deacetylation degree 80% and a density of 0.7 g/cm3 were obtained from Sigma-Aldrich, Oakville, ON, Canada. Chitosan is generally insoluble in pure water and sparingly soluble in acidic water media. Polyvinyl alcohol (molecular weight 78,000) was obtained from Polysciences, Inc., Warrington, PA, USA. with a density of 1.29 g/cm3 and a degree of hydrolysis of 99%. Ammonium persulfate (APS) was an ACS reagent (98%) with a molecular weight of 228.20 g/mol that was obtained from Sigma-Aldrich, Oakville, ON, Canada. Hydrochloric acid (HCl) 36% w/w (aq) solution was obtained from Alfa Aesar, Tewksbury, MA, USA. ACS-grade (95–98%) sulfuric acid (H2SO4, aq) with a density of 1.84 g/cm3 was obtained from Fischer Scientific International, Inc., Ottawa, ON, Canada. Methylene blue (MB) and the sodium salt of fluorescein (FL) sodium were of the highest available purity and obtained from Sigma-Aldrich, Oakville, ON, Canada. All solutions were prepared using highly purity Milli-Q water (resistivity 17–18 MΩ·cm).
2.2. Shorthand Notation
The shorthand notation used herein are defined below, where further details are given in
Table S1 in the Supplementary Materials. In brief, the notation for the binary composites are defined according to the chitosan:PANI (CHT:PANI; w/w%) content indicated in parentheses: CHP25 (25:75), CHP50 (50:50), and CHP75 (75:25). The notation for the ternary composites differ according to the level of polyvinyl alcohol (PVA) content (w/w%), where the relative weight ratio of the aforementioned binary composite with PVA, is denoted by the following notation: CHP25-PVA, CHP50-PVA, and CHP75-PVA, where these composites contain 70% (w/w) PVA. The ternary composites that contain 50% PVA (w/w) are denoted by the following numerical index: CHP25-PVA50, CHP50-PVA50, and CHP75-PVA50.
2.3. Preparation of Polymer Composites
The synthesis of the ternary composite materials involved two steps: (1) copolymerization of aniline with the biopolymer support yielded a binary composite, and (2) preparation of the PVA blend gave a ternary composite material. The ratio of biopolymer/PANI varied from 25 to 75 wt.% biopolymer. The ratio PVA/binary material was constant and equal to 70% PVA by weight. In some cases (swelling and electrical conductivity—see
Section 3.4 and
Section 3.7, respectively), the content of PVA was reduced to 50 wt.%.
A similar procedure for preparation of the samples was adapted from a known method [
9,
29]. An appropriate amount of chitosan, dissolved in 1 M HCl (100 mL), was placed in a sonicator bath for 1 h. After the system was cooled to 0 °C (ice bath), aniline was added drop-wise using a syringe. Then, the APS acidic solution (100 mL, 1M HCl) was added dropwise (0.8:1 molar ratio of APS:aniline) via a burette over a 30 min period (caution: heat evolution). The solution gradually turned to dark blue. After adding APS, the solution was stirred for 1 h. Then, the reactive solution was neutralized by NaOH and filtered. The precipitated residue was washed with water and oven-dried at 60 °C overnight. The resulting binary composite was washed again (to remove residual APS and NaOH), and then dried for 24 h. The greenish-blue colour samples were ball-milled to 32 µm mesh. PVA solutions were prepared by adding dry PVA in hot water (ca. 90 °C) in small portions every 10 min until the PVA content of 8 wt.% was achieved. Finally, the hot solution was poured into a volumetric flask and allowed to cool to 23 °C. The finely ground binary powder was mixed with PVA solution and sonicated at 70 °C for 90 min, periodically stirring with a glass rod. The hot mixture was cast in molds thereafter.
This method yielded the base-neutralized composites of PANI (emeraldine base form; EB). To convert PANI and its composite powders to the emeraldine salt form (ES), acid-doping was used by immersing in 1 M HCl solution and equilibration on a horizontal shaker for 24 h.
2.4. Characterization
2.4.1. 13C Solid State NMR spectroscopy
13C solids NMR spectra were obtained using a Bruker AVANCE III HD spectrometer operating at 125.76 MHz (13C frequency). Samples were packed in 4 mm drop-in rotors and analyzed using a pulse sequence with Cross Polarization with Total Suppression of Spinning Sidebands (CP-TOSS) and a spinning frequency 7.5 kHz with a 1 ms cross-polarization time. Spectra were acquired after variable scans (n = 4 k to 4.4 k) with a recycle delay of 2 s. The 13C chemical shifts (δ) were externally referenced to adamantane (δ = 38.48 ppm). Stack plots were obtained using Topspin 4.0.7 Bruker NMR software.
2.4.2. Fourier Transform Infrared (FTIR) Spectroscopy
FTIR spectra of solids were recorded using a Renishaw IlluminatIR FTIR Microscope (36× Diamond ATR Objective). IR spectra were acquired with the aid of a highly sensitive mercury cadmium telluride (MCT) detector in attenuated total reflectance (ATR) mode. The spectral range spanned from 4000 to 650 cm−1 with a resolution of 4 cm−1. All results were baseline-corrected and normalized using Wire 5.2 software.
2.4.3. Thermal Analysis
Thermogravimetric analysis (TGA) of samples was conducted using a TA Instruments Q50 TGA system. Solid samples (ca. 10 mg) were heated from 23 to 500 °C with a heating rate of 5 °C/min under a nitrogen atmosphere. Differential scanning calorimetry (DSC) curves were acquired using a DSC TA instrument model Q20, where ca. 3 mg of sample was encapsulated in a pan and heated with a scan rate of 20 °C/min from 40 to 250 °C. The integration of the DSC endotherms and TGA peaks was carried using Origin2019 software (OriginLab Corporation, Northampton, MA, USA).
2.4.4. Swelling Tests
Swelling tests were performed by immersing each sample in a separate vial containing either pure water or an aqueous acid solution (hydrochloric or sulfuric). The samples were equilibrated for 24 h by placing them on a horizontal shaker at 150 rpm. After 24 h, they were extracted and gently blotted with a tissue to remove excess solvent. The sample weight was taken before and after equilibration in the solvent. The measurements were replicated five times using different sample portions, where the degree of swelling (%) was calculated according to Equation (1):
is weight of swollen sample, is the weight of the dry sample.
2.4.5. Adsorption Studies
The adsorption properties of the samples were studied at equilibrium and at kinetic conditions, using two types of dyes, cationic (MB) and anionic (FL).
Equilibrium Dye Uptake
Equilibrium adsorption was performed in batch mode (ca. 15 samples) in their film form with a fixed weight (ca. 10.5 ± 0.5 mg). The samples were immersed in dye solutions with variable concentration, where the MB solutions ranged from 0.1 to 5 mM and FL ranged from 20 to 500 µM. Each series of samples were allowed to equilibrate on a horizontal shaker at 150 rpm for 24 h. The optical absorbance of the respective dye solution, before and after adsorption, was measured using a Varian Cary 100 Scan UV−Vis spectrophotometer. Calibration curves were obtained for MB (
= 664 nm) and for FL (
= 489 nm). Equilibrium uptake (mmol/g) was calculated using Equation (2), according to differences between the final and initial concentrations at equilibrium [
30]:
m is the mass of polymer sample, V is the volume of MB solution, C0 and Ce are initial and final concentrations before and after the adsorption process at equilibrium, respectively.
Adsorption isotherms were fitted to three isotherm models (cf.
Figure S2 in Supplementary Materials). The Sips [
31] and Freundlich [
32] isotherm models assume that the surface is heterogeneous, whereas the Langmuir model [
33] accounts for adsorption onto homogeneous surfaces. As seen from
Figure S2 and Table S3, the Sips isotherm model provides the best-fit curve and
values that lie close to unity. According to the Sips isotherm model [
31], the dye uptake at equilibrium conditions was calculated by Equation (3):
is the monolayer adsorption capacity of material (dye uptake at saturated conditions), is the Sips isotherm model constant, is the equilibrium concentration of MB solution, and is the Sips isotherm exponent term.
Dye adsorption was used to estimate the surface area (SA, m
2/g) of the sorbent (Equation (4)):
is the Sips fitting parameter from Equation (1), expressed in mol/g adsorbent,
is Avogadro’s number (6.02 × 10
23 mol
−1),
is the cross-sectional molecular surface area (m
2/molecule) where σ(MB) = 8.72 × 10
−19 and σ(FL) = 7.13 × 10
−19, and
is the coverage factor (non-dimensional quantity) where
Y = 2.0 for MB and FL [
34,
35].
Kinetics of Dye Uptake
Kinetic adsorption studies were conducted for the samples in their powder form, using a one-pot method under dynamic conditions [
36]. The initial dye concentration was known (10 µM in case of MB and 15 µM for FL); its absorbance was measured within the repetitive time spans. As the initial rate of adsorption was always higher, the time spans were shorter at first (2 min) and longer at the end of the kinetic profile (40 min). The experimental data were fitted to the pseudo-first (PFO) or pseudo-second (PSO) order kinetic models (cf. Equations (5) and (6)):
is the amount of sorbate adsorbed by a sample (µmol/g) at a certain time (
),
is the maximum amount of dye (µmol/g), adsorbed at pseudo-equilibrium conditions,
and
are the rate constants according to the PFO and PSO models. Experimental values of
can be obtained by Equation (7), which resembles the equilibrium form of Equation (2) except that
refers to the dye concentration at time (
).
2.4.6. Water Vapour Adsorption
The uptake of water vapour by the sample films was done according to the method described in the literature [
37]. Various atmospheric humidity levels were obtained by preparing a series of saturated salt solutions in closed glass jars. Oven-dried samples were placed inside the jars in ascending order of humidity values that underwent equilibration for 24 h. For example, salt solutions were prepared that correspond to variable relative humidity (%), as follows: LiCl (11), MgCl
2 (33), K
2CO
3 (43), Mg(NO
3)
2 (53), NaCl (71), KCl (84), and K
2SO
4 (97) [
38]. The samples were gradually moved from low to higher humidity every 24 h to obtain the water vapour adsorption properties. After reaching the highest humidity level, the samples were subjected to lower humidity levels to assess their desorption properties. The sample weight was recorded at each humidity level in triplicate. The moisture content was calculated according to Equation (8):
and are the masses of sample in the wet (at respective RH) and dry states, respectively.
2.4.7. Electrical Conductivity of Composite Materials
To investigate the behaviour of samples under an applied potential, the current–voltage behaviour of samples was recorded using a controlled voltage source device (Keithley 2420 source meter). Three specimens for each composite sample were cut out as rectangle shapes with 0.5 cm2 area (0.5 cm × 1.0 cm) and each sample was fixed between two fastener clips. The electric current was measured at ascending tensions from 0 to 20 V with an increment of 1 V at a fixed relative humidity (64%). The raw numerical data were converted to the I–V curves using Origin2019 software and further analyzed.
2.4.8. Mechanical Properties
A dynamic mechanical analyzer (DMA 8000, PerkinElmer, Inc., Waltham, MA, USA) was used to determine the dynamic mechanical properties of the samples. Three specimens of each composite sample had a rectangular shape of ca. 8 × 20 mm2. The tests were done using a single cantilever mode at 5 Hz frequency, and heated from 20 °C to 50 °C with a heating rate of 2 °C/min. The free length of all tests was 10.22 mm. Each sample was measured in dry and hydrated conditions. To obtain the measurements for the hydrated samples at equilibrium, samples were immersed under DI water in individual 50 mL beakers for 24 h. The sample weight and dimensions were recorded before and after water immersion.
3. Results and Discussion
3.1. 13C Solid State NMR Spectroscopy
13C solids NMR spectroscopy provides useful information related to the structure and composition of macromolecules and their noncovalent complexes in the solid state [
39]. Herein,
13C solids NMR spectroscopy was used to characterize the carbon structure of the binary and ternary composite materials. The
13C spectral assignments reported herein are based on previous spectra reported for the single-component systems (chitosan, PVA, and PANI) [
40,
41,
42]. Chitosan has a degree of acetylation (ca. 20%) that is supported by the presence of carbonyl and methyl groups. Pristine chitosan was reported to have the following signatures: C1 at 105.3 ppm, C4 at 83.6 ppm, C3,5 at 75.6 ppm, C6 at 60.8 ppm and C2 at 57.7 ppm [
40]. C7 and C8 relate to the carbonyl and methyl groups due to the presence of acetyl groups in chitosan (see
Figure S1 in the Supplementary Materials). PVA has two groups of signals that correspond to methylene (44–46 ppm) and methine (64–70 ppm) signatures [
41]. It is noteworthy that methine groups of PVA overlap with C2, C3 and C6 carbons of chitosan that reside between 57 and 70 ppm. Polyaniline has aromatic signatures that occur between 120 and 160 ppm [
42] that do not overlap with the spectra of chitosan or PVA.
Figure 2 shows the
13C NMR spectra of PANI–CHT composites with and without PVA.
In
Figure 3, the NMR line intensities of chitosan increased from CHP25 to CHP75, while PANI line intensities showed a concomitant decrease. The C2 and C6 signatures for chitosan had constant intensity that show spectral overlap with the methine carbons of PVA, where it is difficult to assess if there are any chemical versus physical changes. Thus, the spectra do not show any unique signatures that differ from the pristine polymer. However, C3 line intensities for chitosan appear to remain constant, where it is noted that quantitative analysis is limited by the use of the CP-TOSS pulse sequence, in contrast to single-pulse experiments.
3.2. FTIR Spectroscopy
FTIR spectroscopy provides useful information regarding a comparison of the composites versus their pristine components. IR spectral data enable determination of the presence or absence of unique functional groups, before and after composite formation.
Figure 3 shows IR spectra of ternary PANI–CHT–PVA composites. In
Supplementary Materials Table S2, the IR band assignments for the precursor polymer materials are noted among the collective IR spectral signatures.
It is noteworthy that the IR spectrum of the composite with the highest chitosan content did not show notable polyaniline signals, in agreement with the reduced PANI content in the composite (i.e., benzenoid stretching at 1495–1500 cm−1 and quinoid stretching at 1588 cm−1). By contrast, chitosan signatures show incremental intensity due to the greater chitosan content (C–H symmetric stretching at 2910 cm−1 and overlapping CHT–PVA signals at 1034–1086 cm−1). In addition, a weak NHCOCH3 signal (carbonyl from acetylated amino group) is observed. While IR results do not clearly reveal chemical interactions between the composite components, H-bonding between PANI and CHT is the most favourable interaction. PVA shows only two bands (alkyl CH and CO from alcohol) that are of similar intensity. The foregoing suggests that PVA is not involved in covalent bonding with chitosan or PANI.
3.3. Thermal Analysis
3.3.1. Thermogravimetric Analysis (TGA)
TGA is a useful tool to study the thermal stability of polymers and to estimate the composition of materials, degradation temperatures of components, and to the prevalence of cohesive interactions among components in mixed systems.
Chitosan and PVA collectively exhibit a single DTG event between 250–300 °C, [
43,
44] as observed in the TGA plot (
Figure 4a,
Figure S3). A prominent thermal event occurs between 325–400 °C due to the enhanced thermal stability of CHT–PANI–PVA composite. A higher temperature thermal event (425–500 °C) is noted with relatively low intensity that denotes the presence of PANI [
29].
Thermal events occur in several regions as denoted by bold numbers: Region 1 (loss of water), Region 2 (decomposition of CHT and PVA), Region 3 (decomposition of polymer composite materials), and Region 4 (decomposition of PANI). The occurrence of evaporative loss of water may indicate occluded or micropore water according to a range of temperatures up to ca. 200 °C. Insight into the thermodynamic state of water can be determined from the differential scanning calorimetry (DSC) results, in addition to estimates of the enthalpy of desorption.
3.3.2. Differential Scanning Calorimetry (DSC)
Figure 4b illustrates the DSC profiles of the polymer composite materials. The profiles are characterized by two prominent endotherms; a sharp transition at ca. 230 °C and a broader thermal event from 50 to 200 °C. The sharp transition corresponds to sample melting, whereas the broad transition relates to bound water as denoted by Region
1 (cf.
Figure 4a). The broad transition for the bound water relates to the different thermodynamic states of water, as evidenced by the broad DSC transition.
Figure 4b shows that the peak area is greater, especially for materials with higher chitosan content. This suggests that the enthalpy of dehydration increases as the mass fraction of chitosan increases. The trend can be accounted for, as follows: (1) stronger interactions between water molecules and the polymer components; or (2) greater levels of water incorporation within the material. These effects are indirectly supported because chitosan is hygroscopic and hydrophilic, whereas CHP75–PVA samples are observed to retain more water than CHP25–PVA (vide infra).
3.4. Solvent Swelling Tests
Solvent swelling of the single-component materials and composites occur due to sorption processes (adsorption + absorption) between the solvent and the polymer sorbent. Solvent (water) molecules penetrate into the composite microstructure and onto surface sites, where the results of solvent swelling in water or aqueous acid (HCl, H
2SO
4) solutions are listed in
Table 1. Undoped samples refer to emeraldine base (EB), while doped samples correspond to the emeraldine salt (ES) form of PANI. ES samples were obtained by equilibrating the corresponding base-neutralized form of the composites in 1 M HCl solution for 24 h.
A comparison between two acid systems, HCl and H2SO4, was carried out to ascertain any notable differences that may account for dissolution effects, since chitosan is soluble in HCl (aq) but it is generally insoluble in H2SO4 (aq). The comparable swelling observed for both acid systems indicate that chitosan does not dissolve in HCl (aq).
The level of swelling (%) should increase for samples that possess more accessible hydrophilic groups such as –OH, –COOH, –NH
2, –NHCOCH
3, etc. Accordingly, the components of the composites listed in
Table 1 possess hydrophilic character, where chitosan and PVA are among the most hydrophilic components in the composite formulation. A literature report reveals that PVA has a greater swelling contribution than chitosan [
45]. For this reason, it is convenient to compare the following samples: (1) according to CHT content (series of three samples in
Table 1a/b) and (2) by PVA content (cf. sections (a) and (b) in
Table 1). The overall trend shows that materials with a greater CHT content show greater solvent swelling. However, there are some exclusions, as follows: CHP50 and CHP75 have 70% PVA content for swelling in water, and 50% PVA content for swelling in 1 M acid solution. The respective swelling results in acid and water are nearly equal within the limits of error. Moreover, the swelling of CHP50–PVA50 undoped samples in water is notably higher than corresponding CHP75–PVA50 samples (129% vs 97%). The variability in swelling can be accounted for by the excess content of CHT that prevents the formation of a branched and cross-linked structure that has reduced swelling. To summarize the swelling results, there are eight sets of samples (each of three specimens) that show the following trends: 83%–119%–109% (↑ and = , means to be nearly equal within an error); 77%–122%–228% (all ↑); 92%–98%–119% (↑); 93%–98%–114% (↑), 68%–129%–97% (↑ and ↓, falls out of the general trend), 131%–195%–210% (↑); 77%–119%–116% (↑ and =); 82%–119%–117% (↑ and =). Half of the samples (4) show a clear trend: higher CHT content yields greater swelling, three samples firstly increase and then remain constant, and only one sample does not obey this rule (and even then, not for the whole set of three samples). In 75% of the cases (12 out of 16), the swelling of samples exhibit general trends, where greater CHT and PVA content yield greater swelling, along with greater cationic dye adsorption (vide infra). A comparison of doped versus undoped samples reveal greater swelling in water and moderate swelling in 1 M acid solutions. Comparable swelling was observed in aqueous HCl and H
2SO
4, where doped samples had greater swelling in pure water. This is understood due to protonation effects for the swelling process when comparing the results for water vs. aqueous acid solutions. Chitosan is insoluble in H
2SO
4 (
aq), but it may have partial solubility in HCl (
aq). The similar level of solvent swelling in both acid media rules out the dissolution of chitosan.
3.5. Equilibrium and Kinetic Dye Uptake
Equilibrium swelling was carried out for the ternary composite films, while the kinetic uptake for the binary composites was studied for powdered samples. This approach was undertaken due to the following: (1) effective conversion of films into uniform powders with a conventional mortar and pestle has limitations; (2) batch adsorption requires an additional phase separation step with powders, involving an additional operation (e.g., filtration or centrifugation); (3) the one-pot kinetic method is suitable for the study of powdered materials. The adsorption results for samples in film and powder forms should follow parallel trends in adsorption.
3.5.1. Methylene Blue (MB) Adsorption
Equilibrium Dye Uptake
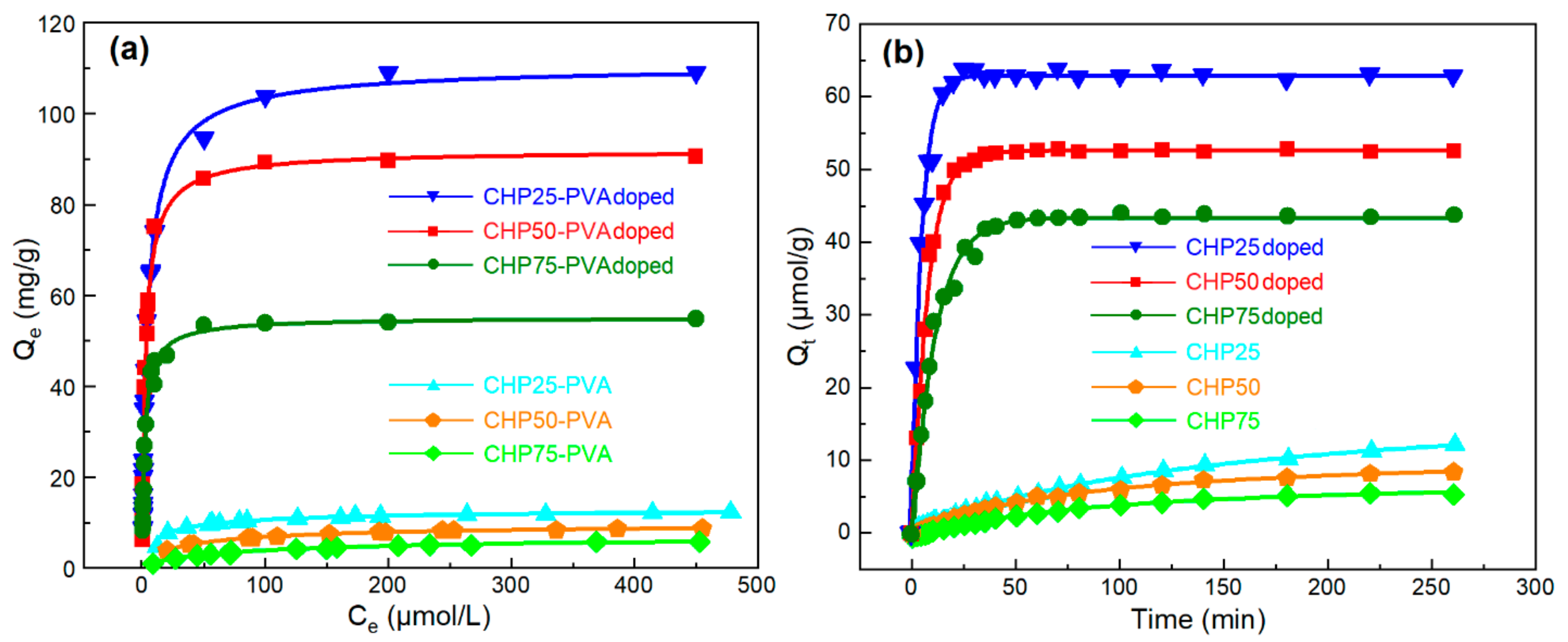
MB was used as a dye probe to study the adsorption properties of materials under equilibrium and kinetic conditions. The ternary sorbents in their film form adsorbed MB according to the isotherm profiles shown in
Figure 5a, where MB solutions with greater decolorization correspond to samples with greater dye uptake.
Figure 5a shows that composite samples with variable chitosan content had greater adsorption capacity, as follows: CHP75–PVA > CHP50–PVA > CHP25–PVA.
The monolayer uptake capacity (
) can be calculated according to the type of adsorption isotherm (Langmuir, Freundlich and Sips) models in
Supplementary Materials Table S3. The best-fit results are shown by the Sips isotherm model with the correlation coefficients (
) that lie closer to unity (
Table 2). The SA of the materials was estimated from the best-fit adsorption parameters by the Sips model (
Table 2).
Table 2 lists a comparison of the adsorption capacities of single components (CHT and PANI) and binary composite CHT/PANI materials [
29,
46], where the
values for ternary films are slightly higher than the
values for the pristine components.
A clear trend for the MB adsorption data is observed, whereby the values for
and SA of the composites increase as the chitosan content increases. This trend can be explained by the role of cross-linking and the favourable MB dye adsorption properties of chitosan. The increase in the SA of the adsorbent parallels the trend in results for the estimated density values. As seen from
Supplementary Materials Table S4, the density of the composite gradually decreases with increasing chitosan content. The decreasing trend in composite density may add further evidence that the greater porosity noted for CHP75–PVA provides an account of its greater SA and dye uptake properties, as compared with the other ternary composites.
By contrast, the theoretical densities listed in
Table S4 have greater values, as compared with the experimental results. The density results indicate that the ternary composite film is not just a physical mixture but also forms a uniquely structured composite with a well-defined pore structure.
Dye Uptake Kinetics
The kinetic uptake of MB by the PANI-based composites is shown in
Figure 5b. The overall trend reveals that rapid adsorption occurs during the first 25 min of the isotherm profile, where reduced uptake occurs thereafter. The greater adsorption during the first stage is accounted for by the presence of more accessible surface sites, whereas subsequent uptake is reduced due to saturation of the remaining surface and/or uptake at micropore domains where slower diffusion occurs.
The kinetic profiles are well-described by the pseudo-second order (PSO) model (cf.
Figure S4 in Supplementary Materials), in agreement with correlation coefficients (
) near unity. As shown in
Table 3, the general trends for the maximum kinetic uptake (
) are paralleled by the trends for the equilibrium uptake, where greater adsorption occurs as the mass fraction of chitosan in the composite increases.
The adsorption capacities of pristine components at pseudo-equilibrium conditions from the kinetic isotherms follow similar trends as the isotherms at equilibrium conditions. Pristine chitosan displays relatively poor adsorption with respect to its binary composites. In a study reported by Mohamed et al. [
29], a binary composite of CHT and PANI showed similar adsorptive capacity to those prepared herein. The values of
for the composites (CHP25–CHP50–CHP75) gradually increase in a parallel fashion, similar to the adsorption capacities listed in
Table 3.
The trends for the equilibrium and kinetic uptake of MB revealed several effects: greater cross-linking and chitosan content increased the textural properties; diffusion effects in a pore system were reduced as the contact time of the adsorbent-adsorbate system increased.
3.5.2. Fluorescein (FL) Adsorption
Equilibrium Dye Uptake
PANI-based composites display favourable adsorption properties toward MB, a cationic dye that is suitable for probing heteroatom sites with abundant electron density. Complementary adsorption isotherms were obtained using an anionic dye (fluorescein; FL) to assess the presence of active sites with Lewis acid character in the composite materials. Ternary PANI-based films were prepared in their base-neutralized form that were initially isolated as the emeraldine base (EB) form. Thus, it was of particular interest to acid-dope the composites to assess whether protonation would contribute enhanced dye adsorption of anions. The contribution of electrostatic effects between acid-doped PANI was hypothesized to result in greater dye uptake of FL relative to the undoped composite. The effect of acid doping was confirmed by visual observation, where fluorescein at variable concentration (up to 150 µM; samples 501–509) underwent complete decolorization (cf.
Figure S5 in Supplementary Materials).
Furthermore, the undoped (base-neutralized) films were also tested. The comparative chart depicts the FL uptake by the doped films (indexed as “doped”) versus undoped samples at equilibrium conditions—shown in
Figure 6a. Accordingly, the corresponding surface area of the sorbent was estimated from the results in
Table 4.
As shown in
Table 2 and
Table 4, there was a reversal in the trend for FL adsorption, as compared to that for MB adsorption. A lower chitosan content led to a greater uptake of FL, in accordance with the higher PANI content for samples with low-chitosan content. This indicates that the greater number of positively charged amine sites attract negatively charged dye molecules. However, both PANI and chitosan are susceptible to acid doping, where similar trends for uptake of MB may not apply, as noted for fluorescein. On the other hand, undoped PANI-based species reveal higher uptake of a MB cation rather than a FL anion, where a maximum 7-fold difference is noted in
Table 2 and
Table 4, where CHP25 displayed the highest FL uptake in either its doped and undoped forms. In contrast, the undoped species display ca. 10-fold lower
values versus the doped composites. By contrast, pristine chitosan has negligible adsorption, in agreement with other reports [
29,
35]. Regarding the adsorptive properties towards FL with pristine PANI, no relevant literature data were found, revealing a knowledge gap for such dye systems in this field of polymer adsorbents. However, other reports [
47,
48] reveal that undoped PANI favours adsorption of cationic dyes over anionic dyes, in parallel agreement with the trends reported herein.
Kinetic Dye Uptake
Figure 6b outlines the uptake of FL by the PANI–CHT binary powders at kinetic conditions. While these composites do not contain PVA, the general trend shows a similar correspondence with the isotherm results obtained at equilibrium conditions.
Table 5 shows kinetic parameters of the doped powders versus undoped binary composites. In the case of doped composites, the PFO model was tested, whereas the PSO kinetic model was found to be more suitable for describing the kinetic profiles of the undoped composite materials.
At a glance, the kinetic adsorption models indicate that the acid-doped samples favour physisorption (PFO), while the undoped samples obey the chemisorption mechanism (PSO) [
49]. However, the intermolecular interactions are stronger in the case of doped samples (due to electrostatic forces) and should facilitate chemisorption. The origin of this discrepancy is understood since the adsorption mechanism cannot be determined solely by the fitting results. The adsorption process is far more complex and altered by diffusion effects of the solute into the adsorbent.
3.6. Water Vapour Adsorption
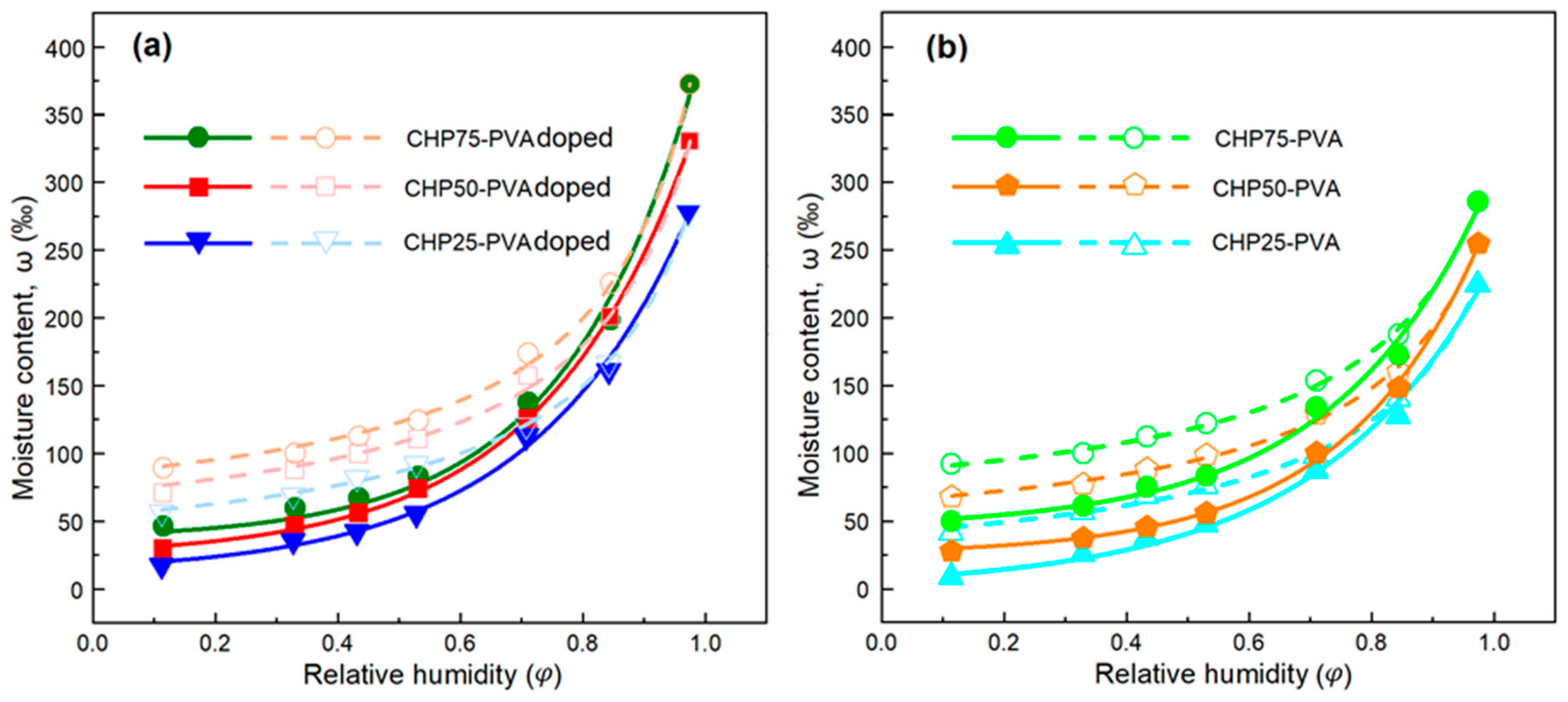
PANI-based films have hydrophilic character that should be sensitive towards ambient humidity. The sample weight gain of film varies as the relative humidity (RH) increased, according to the presence of different inorganic salt solutions. The equilibrium vapour pressure (RH) over the salt solution depends on the nature of salt, resulting in variable RH values for different salt solutions, as described elsewhere [
37]. The saturated salts solutions were used to cover a range of RH values. The samples were initially dried prior to exposure to an RH vapour of 11% over a LiCl solution. After 24 h of equilibration, the sample was removed and equilibrated at the next incremental RH, along with the change in sample weight, until the maximal value was achieved. The samples were likewise exposed to lower RH values in descending order to establish the desorption profile over a range of humidity values from 11% to 97% (
Figure 7).
Equation (9) represents the corresponding exponential correlation of the moisture adsorption by films.
is the moisture content in %; and are parameters of the exponential function; and is a relative humidity expressed in non-dimensional relative units (not as % values).
The moisture desorption law somewhat differs from the adsorption relation since it is represented by the reciprocal function denoted by Equation (10).
The terms, and , have similar meaning as in Equation (9); where and are non-dimensional parameters of the reciprocal equation.
Supplementary Materials Table S6 represents parameters for the fitting curves and the moisture content
at the highest RH (97%), depending on the formulation of the composite films. The values of the free parameters are similar, where the approximate magnitude ranges, as follows:
and
for the adsorption process. By comparison,
and
is always negative (≈ −0.02) for the desorption process. This implies that the moisture adsorption–desorption mechanism for all of the films are similar. On the other hand, the quantity of adsorbed water vapour increases as the chitosan content increases that parallels the trend noted for the MB adsorption. This can be accounted for by increasing the number of active sites (surface area) of the sorbent, where an increased chitosan content yields a more branched and cross-linked structure. Doped films adsorb ca. 30 to 100% more moisture, relative to the undoped analogues. This result is in good correspondence with the trends in FL adsorption herein. As the water molecules possess partial negative electrostatic charge on oxygen, adsorption occurs at the positively charged nitrogen of PANI for its emeraldine salt form.
3.7. Electrical Conductivity
Electrical conductivity of samples was estimated from the I–V curve results that illustrate the relationship between the current and the applied voltage through the sample. PANI-based samples in their film form were produced with two types of PANI: EB and ES. The EB form resulted in non-conductive films, whereas ES showed a weak conductivity at low current values. The main conductive component in these films is the PANI ES fraction that has been widely reported [
14]. By contrast, PANI in its EB form is a semiconductor, while the ES form of PANI is conductive [
50,
51]. In this work, the EB form of PANI was non-conductive due to the high content of PVA, a non-conductive polymer used in ternary film preparation. The presence of PVA led to greater electrical resistance of the film. A conductive polymer should reach a certain value of the “percolation threshold” [
52] in order to impart conductivity. For a significant quantity of non-conductive polymer, this threshold is also high. Thus, only small electrical currents can pass through ES samples. However, altering the PVA content in the composites led to a marked change in conductivity, as shown in
Figure 8.
When the PVA content was reduced by 20%, the conductivity showed a 100-fold improvement. On the other hand, when the PVA content decreased, the dependence appeared to be more linear. For ideal cases, the current is described by Equation (11):
is the current in Amps and
is the tension measured in Volts.
is a constant referred to as the conductance, where the magnitude of this physical quantity is the inverse of resistance, where
and
. When a material exhibits a non-linear behaviour for
, Equation (11) transforms to the power function (cf. Equation (12)):
is a non-linear factor, where
Supplementary Materials Table S7 shows non-linear factors together with the correlation coefficients
for each curve. In case (a), the non-linear factor is significant and varies from 1.3 to 2.4; whereas for case (b), the I–V curve is subject to a linear behaviour with
values between 1.0 and 1.2. This suggests that with a lower PVA content, the I–V dependence becomes almost linear and is typical for conductors such as metals. When the PVA content increases, the line significantly deviates from the linear form, where such behaviour is typical for semiconductors such as diodes and thyristors. Comparing the current flow through the samples with various CHT content, it is noted that higher content of chitosan (with lower content of PANI) resulted in decreased conductivity
, as listed in
Table 6. This concurs with the non-conductive nature of chitosan and the conducting properties of PANI.
3.8. Mechanical Properties
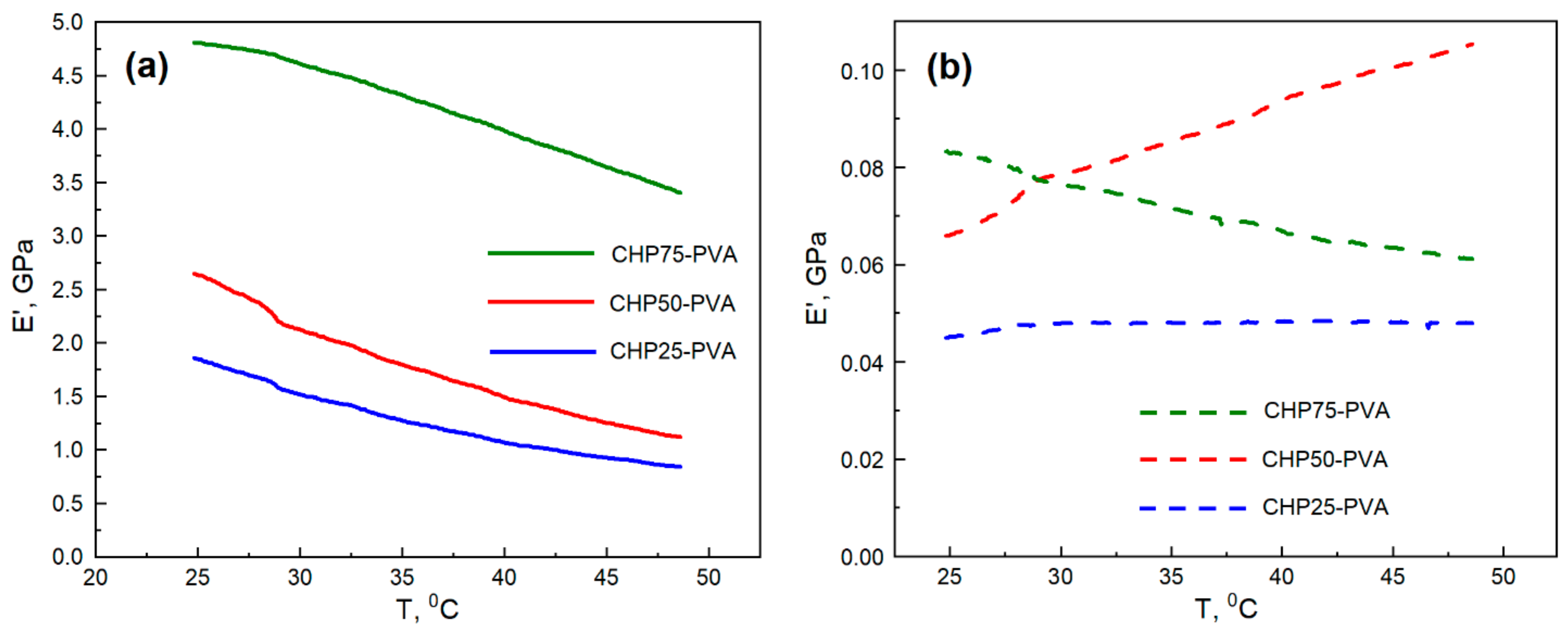
Mechanical properties of the composite materials were measured using Dynamic Mechanical Analysis (DMA). DMA is a powerful technique which allows for determination of the viscoelastic behaviour of a sample under variable temperature conditions. The general ability to resist the deformation is measured by a complex modulus that consists of the Young’s storage and loss moduli (E’ and E’’), respectively. The storage modulus (E’) is a measure of elastic behaviour of a material, and provides a measure of the stored energy that is responsible for the elastic portion. The loss modulus (E’’) is responsible for the viscous portion in the material and measures the energy dissipated during the sinusoidal deformation. Both loss and storage moduli tend to decrease with increased temperature, where less force is required to deform the sample. However, it is the storage modulus that has direct relation to the material stiffness (higher E’—higher stiffness). In contrast, the loss modulus is not directly proportional to the material stiffness. When the material reaches its glass transition temperature (
), the molecular friction increases and dissipates the applied force. At the
value of the material, the loss modulus increases while the storage modulus rapidly declines (as the dissipated energy surpasses the stored energy). This can be observed for the sample CHP75–PVA (cf.
Figure 9a and
Figure S6, Ia).
Figure 9 illustrates the DMA properties of the CHP–PVA films, based on their chitosan content, and in their dry or wet states. The general trend showed the elastic modulus E’ increased (with an increase in the material stiffness) as the chitosan content was augmented. This may imply that when the chitosan sample content increased, a more cross-linked network structure is created that results in the so-called “pillaring effect” [
53]. As the sample is exposed to moisture, the trends in the change of the storage modulus become altered. For example, the CHP75–PVA composite samples decreased their storage modulus, while the CHP50–PVA composites showed an increase in storage modulus, while CHP25–PVA composites have a relatively constant storage modulus. Such behaviour can be explained when water is imbibed within the sample structure it diminishes its stiffness, resulting in the storage of more energy and an increase in the elastic modulus. The CHP50–PVA composite sample displayed similar behaviour in its wet state, where both the loss and storage moduli increased with elevated temperature.
Another interesting observation relates to the maxima in the loss modulus graphs, as shown in
Figure S6 (I). The sharp energy dissipation emphasizes that the sample reached its respective
value. For instance, the
value for CHP50–PVA (dry state, one sample) and CHP75–PVA (wet state, two samples) were 41 °C and (27 °C and 31 °C), respectively. The glass transitions can be seen for both E′ and tanδ (tan delta) graphs, as shown in
Figure S6 (II). The tan delta, or the damping factor, is measured as the ratio of E″/E′ and its trends are depicted in
Figure S6, Section II. In general, the loss tangent factor is inversely proportional to the storage modulus. For this reason, CHP25–PVA had the lowest E’ (and stiffness) and the highest tan delta (cf.
Figure 9a, and
Figure S6, IIa in the Supplementary Materials).
Regardless of the behaviour of the wet composite samples at elevated temperature, the room temperature storage modulus had a similar performance to the dry samples, as shown in
Figure 9b. For example, the storage modulus trend was CHP75–PVA > CHP50–PVA > CHP25–PVA. The loss tangent factor shows a reverse trend, as follows: CHP75–PVA < CHP50–PVA < CHP25–PVA. It is also noteworthy that the loss and storage moduli for wet composite samples were almost two orders of magnitude less than that for dry composites. This comparison suggests that the rigidity dropped by almost 100-fold when the samples became hydrated.
4. Conclusions
Several unique ternary PANI–CHT–PVA composites were prepared via a simple and low-cost synthetic method that employed noncovalent cross-linking and physical blending. NMR and IR spectroscopy confirmed the presence of the components, where it was inferred that cross-linking occurs between CHT and PANI via hydrogen bonding, while PVA forms weak nonspecific interactions. TGA profiles of the composites displayed a unique thermal event that was absent for the pristine components, in agreement with the formation of polymer composites. DSC showed a broad endotherm region related to the water sorption at micropore and surface sites, where the effect was more pronounced at higher CHT content. Cationic and anionic dye adsorption showed a reversal of trends for dye uptake by the composites: MB uptake increased as the chitosan content of the composites increased, whereas a decreased adsorption capacity was shown for FL uptake. Acid-doped films showed greater FL uptake, in comparison with their undoped analogues. Water swelling for CHP75–PVA samples was the highest, where the trend for incremental swelling revealed by DSC profiles parallel the MB uptake results. The acid-doped samples showed a clear incremental trend in conductivity with decreased weight fraction of chitosan. Reducing the incorporation of the non-conductive PVA (by 20 wt.%) resulted in ca. 100-fold increase in the conductivity (and in some cases significantly reduced swelling), whereas the undoped samples are generally non-conductive. Mechanical properties reveal that the highest modulus for the composites with greater chitosan content concur with the trends in solvent swelling and MB adsorption. On the other hand, the storage modulus of wet samples dramatically decreased in comparison to their dry analogues.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}